Low-Density Polybutylene Terephthalate Foams with Enhanced Compressive Strength via a Reactive-Extrusion Process


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
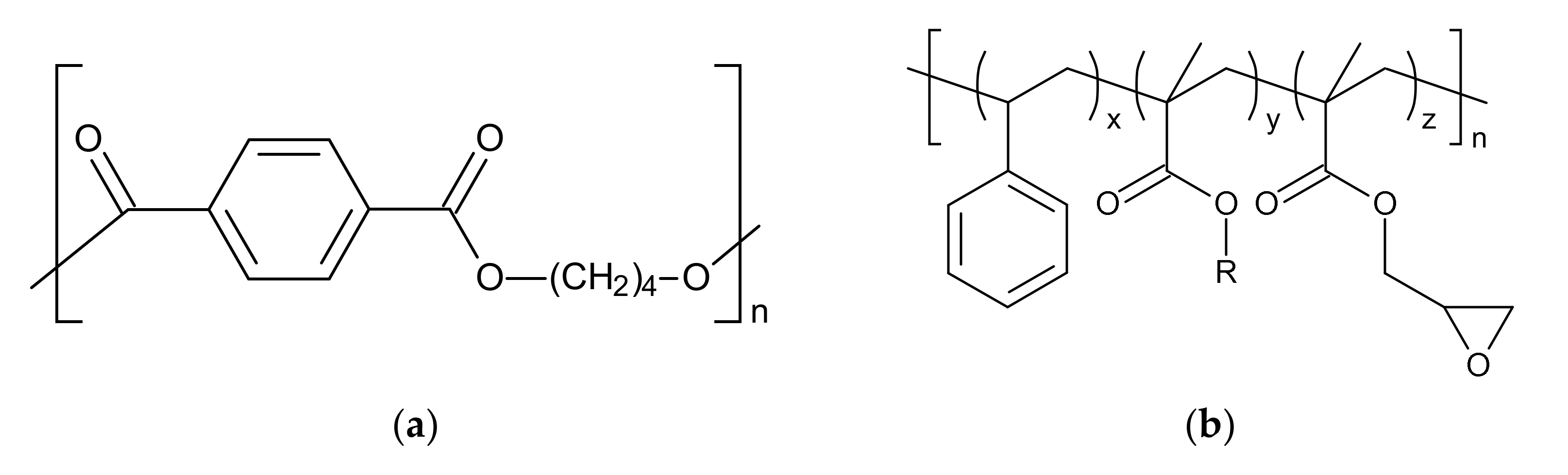
2.1. Materials
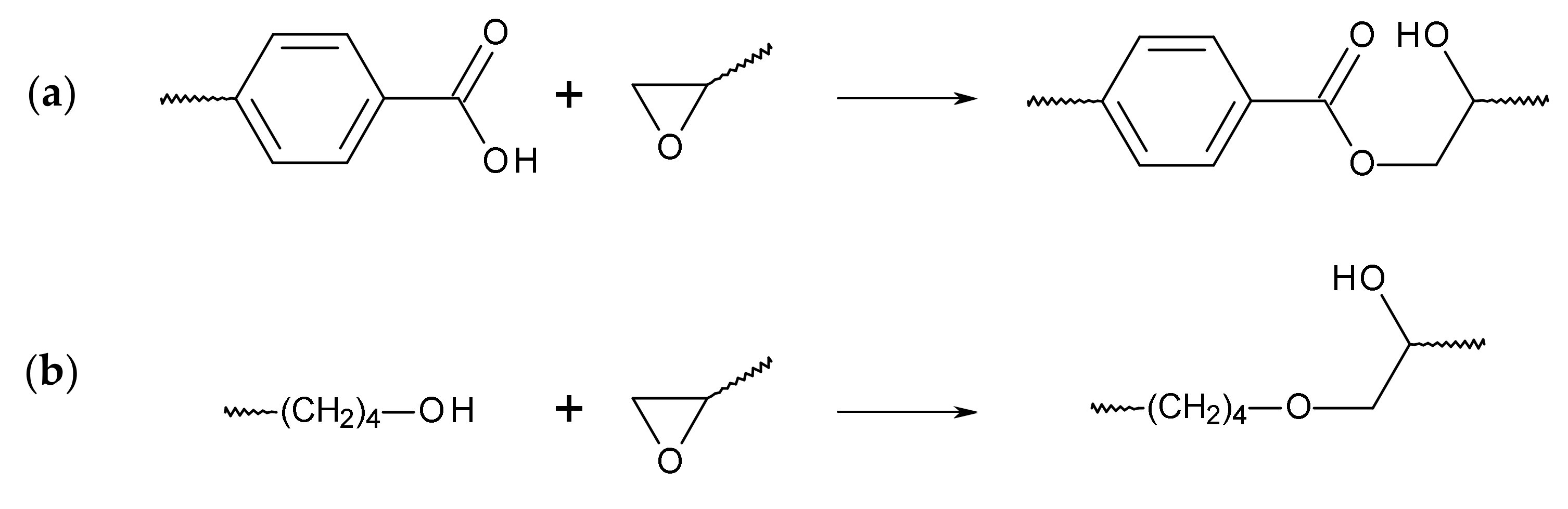
2.2. Reactive Foam Extrusion Process
2.3. Rheological Analysis
2.4. Foam Characterization
2.4.1. Foam Density
2.4.2. Foam Morphology
2.4.3. Compressive Properties
2.4.4. Degree of Crystallinity
2.4.5. Open Cell Content
3. Results and Discussions
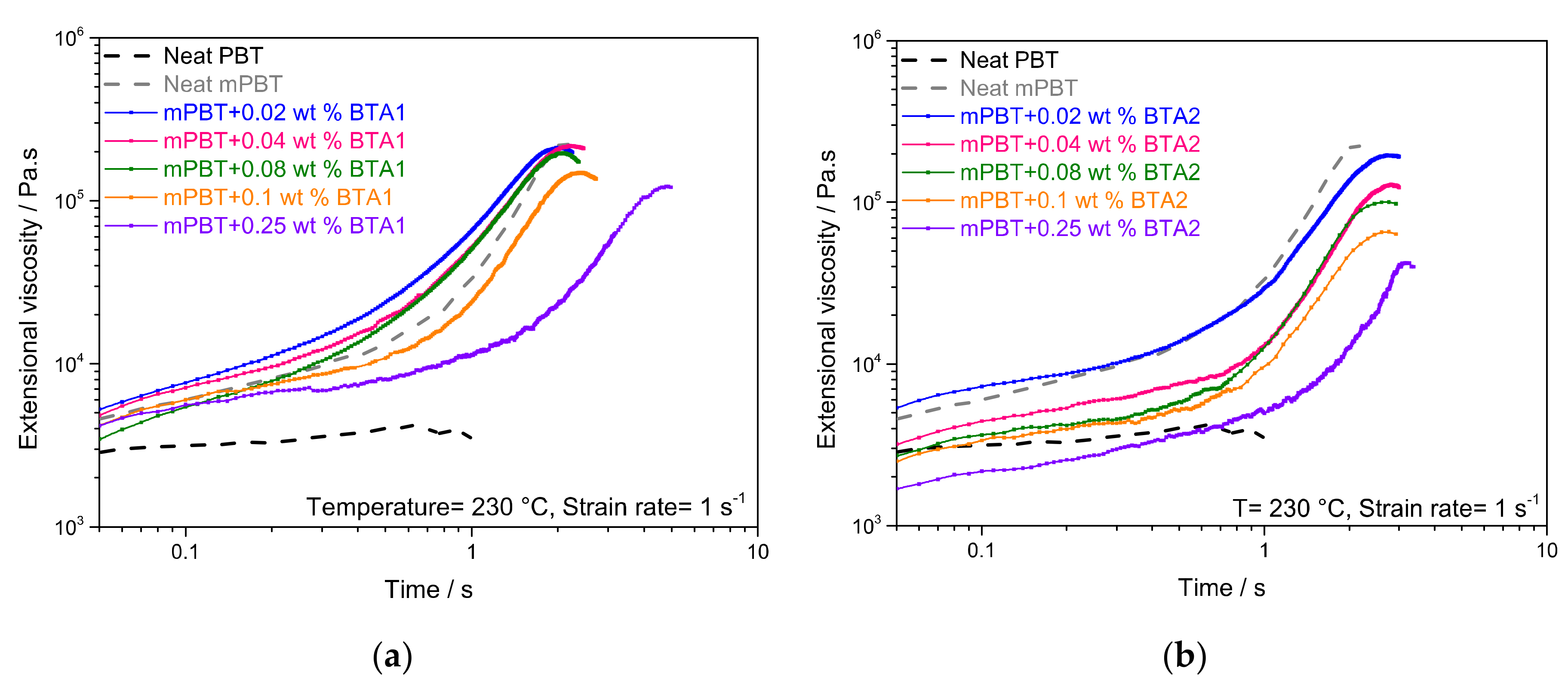
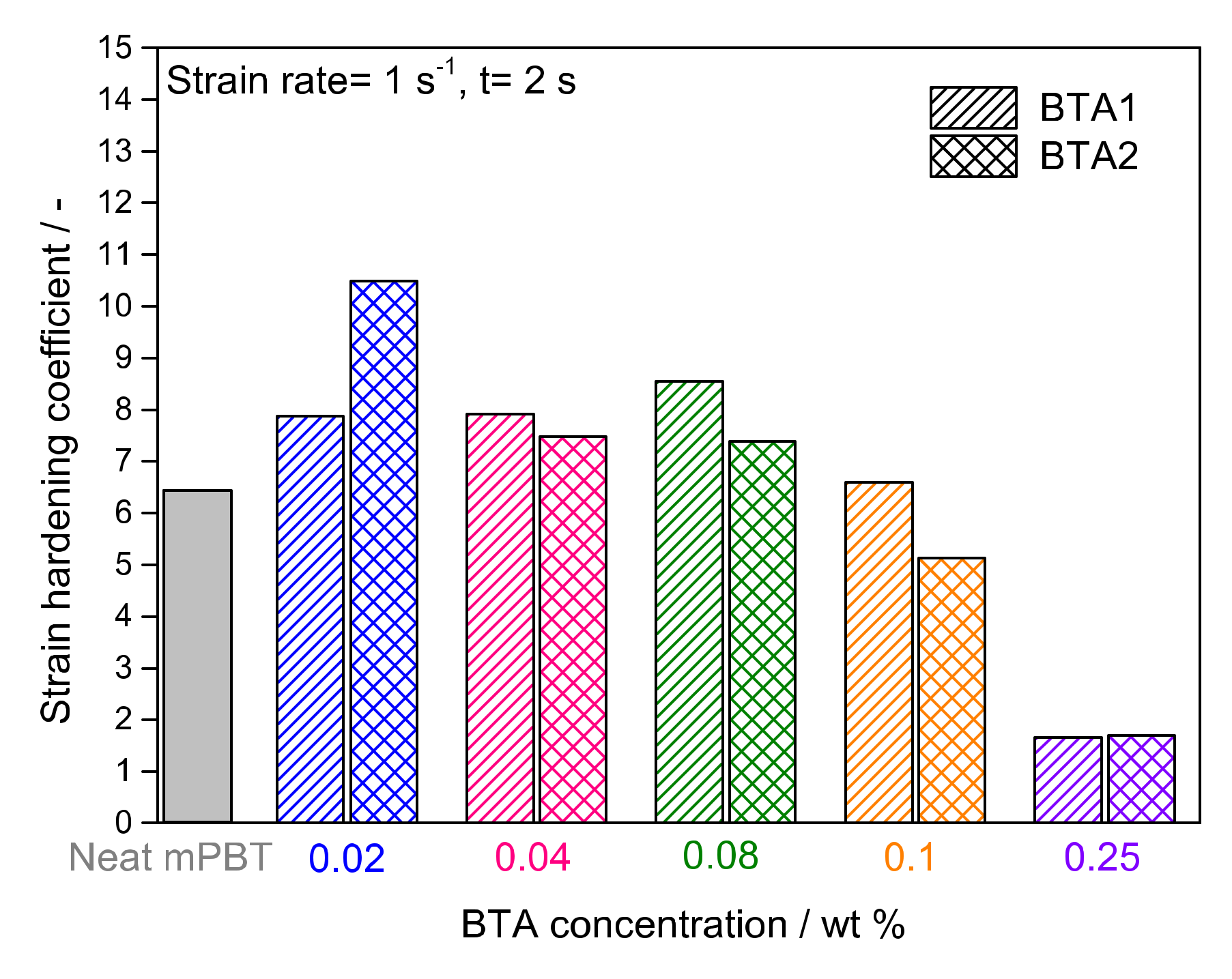
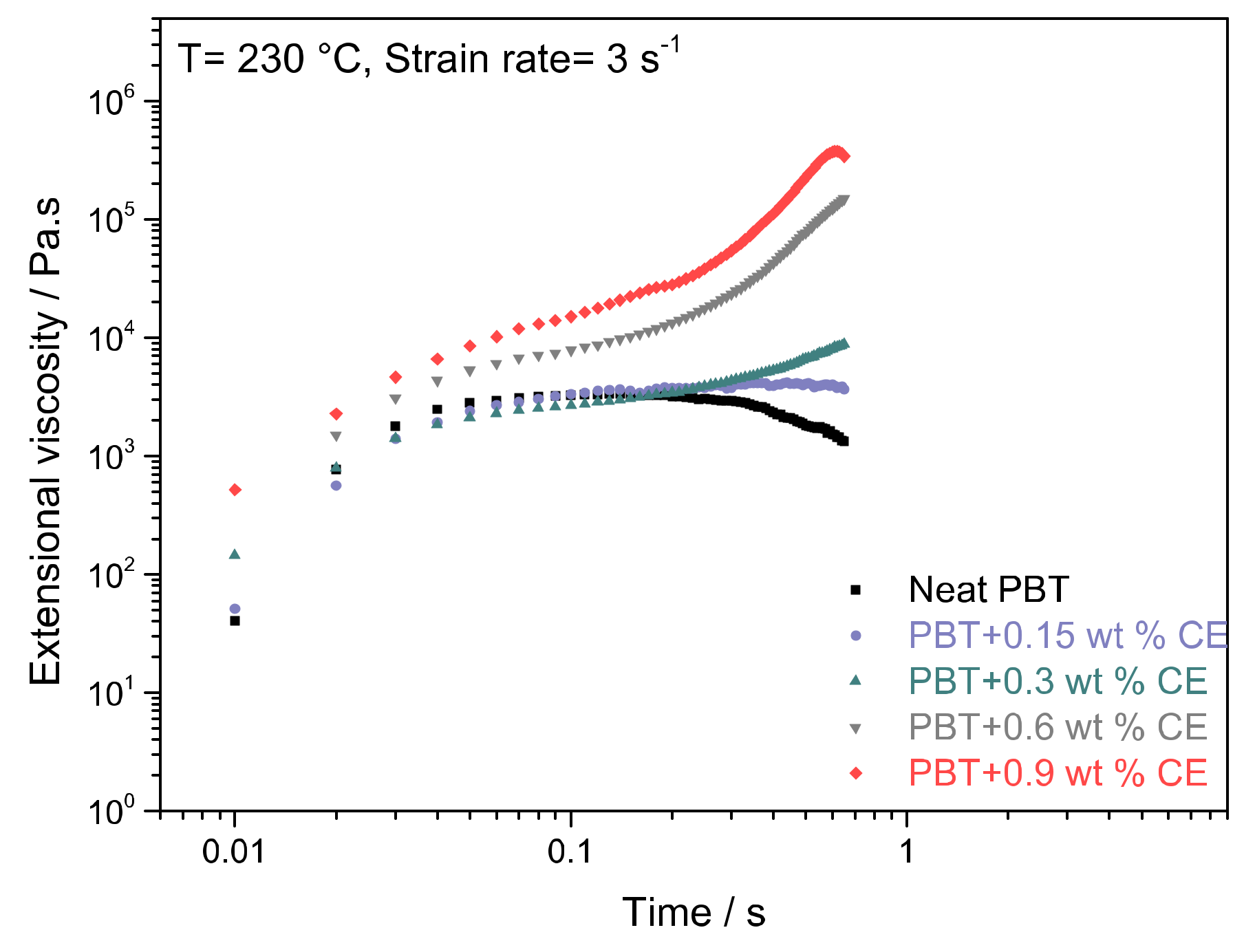
3.1. Influence of BTAs on Extensional Viscosity of mPBT
3.2. Optimization Study of the Process Parameters
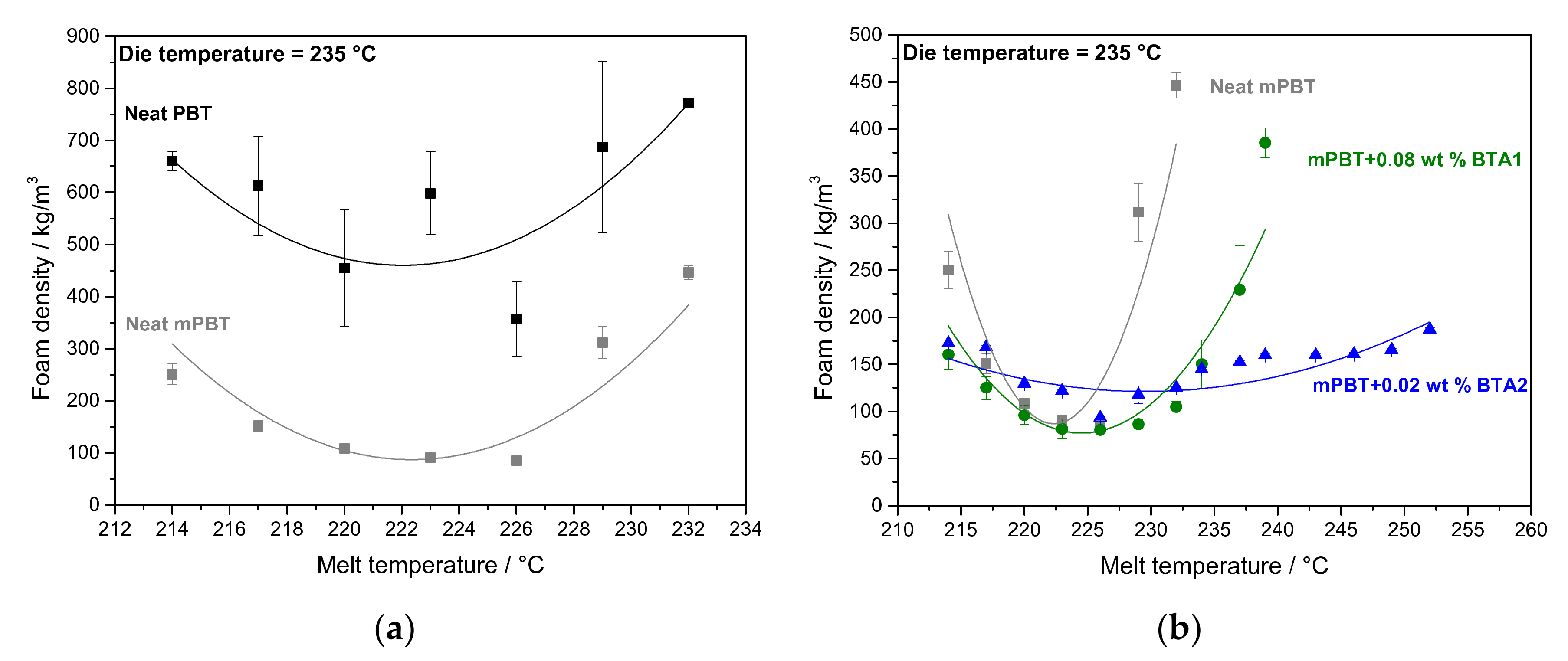
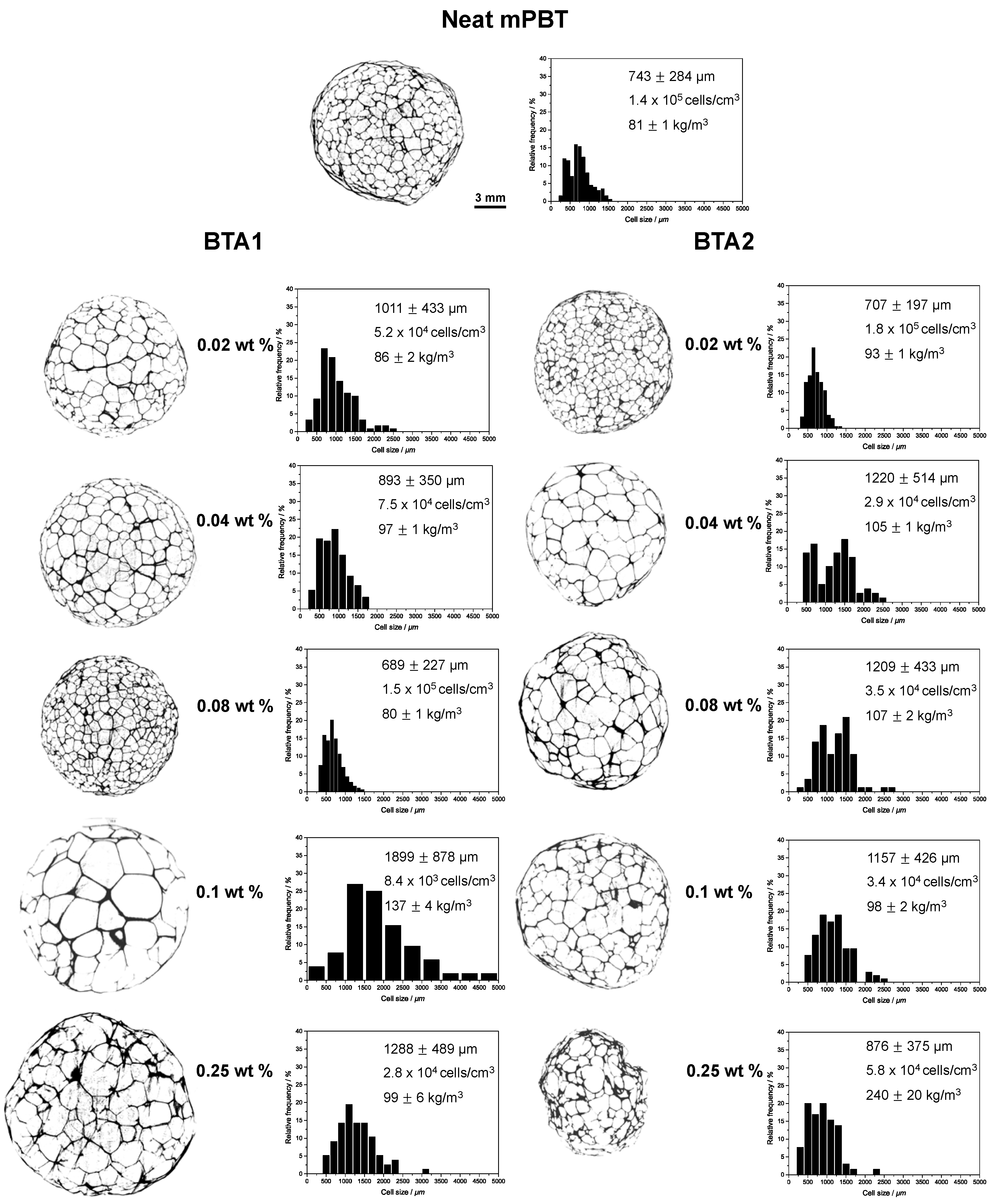
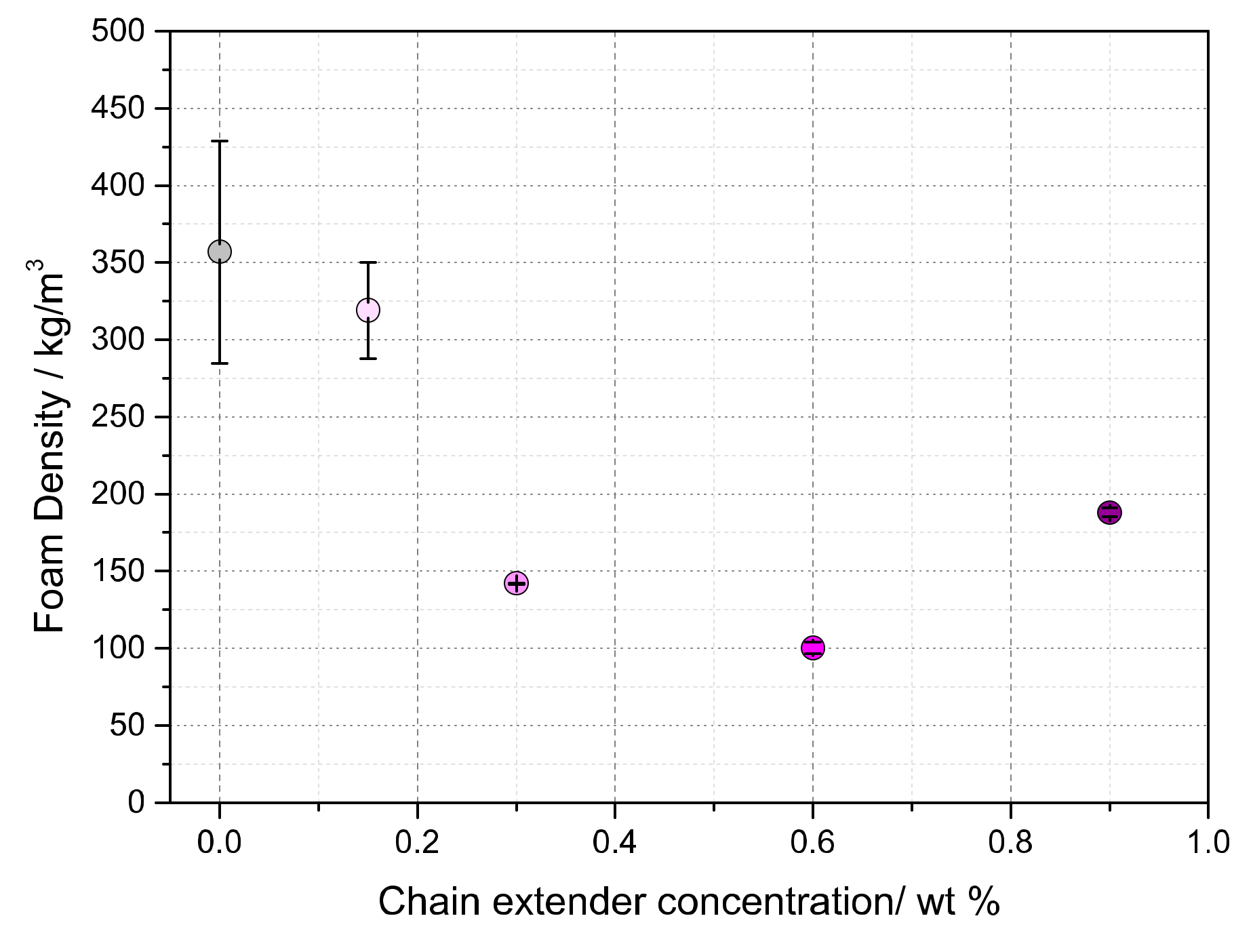
3.3. Influence of BTAs on the Density and Morphology of Foams
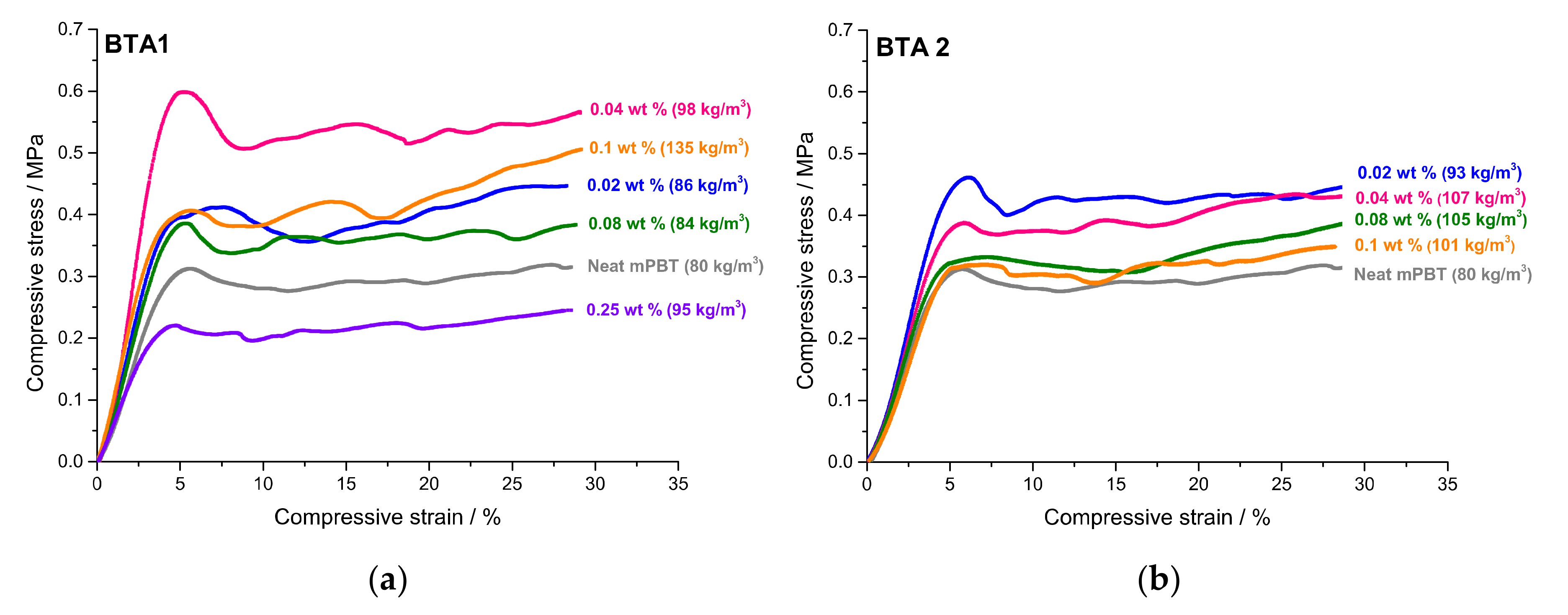
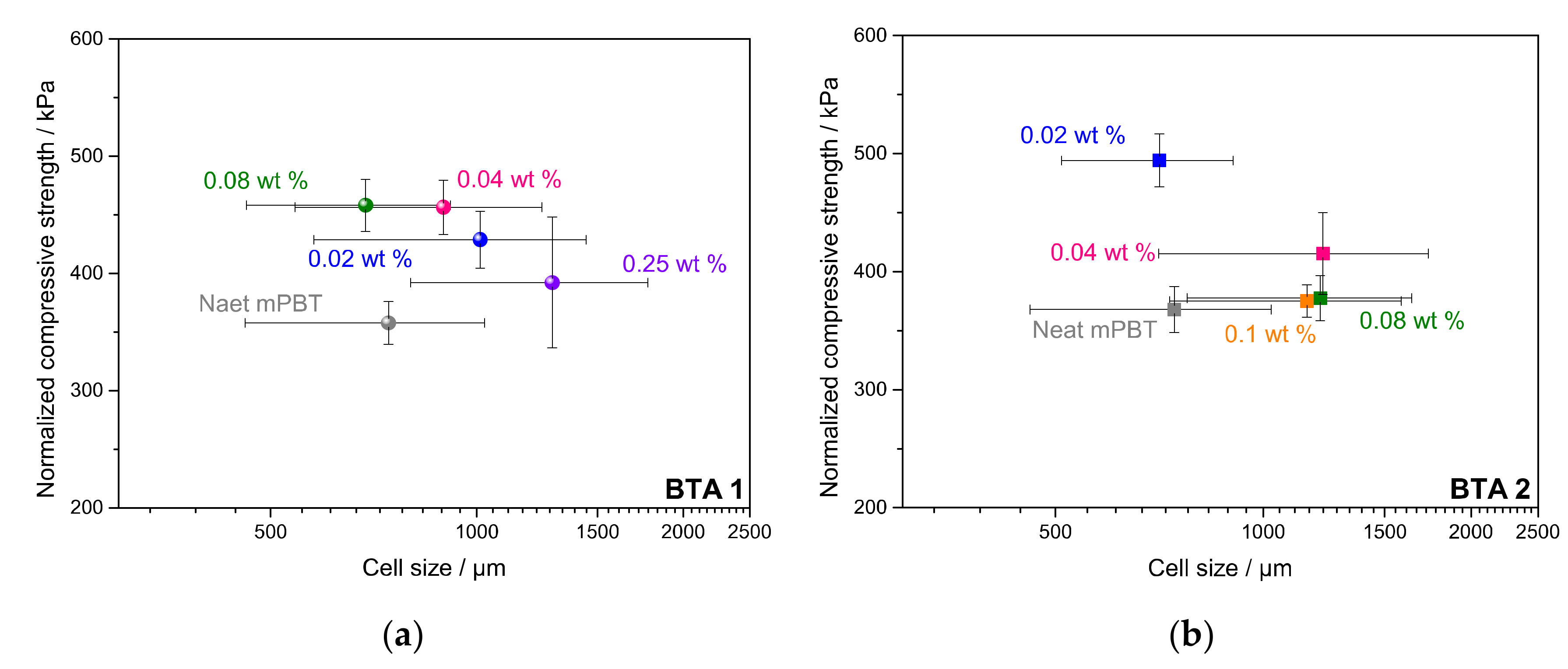
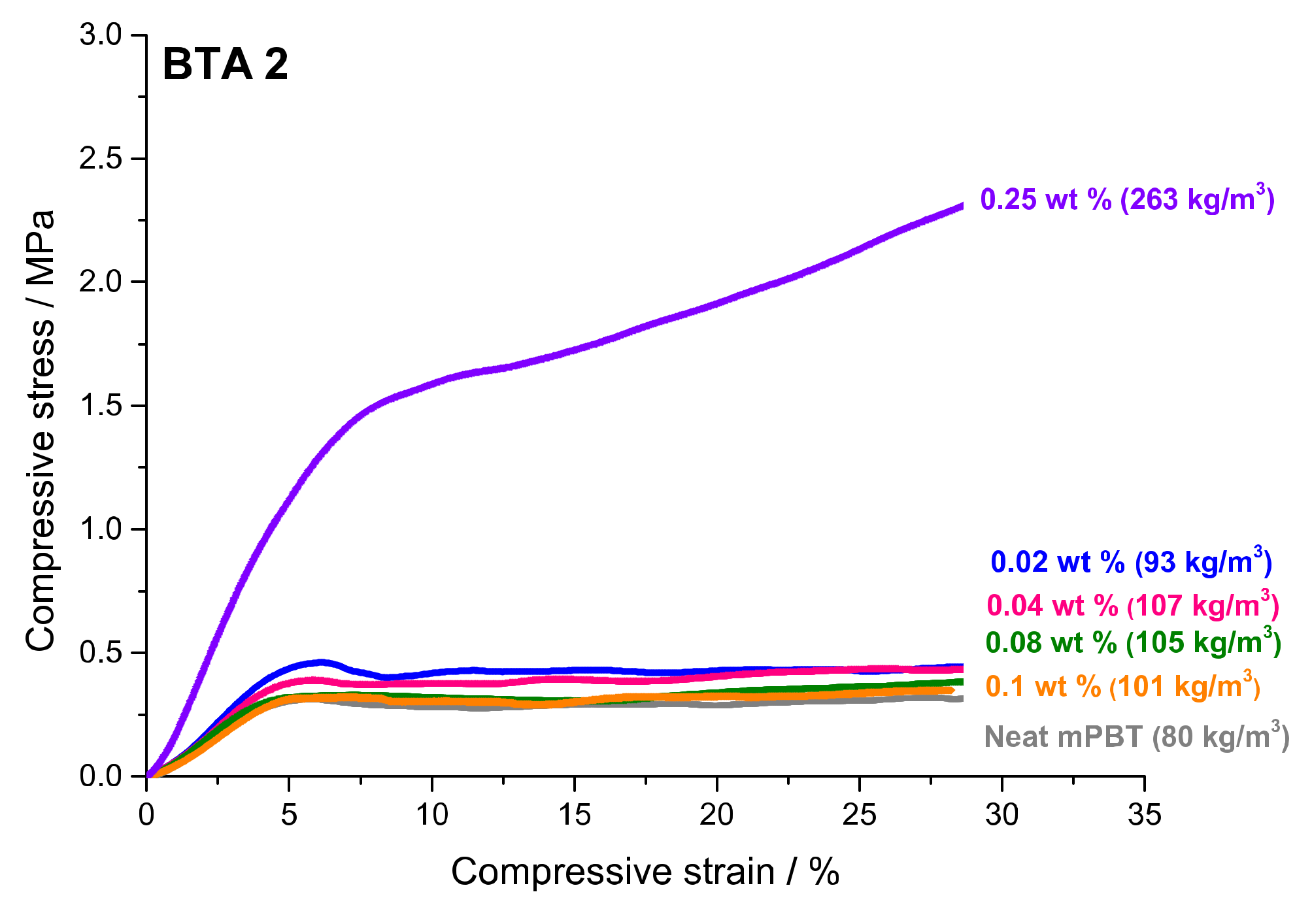
3.4. Influence of BTAs on Compression Strength and Other Properties of Foams
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A






References
- Wood, L. Global Polyethylene Terephthalate (PET) Foam Market Research Report 2016-2021—Analysis of Wind Energy, Transportation, Marine, Packaging, Building & Construction & Others; Research and Markets: Dublin, UK, 2016. [Google Scholar]
- Köppl, T.; Raps, D.; Altstädt, V. E-PBT—Bead foaming of poly(butylene terephthalate) by underwater pelletizing. J. Cell. Plast. 2014, 50, 475–487. [Google Scholar] [CrossRef]
- Mörl, M.; Steinlein, C.; Kreger, K.; Schmidt, H.-W.; Altstädt, V. Improved compression properties of polypropylene extrusion foams by supramolecular additives. J. Cell. Plast. 2018, 54, 483–498. [Google Scholar] [CrossRef]
- Rosato, D.V. Extruding Plastics; Springer US: Boston, MA, USA, 1998; ISBN 978-0-412-82810-2. [Google Scholar]
- Lee, S.-T.; Park, C.B. Foam Extrusion: Principles and Practice, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2014; ISBN 9781439898598. [Google Scholar]
- Standau, T.; Hädelt, B.; Schreier, P.; Altstädt, V. Development of a Bead Foam from an Engineering Polymer with Addition of Chain Extender: Expanded Polybutylene Terephthalate. Ind. Eng. Chem. Res. 2018, 57, 17170–17176. [Google Scholar] [CrossRef]
- Romani, F.; Corrieri, R.; Braga, V.; Ciardelli, F. Monitoring the chemical crosslinking of propylene polymers through rheology. Polymer 2002, 43, 1115–1131. [Google Scholar] [CrossRef]
- Sugimoto, M.; Tanaka, T.; Masubuchi, Y.; Takimoto, J.-I.; Koyama, K. Effect of chain structure on the melt rheology of modified polypropylene. J. Appl. Polym. Sci. 1999, 73, 1493–1500. [Google Scholar] [CrossRef]
- Tian, J.; Yu, W.; Zhou, C. The preparation and rheology characterization of long chain branching polypropylene. Polymer 2006, 47, 7962–7969. [Google Scholar] [CrossRef]
- Gotsis, A.D.; Zeevenhoven, B.L.F.; Tsenoglou, C. Effect of long branches on the rheology of polypropylene. J. Rheol. 2004, 48, 895–914. [Google Scholar] [CrossRef]
- Zhai, W.; Wang, H.; Yu, J.; Dong, J.-Y.; He, J. Foaming behavior of isotactic polypropylene in supercritical CO2 influenced by phase morphology via chain grafting. Polymer 2008, 49, 3146–3156. [Google Scholar] [CrossRef]
- Bimestre, B.H.; Saron, C. Chain extension of poly (ethylene terephthalate) by reactive extrusion with secondary stabilizer. Mater. Res. 2012, 15, 467–472. [Google Scholar] [CrossRef]
- Bethke, C.; Goedderz, D.; Weber, L.; Standau, T.; Döring, M.; Altstädt, V. Improving the flame-retardant property of bottle-grade PET foam made by reactive foam extrusion. J. Appl. Polym. Sci. 2020, 137, e49042. [Google Scholar] [CrossRef] [Green Version]
- Jeong, B.J.; Xanthos, M. Reactive modification of PBT with applications in low density extrusion foaming. Polym. Eng. Sci. 2007, 47, 244–253. [Google Scholar] [CrossRef]
- Leslie, J.; Lane, C.; Grant, R. Melt Strength Improvement of PET. U.S. Patent US4176101A, 27 November 1979. [Google Scholar]
- Phobos, N. Process for the Continuous Production of High Molecular Weight Polyester Resin. European Patent EP0422282A1, 5 April 1995. [Google Scholar]
- Thomas, N.W.; Berardinelli, F.M.; Edelman, R. Polycarbodiimide Modification of Polyesters for Extrusion Applications. U.S. Patent US4071503A, 31 January 1978. [Google Scholar]
- Raffa, P.; Coltelli, M.-B.; Savi, S.; Bianchi, S.; Castelvetro, V. Chain extension and branching of poly(ethylene terephthalate) (PET) with di- and multifunctional epoxy or isocyanate additives: An experimental and modelling study. React. Funct. Polym. 2012, 72, 50–60. [Google Scholar] [CrossRef]
- Gallucci, R.R.; Nelson, L.H.; Shannon, T.G. Stabilization of Polyesters Using Epoxy Compounds in Combination with a Catalyst. U.S. Patent US5596049A, 21 January 1997. [Google Scholar]
- Köppl, T. Halogen-Free Flame-Retarded Polybutylene Terephthalate and its Processing into Polymer Foams. Ph.D. Thesis, University of Bayreuth, Bayreuth, Germany, December 2014. ISBN:978-3-941492-83-7. [Google Scholar]
- Guo, B.; Chan, C.-M. Chain extension of poly(butylene terephthalate) by reactive extrusion. J. Appl. Polym. Sci. 1999, 71, 1827–1834. [Google Scholar] [CrossRef]
- Bikiaris, D.; Karayannidis, G. Synthesis and characterisation of branched and partially crosslinked poly(ethylene terephthalate). Polym. Int. 2003, 52, 1230–1239. [Google Scholar] [CrossRef]
- Bikiaris, D.N.; Karayannidis, G.P. Chain extension of polyesters PET and PBT with two new diimidodiepoxides. II. J. Polym. Sci. Part A Polym. Chem. 1996, 34, 1337–1342. [Google Scholar] [CrossRef]
- Bikiaris, D.N.; Karayannidis, G.P. Chain extension of polyesters PET and PBT with N,N′-bis (glycidyl ester) pyromellitimides. I. J. Polym. Sci. Part A Polym. Chem. 1995, 33, 1705–1714. [Google Scholar] [CrossRef]
- Jeong, B.; Xanthos, M.; Seo, Y. Extrusion Foaming Behavior of PBT Resins. J. Cell. Plast. 2006, 42, 165–176. [Google Scholar] [CrossRef]
- Nofar, M.; Oğuz, H. Development of PBT/Recycled-PET Blends and the Influence of Using Chain Extender. J. Polym. Environ. 2019, 27, 1404–1417. [Google Scholar] [CrossRef]
- Song, K.; Li, W.; Eckert, J.O.; Wu, D.; Apfel, R.E. Generation of novel microstructures in rapidly foamed polybutylene terephthalate. J. Mater. Sci. 1999, 34, 5387–5395. [Google Scholar] [CrossRef]
- Klotzer, R.; Paul, D.; Seibig, B. Extrusion of microcellular foams and applications. In Proceedings of the SPE ANTEC‘97, Toronto, OT, Canada, 27 April–2 May 1997; pp. 2042–2045. [Google Scholar]
- Köppl, T.; Fischer, B.; Altstädt, V. Challenges and possibilities in foaming of semicrystalline poly(butylene terephthalate). In Proceedings of the Blowing Agents and Foaming Processes 2012 Conference Proceedings, Berlin, Germany, 8–9 May 2012; Shawbury, U., Ed.; Smithers Rapra Publishing: Berlin, Germany, 2012; pp. 175–180. [Google Scholar]
- Standau, T.; Hädelt, B.; Fafara, M.; Altstädt, V. Development of a bead foam based on the engineering polymer polybutylene terephthalate. In AIP Conference Proceedings; AIP Publishing LLC: Melville, NY, USA, 2019; Volume 2055, p. 060004. [Google Scholar]
- Yetgin, S.; Unal, H.; Mimaroglu, A. Influence of foam agent content and talc filler on the microcellular and mechanical properties of injection molded polypropylene and talc filled polypropylene composite foams. J. Cell. Plast. 2014, 50, 563–576. [Google Scholar] [CrossRef]
- Lee, Y.H.; Wang, K.H.; Park, C.B.; Sain, M. Effects of clay dispersion on the foam morphology of LDPE/clay nanocomposites. J. Appl. Polym. Sci. 2007, 103, 2129–2134. [Google Scholar] [CrossRef]
- Yousefzade, O.; Hemmati, F.; Garmabi, H.; Mahdavi, M. Assisted heterogeneous multinucleation and bubble growth in semicrystalline ethylene-vinyl acetate copolymer/expanded graphite nanocomposite foams: Control of morphology and viscoelastic properties. Express Polym. Lett. 2015, 9, 932–944. [Google Scholar] [CrossRef]
- Werner, P.; Verdejo, R.; Wöllecke, F.; Altstädt, V.; Sandler, J.K.W.; Shaffer, M.S.P. Carbon Nanofibers Allow Foaming of Semicrystalline Poly(ether ether ketone). Adv. Mater. 2005, 17, 2864–2869. [Google Scholar] [CrossRef]
- Mahdavi, M.; Yousefzade, O.; Garmabi, H. A simple method for preparation of microcellular PLA/calcium carbonate nanocomposite using super critical nitrogen as a blowing agent: Control of microstructure. Adv. Polym. Technol. 2018, 37, 3017–3026. [Google Scholar] [CrossRef]
- Barboza, A.C.R.N.; De Paoli, M.A. Polipropileno Carregado com Microesferas Ocas de Vidro (Glass BubblesTM): Obtenção de Espuma Sintática. Polímeros 2002, 12, 130–137. [Google Scholar] [CrossRef] [Green Version]
- García-Casas, I.; Montes, A.; Valor, D.; Pereyra, C.; Martínez de la Ossa, E.J. Foaming of Polycaprolactone and Its Impregnation with Quercetin Using Supercritical CO2. Polymers 2019, 11, 1390. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Wu, F.; Zhai, W.; Zheng, W. Effect of polytetrafluoroethylene on the foaming behaviors of linear polypropylene in continuous extrusion. J. Appl. Polym. Sci. 2013, 129, 2253–2260. [Google Scholar] [CrossRef]
- Rizvi, A.; Park, C.B.; Favis, B.D. Tuning viscoelastic and crystallization properties of polypropylene containing in-situ generated high aspect ratio polyethylene terephthalate fibrils. Polymer 2015, 68, 83–91. [Google Scholar] [CrossRef]
- Rizvi, A.; Park, C.B. Dispersed polypropylene fibrils improve the foaming ability of a polyethylene matrix. Polymer 2014, 55, 4199–4205. [Google Scholar] [CrossRef]
- Mohmeyer, N.; Müller, B.; Behrendt, N.; Hillenbrand, J.; Klaiber, M.; Zhang, X.; Smith, P.; Altstädt, V.; Sessler, G.M.; Schmidt, H.-W. Nucleation of isotactic polypropylene by triphenylamine-based trisamide derivatives and their influence on charge-storage properties. Polymer 2004, 45, 6655–6663. [Google Scholar] [CrossRef]
- Mohmeyer, N.; Behrendt, N.; Zhang, X.; Smith, P.; Altstädt, V.; Sessler, G.M.; Schmidt, H.-W. Additives to improve the electret properties of isotactic polypropylene. Polymer 2007, 48, 1612–1619. [Google Scholar] [CrossRef]
- Abraham, F.; Schmidt, H.-W. 1,3,5-Benzenetrisamide based nucleating agents for poly(vinylidene fluoride). Polymer 2010, 51, 913–921. [Google Scholar] [CrossRef]
- Kersch, M.; Schmidt, H.-W.; Altstädt, V. Influence of different beta-nucleating agents on the morphology of isotactic polypropylene and their toughening effectiveness. Polymer 2016, 98, 320–326. [Google Scholar] [CrossRef]
- Blomenhofer, M.; Ganzleben, S.; Hanft, D.; Schmidt, H.-W.; Kristiansen, M.; Smith, P.; Stoll, K.; Mäder, D.; Hoffmann, K. “Designer” Nucleating Agents for Polypropylene. Macromolecules 2005, 38, 3688–3695. [Google Scholar] [CrossRef]
- Aksit, M.; Klose, B.; Zhao, C.; Kreger, K.; Schmidt, H.-W.; Altstädt, V. Morphology control of extruded polystyrene foams with benzene-trisamide-based nucleating agents. J. Cell. Plast. 2019, 55, 249–261. [Google Scholar] [CrossRef]
- Kersch, M.; Pischke, L.; Schmidt, H.W.; Altstädt, V. Influence of trisamide-based additives on the morphological and mechanical properties of isotactic polypropylene. Polymer 2014, 55, 3227–3233. [Google Scholar] [CrossRef]
- Stumpf, M.; Spörrer, A.; Schmidt, H.-W.; Altstädt, V. Influence of supramolecular additives on foam morphology of injection-molded i-PP. J. Cell. Plast. 2011, 47, 519–534. [Google Scholar] [CrossRef]
- Aksit, M.; Zhao, C.; Klose, B.; Kreger, K.; Schmidt, H.; Altstädt, V. Extruded Polystyrene Foams with Enhanced Insulation and Mechanical Properties by a Benzene-Trisamide-Based Additive. Polymers 2019, 11, 268. [Google Scholar] [CrossRef] [Green Version]
- Richter, F.; Schmidt, H. Supramolecular Nucleating Agents for Poly(butylene terephthalate) Based on 1,3,5-Benzenetrisamides. Macromol. Mater. Eng. 2013, 298, 190–200. [Google Scholar] [CrossRef]
- BASF SE. Irgaclear ® XT 386; Product Brochure; BASF: Ludwigshafen, Germany, 2012; pp. 1–2. [Google Scholar]
- Schmidt, H.-W.; Blomenhofer, M.; Stoll, K.; Meier, H.-R. Resin Compositions Containing Amides as Nucleating Agents. Canadian Patent CA2514034C, 16 April 2013. [Google Scholar]
- Mäder, D.; Hoffmann, K.; Schmidt, H.-W. Beta Crystalline Polypropylenes. Canadian Patent CA2486940C, 8 March 2011. [Google Scholar]
- Schmidt, H.-W.; Smith, P.; Blomenhofer, M. Polypropylene Resin Compositions. U.S. Patent US7479515B2, 20 January 2009. [Google Scholar]
- BASF SE Ultradur® B 6550 Product Brochure. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwjlvMeehovqAhVji8MKHUgxBCYQFjACegQIARAB&url=https%3A%2F%2Fwww.basf.com%2Fglobal%2Fdocuments%2Fen%2Fproducts-and-industries%2Fcar-interior-ideal%2F2019%2FBASF_Ultradur_brochure.pdf&usg=AOvVaw3W9e-WjaKr84FCOaWeBPmF (accessed on 14 January 2020).
- BASF SE. JONCRYL® Acrylic Resins and Polymer Emulsions, Product Brochure; BASF: Ludwigshafen, Germany, 2014. [Google Scholar]
- Spitael, P.; Macosko, C.W. Strain hardening in polypropylenes and its role in extrusion foaming. Polym. Eng. Sci. 2004, 44, 2090–2100. [Google Scholar] [CrossRef]
- Gendron, R.; Daigneault, L.E. Rheology of Thermoplastic Foam Extrusion Process. In Foam Extrusion: Principles and Practice (Polymeric Foams); Lee, S.-T., Ed.; CRC Press Technomic Publishing: Florida, FL, USA, 2000; p. 35. ISBN 1566768799. [Google Scholar]
- Stadler, F.J.; Kaschta, J.; Münstedt, H.; Becker, F.; Buback, M. Influence of molar mass distribution and long-chain branching on strain hardening of low density polyethylene. Rheol. Acta 2009, 48, 479–490. [Google Scholar] [CrossRef]
- Wunderlich, B. Thermal Analysis of Polymeric Materials; Springer: Berlin/Heidelberg, Germany, 2005; ISBN 3-540-23629-5. [Google Scholar]
- Zepnik, S.; Kabasci, S.; Kopitzky, R.; Radusch, H.; Wodke, T. Extensional Flow Properties of Externally Plasticized Cellulose Acetate: Influence of Plasticizer Content. Polymers 2013, 5, 873–889. [Google Scholar] [CrossRef]
- Naguib, H.E.; Park, C.B.; Reichelt, N. Fundamental foaming mechanisms governing the volume expansion of extruded polypropylene foams. J. Appl. Polym. Sci. 2004, 91, 2661–2668. [Google Scholar] [CrossRef]
- Gibson, I.; Ashby, M.F. The mechanics of three-dimensional cellular materials. Proc. R. Soc. London A. Math. Phys. Sci. 1982, 382, 43–59. [Google Scholar]
- Chen, Y.; Das, R.; Battley, M. Effects of cell size and cell wall thickness variations on the stiffness of closed-cell foams. Int. J. Solids Struct. 2015, 52, 150–164. [Google Scholar] [CrossRef]
- Santo, L.; Bellisario, D.; Quadrini, F. Shape Memory Behavior of PET Foams. Polymers 2018, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Lyon, C.K.; Garrett, V.H.; Goldblatt, L.A. Solvent-blown rigid urethane foams from castor-based polyols. J. Am. Oil Chem. Soc. 1961, 38, 262–266. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Melt Temperature [°C] | Die Temperature [°C] | Screw Speed [rpm] | Throughput [kg/h] | s-CO2 Content [wt %] |
|---|---|---|---|---|
| 226 | 235 | 6–7 | 4 | 2 |
| Sample | Compressive Strength (CS) [kPa] | Normalized CS at Minimum Density of 80 kg/m3 [kPa] | Normalized CS at Average Density of 88 kg/m3 [kPa] | Normalized CS at Maximum Density of 99 kg/m3 [kPa] |
| Neat mPBT | 368.1 21.2 | 357.8 18.4 | 428.1 22.1 | 534.1 27.5 |
| 0.02 wt % BTA1 | 486.2 22.4 | 428.8 24.3 | 512.9 29.1 | 640.2 36.2 |
| 0.04 wt % BTA1 | 650.2 33.2 | 456.3 23.3 | 545.9 27.8 | 681.2 34.7 |
| 0.08 wt % BTA1 | 460.4 15.5 | 458.1 22.1 | 547.9 26.4 | 683.8 33.1 |
| 0.25 wt % BTA1 | 584.9 54.4 | 392.2 55.9 | 469.2 66.8 | 585.4 83.4 |
| Sample | Compressive Strength (CS) [kPa] | Normalized CS at Minimum Density of 80 kg/m3 [kPa] | Normalized CS at Average Density of 97 kg/m3 [kPa] | Normalized CS at Maximum Density of 107 kg/m3 [kPa] |
| Neat mPBT | 368.1 21.2 | 367.9 21.2 | 371.5 21.3 | 373.3 21.5 |
| 0.02 wt % BTA2 | 497.8 22.6 | 494.2 22.4 | 499.1 22.6 | 501.5 22.7 |
| 0.04 wt % BTA2 | 421.1 35.4 | 415.3 34.9 | 419.4 35.2 | 421.4 35.4 |
| 0.08 wt % BTA2 | 383.1 19.5 | 377.6 19.1 | 381.3 19.2 | 383.2 19.4 |
| 0.1 wt % BTA2 | 381.8 12.4 | 375.1 13.9 | 378.7 14.1 | 380.5 14.1 |
| Sample | Degree of Crystallinity [%] | Open Cell Content [%] |
|---|---|---|
| Neat mPBT | 32.4 0.3 | 42.2 0.8 |
| 0.08 wt % BTA1 | 28.8 1.4 | 42.3 0.5 |
| 0.02 wt % BTA2 | 30.1 1.1 | 29.9 1.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aksit, M.; Gröschel, S.; Kuhn, U.; Aksit, A.; Kreger, K.; Schmidt, H.-W.; Altstädt, V. Low-Density Polybutylene Terephthalate Foams with Enhanced Compressive Strength via a Reactive-Extrusion Process. Polymers 2020, 12, 2021. https://doi.org/10.3390/polym12092021
Aksit M, Gröschel S, Kuhn U, Aksit A, Kreger K, Schmidt H-W, Altstädt V. Low-Density Polybutylene Terephthalate Foams with Enhanced Compressive Strength via a Reactive-Extrusion Process. Polymers. 2020; 12(9):2021. https://doi.org/10.3390/polym12092021
Chicago/Turabian StyleAksit, Merve, Sebastian Gröschel, Ute Kuhn, Alper Aksit, Klaus Kreger, Hans-Werner Schmidt, and Volker Altstädt. 2020. "Low-Density Polybutylene Terephthalate Foams with Enhanced Compressive Strength via a Reactive-Extrusion Process" Polymers 12, no. 9: 2021. https://doi.org/10.3390/polym12092021
APA StyleAksit, M., Gröschel, S., Kuhn, U., Aksit, A., Kreger, K., Schmidt, H. -W., & Altstädt, V. (2020). Low-Density Polybutylene Terephthalate Foams with Enhanced Compressive Strength via a Reactive-Extrusion Process. Polymers, 12(9), 2021. https://doi.org/10.3390/polym12092021