
Protein-, (Poly)peptide-, and Amino Acid-Based Nanostructures Prepared via Polymerization-Induced Self-Assembly



Abstract
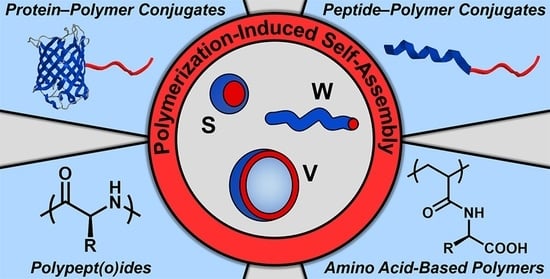
:
1. Introduction
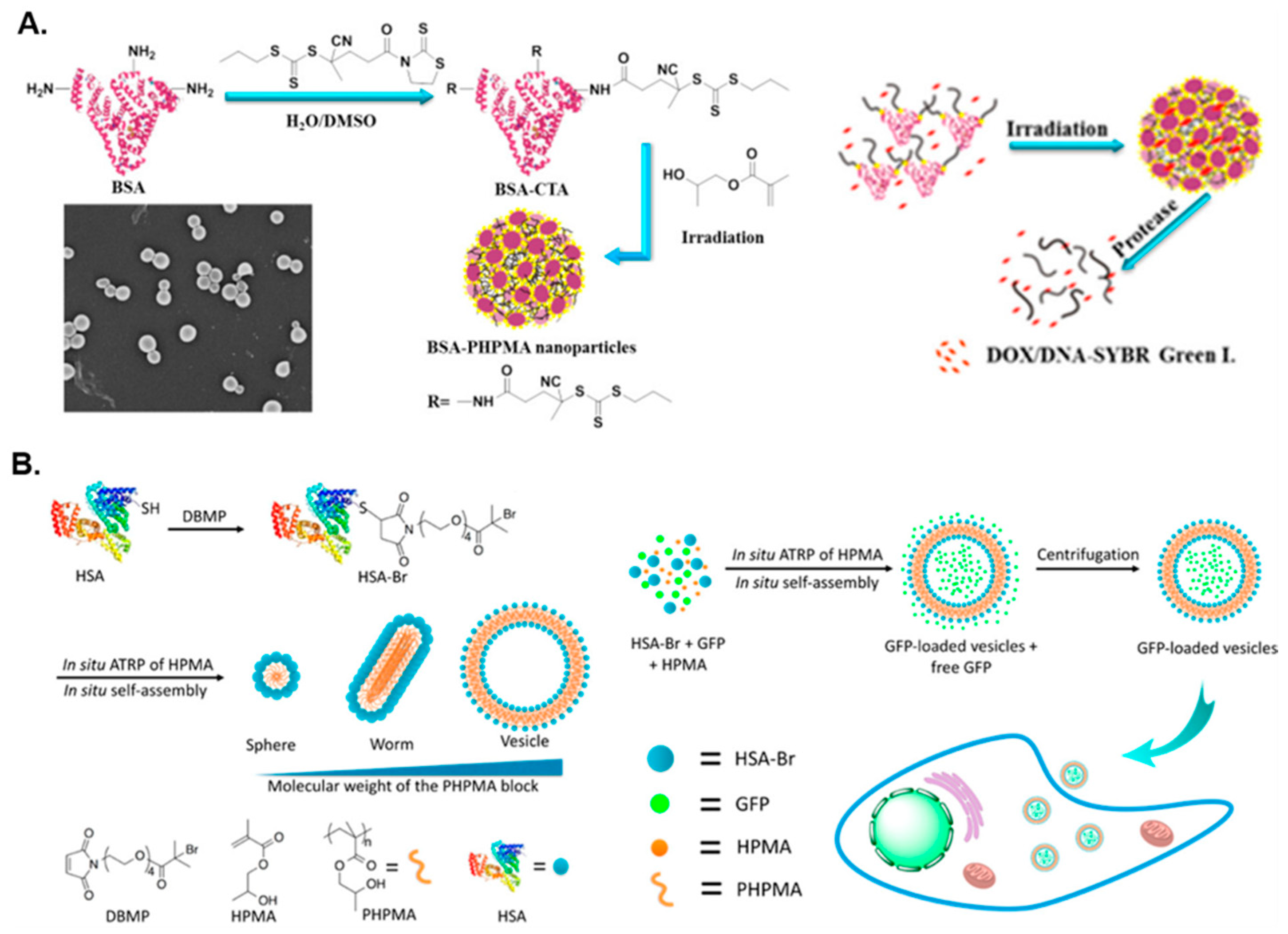
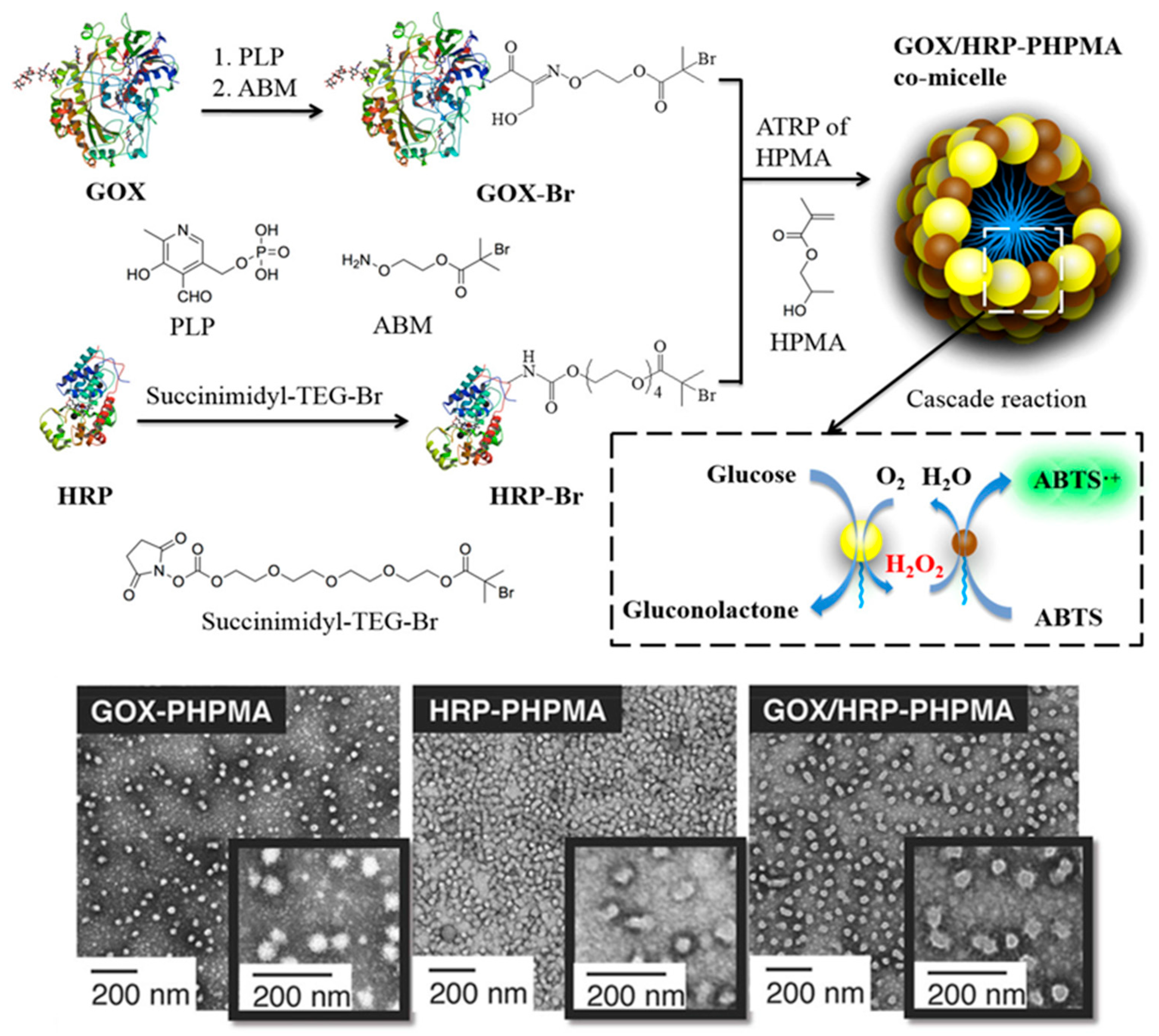
2. Protein–Polymer Hybrid Nanostructures via PISA
3. Peptide–Polymer Hybrid Nanostructures via PISA
4. Polypeptide- and Polypeptoid-Based Nanostructures via PISA
5. Amino Acid-Based Nanostructures via PISA
6. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef]
- Groß, A.; Hashimoto, C.; Sticht, H.; Eichler, J. Synthetic Peptides as Protein Mimics. Front. Bioeng. Biotechnol. 2016, 3, 211. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-X.; Eden, H.S.; Chen, X. Peptides in cancer nanomedicine: Drug carriers, targeting ligands and protease substrates. J. Control. Release 2012, 159, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhi, K.; Ding, Z.; Sun, Y.; Li, S.; Li, M.; Pu, K.; Zou, J. Emergence in protein derived nanomedicine as anticancer therapeutics: More than a tour de force. Semin. Cancer Biol. 2021, 69, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, J.; Song, J.; Liu, Y.; Ren, X.; Zhao, Y. Protein-Based Nanomedicine for Therapeutic Benefits of Cancer. ACS Nano 2021, 15, 8001–8038. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cirino, P.C. Recent advances in engineering proteins for biocatalysis. Biotechnol. Bioeng. 2014, 111, 1273–1287. [Google Scholar] [CrossRef]
- Abdelraheem, E.M.M.; Busch, H.; Hanefeld, U.; Tonin, F. Biocatalysis explained: From pharmaceutical to bulk chemical production. React. Chem. Eng. 2019, 4, 1878–1894. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; McLeod, H.L. Strategies for Enzyme/Prodrug Cancer Therapy. Clin. Cancer Res. 2001, 7, 3314. [Google Scholar]
- Malekshah, O.M.; Chen, X.; Nomani, A.; Sarkar, S.; Hatefi, A. Enzyme/Prodrug Systems for Cancer Gene Therapy. Curr. Pharmacol. Rep. 2016, 2, 299–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-J.; Di, L.; Ren, Q.-S.; Wang, J.-Y. Applications and Degradation of Proteins Used as Tissue Engineering Materials. Materials 2009, 2, 613–635. [Google Scholar] [CrossRef] [Green Version]
- Klimek, K.; Ginalska, G. Proteins and Peptides as Important Modifiers of the Polymer Scaffolds for Tissue Engineering Applications—A Review. Polymers 2020, 12, 844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.A.; Kaur, K.; Klok, H.-A. Self-assembly of protein-polymer conjugates for drug delivery. Adv. Drug Deliv. Rev. 2021, 174, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.A.; Klok, H.-A. Peptide/protein–polymer conjugates: Synthetic strategies and design concepts. Chem. Commun. 2008, 23, 2591–2611. [Google Scholar] [CrossRef] [PubMed]
- Grover, G.N.; Maynard, H.D. Protein–polymer conjugates: Synthetic approaches by controlled radical polymerizations and interesting applications. Curr. Opin. Chem. Biol. 2010, 14, 818–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer–drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov. 2019, 18, 273–294. [Google Scholar] [CrossRef]
- Olson, R.A.; Korpusik, A.B.; Sumerlin, B.S. Enlightening advances in polymer bioconjugate chemistry: Light-based techniques for grafting to and from biomacromolecules. Chem. Sci. 2020, 11, 5142–5156. [Google Scholar] [CrossRef]
- González-Henríquez, C.M.; Sarabia-Vallejos, M.A.; Rodríguez-Hernández, J. Strategies to Fabricate Polypeptide-Based Structures via Ring-Opening Polymerization of N-Carboxyanhydrides. Polymers 2017, 9, 551. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Lahasky, S.H.; Guo, L.; Lee, C.-U.; Lavan, M. Polypeptoid Materials: Current Status and Future Perspectives. Macromolecules 2012, 45, 5833–5841. [Google Scholar] [CrossRef]
- Rasines Mazo, A.; Allison-Logan, S.; Karimi, F.; Chan, N.J.-A.; Qiu, W.; Duan, W.; O’Brien-Simpson, N.M.; Qiao, G.G. Ring opening polymerization of α-amino acids: Advances in synthesis, architecture and applications of polypeptides and their hybrids. Chem. Soc. Rev. 2020, 49, 4737–4834. [Google Scholar] [CrossRef]
- O’Reilly, R.K. Using controlled radical polymerisation techniques for the synthesis of functional polymers containing amino acid moieties. Polym. Int. 2010, 59, 568–573. [Google Scholar] [CrossRef]
- Mori, H.; Endo, T. Amino-Acid-Based Block Copolymers by RAFT Polymerization. Macromol. Rapid Commun. 2012, 33, 1090–1107. [Google Scholar] [CrossRef]
- Warren, N.J.; Armes, S.P. Polymerization-Induced Self-Assembly of Block Copolymer Nano-objects via RAFT Aqueous Dispersion Polymerization. J. Am. Chem. Soc. 2014, 136, 10174–10185. [Google Scholar] [CrossRef] [PubMed]
- Derry, M.J.; Fielding, L.A.; Armes, S.P. Polymerization-induced self-assembly of block copolymer nanoparticles via RAFT non-aqueous dispersion polymerization. Prog. Polym. Sci. 2016, 52, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Penfold, N.J.W.; Yeow, J.; Boyer, C.; Armes, S.P. Emerging Trends in Polymerization-Induced Self-Assembly. ACS Macro Lett. 2019, 8, 1029–1054. [Google Scholar] [CrossRef] [Green Version]
- Derry, M.J.; Fielding, L.A.; Armes, S.P. Industrially-relevant polymerization-induced self-assembly formulations in non-polar solvents: RAFT dispersion polymerization of benzyl methacrylate. Polym. Chem. 2015, 6, 3054–3062. [Google Scholar] [CrossRef] [Green Version]
- Canning, S.L.; Smith, G.N.; Armes, S.P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2016, 49, 1985–2001. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.C.; Varlas, S.; Couturaud, B.; Coe, Z.; O’Reilly, R.K. Getting into Shape: Reflections on a New Generation of Cylindrical Nanostructures’ Self-Assembly Using Polymer Building Blocks. J. Am. Chem. Soc. 2019, 141, 2742–2753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Hong, C.-Y.; Pan, C.-Y. Polymerization techniques in polymerization-induced self-assembly (PISA). Polym. Chem. 2020, 11, 3673–3689. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Hong, C.-Y.; Pan, C.-Y. Polymerization-Induced Self-Assembly of Functionalized Block Copolymer Nanoparticles and Their Application in Drug Delivery. Macromol. Rapid Commun. 2019, 40, 1800279. [Google Scholar] [CrossRef]
- Phan, H.; Taresco, V.; Penelle, J.; Couturaud, B. Polymerisation-induced self-assembly (PISA) as a straightforward formulation strategy for stimuli-responsive drug delivery systems and biomaterials: Recent advances. Biomater. Sci. 2021, 9, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Blackman, L.D.; Varlas, S.; Arno, M.C.; Houston, Z.H.; Fletcher, N.L.; Thurecht, K.J.; Hasan, M.; Gibson, M.I.; O’Reilly, R.K. Confinement of Therapeutic Enzymes in Selectively Permeable Polymer Vesicles by Polymerization-Induced Self-Assembly (PISA) Reduces Antibody Binding and Proteolytic Susceptibility. ACS Cent. Sci. 2018, 4, 718–723. [Google Scholar] [CrossRef]
- Varlas, S.; Foster, J.C.; Georgiou, P.G.; Keogh, R.; Husband, J.T.; Williams, D.S.; O’Reilly, R.K. Tuning the membrane permeability of polymersome nanoreactors developed by aqueous emulsion polymerization-induced self-assembly. Nanoscale 2019, 11, 12643–12654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Pérez-Mercader, J. Polymerization-Induced Self-Assembly for Artificial Biology: Opportunities and Challenges. Macromol. Rapid Commun. 2019, 40, 1800513. [Google Scholar] [CrossRef] [PubMed]
- Derry, M.J.; Smith, T.; O’Hora, P.S.; Armes, S.P. Block Copolymer Nanoparticles Prepared via Polymerization-Induced Self-Assembly Provide Excellent Boundary Lubrication Performance for Next-Generation Ultralow-Viscosity Automotive Engine Oils. ACS Appl. Mater. Interfaces 2019, 11, 33364–33369. [Google Scholar] [CrossRef] [PubMed]
- Derry, M.J.; Mykhaylyk, O.O.; Armes, S.P. A Vesicle-to-Worm Transition Provides a New High-Temperature Oil Thickening Mechanism. Angew. Chem. Int. Ed. 2017, 56, 1746–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsman, I.R.; Derry, M.J.; Cunningham, V.J.; Brown, S.L.; Williams, C.N.; Armes, S.P. Tuning the vesicle-to-worm transition for thermoresponsive block copolymer vesicles prepared via polymerisation-induced self-assembly. Polym. Chem. 2021, 12, 1224–1235. [Google Scholar] [CrossRef]
- Deng, R.; Derry, M.J.; Mable, C.J.; Ning, Y.; Armes, S.P. Using Dynamic Covalent Chemistry To Drive Morphological Transitions: Controlled Release of Encapsulated Nanoparticles from Block Copolymer Vesicles. J. Am. Chem. Soc. 2017, 139, 7616–7623. [Google Scholar] [CrossRef]
- James, A.M.; Derry, M.J.; Train, J.S.; Dawson, R. Dispersible microporous diblock copolymer nanoparticles via polymerisation-induced self-assembly. Polym. Chem. 2019, 10, 3879–3886. [Google Scholar] [CrossRef]
- Albigès, R.; Klein, P.; Roi, S.; Stoffelbach, F.; Creton, C.; Bouteiller, L.; Rieger, J. Water-based acrylic coatings reinforced by PISA-derived fibers. Polym. Chem. 2017, 8, 4992–4995. [Google Scholar] [CrossRef] [Green Version]
- Thompson, K.L.; Fielding, L.A.; Mykhaylyk, O.O.; Lane, J.A.; Derry, M.J.; Armes, S.P. Vermicious thermo-responsive Pickering emulsifiers. Chem. Sci. 2015, 6, 4207–4214. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.J.; Armes, S.P. Pickering Emulsifiers Based on Block Copolymer Nanoparticles Prepared by Polymerization-Induced Self-Assembly. Langmuir 2020, 36, 15463–15484. [Google Scholar] [CrossRef]
- Mitchell, D.E.; Lovett, J.R.; Armes, S.P.; Gibson, M.I. Combining Biomimetic Block Copolymer Worms with an Ice-Inhibiting Polymer for the Solvent-Free Cryopreservation of Red Blood Cells. Angew. Chem. Int. Ed. 2016, 55, 2801–2804. [Google Scholar] [CrossRef]
- Georgiou, P.G.; Marton, H.L.; Baker, A.N.; Congdon, T.R.; Whale, T.F.; Gibson, M.I. Polymer Self-Assembly Induced Enhancement of Ice Recrystallization Inhibition. J. Am. Chem. Soc. 2021, 143, 7449–7461. [Google Scholar] [CrossRef]
- Canton, I.; Warren, N.J.; Chahal, A.; Amps, K.; Wood, A.; Weightman, R.; Wang, E.; Moore, H.; Armes, S.P. Mucin-Inspired Thermoresponsive Synthetic Hydrogels Induce Stasis in Human Pluripotent Stem Cells and Human Embryos. ACS Cent. Sci. 2016, 2, 65–74. [Google Scholar] [CrossRef]
- Li, P.; Sun, M.; Xu, Z.; Liu, X.; Zhao, W.; Gao, W. Site-Selective in Situ Growth-Induced Self-Assembly of Protein–Polymer Conjugates into pH-Responsive Micelles for Tumor Microenvironment Triggered Fluorescence Imaging. Biomacromolecules 2018, 19, 4472–4479. [Google Scholar] [CrossRef]
- Liu, X.; Sun, M.; Sun, J.; Hu, J.; Wang, Z.; Guo, J.; Gao, W. Polymerization Induced Self-Assembly of a Site-Specific Interferon α-Block Copolymer Conjugate into Micelles with Remarkably Enhanced Pharmacology. J. Am. Chem. Soc. 2018, 140, 10435–10438. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Dong, C.; Wang, X.; Wang, H.; Li, W.; Tan, J.; Chang, J. Self-Assembled Biodegradable Protein–Polymer Vesicle as a Tumor-Targeted Nanocarrier. ACS Appl. Mater. Interfaces 2014, 6, 2393–2400. [Google Scholar] [CrossRef] [PubMed]
- Vandermeulen, G.W.M.; Klok, H.-A. Peptide/Protein Hybrid Materials: Enhanced Control of Structure and Improved Performance through Conjugation of Biological and Synthetic Polymers. Macromol. Biosci. 2004, 4, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Jutz, G.; Böker, A. Bionanoparticles as functional macromolecular building blocks—A new class of nanomaterials. Polymer 2011, 52, 211–232. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Gao, W. Precision Conjugation: An Emerging Tool for Generating Protein–Polymer Conjugates. Angew. Chem. Int. Ed. 2021, 60, 11024–11035. [Google Scholar] [CrossRef]
- Obermeyer, A.C.; Olsen, B.D. Synthesis and Application of Protein-Containing Block Copolymers. ACS Macro Lett. 2015, 4, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.H.; Maynard, H.D. A guide to maximizing the therapeutic potential of protein–polymer conjugates by rational design. Chem. Soc. Rev. 2018, 47, 8998–9014. [Google Scholar] [CrossRef]
- Le Droumaguet, B.; Velonia, K. In Situ ATRP-Mediated Hierarchical Formation of Giant Amphiphile Bionanoreactors. Angew. Chem. Int. Ed. 2008, 47, 6263–6266. [Google Scholar] [CrossRef]
- Theodorou, A.; Liarou, E.; Haddleton, D.M.; Stavrakaki, I.G.; Skordalidis, P.; Whitfield, R.; Anastasaki, A.; Velonia, K. Protein-polymer bioconjugates via a versatile oxygen tolerant photoinduced controlled radical polymerization approach. Nat. Commun. 2020, 11, 1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorou, A.; Mandriotis, P.; Anastasaki, A.; Velonia, K. Oxygen tolerant, photoinduced controlled radical polymerization approach for the synthesis of giant amphiphiles. Polym. Chem. 2021, 12, 2228–2235. [Google Scholar] [CrossRef]
- Ma, C.; Liu, X.; Wu, G.; Zhou, P.; Zhou, Y.; Wang, L.; Huang, X. Efficient Way to Generate Protein-Based Nanoparticles by in-Situ Photoinitiated Polymerization-Induced Self-Assembly. ACS Macro Lett. 2017, 6, 689–694. [Google Scholar] [CrossRef]
- Liu, X.; Gao, W. In Situ Growth of Self-Assembled Protein–Polymer Nanovesicles for Enhanced Intracellular Protein Delivery. ACS Appl. Mater. Interfaces 2017, 9, 2023–2028. [Google Scholar] [CrossRef] [PubMed]
- Rucco, D.J.; Barnes, B.E.; Garrison, J.B.; Sumerlin, B.S.; Savin, D.A. Modular Genetic Code Expansion Platform and PISA Yield Well-Defined Protein-Polymer Assemblies. Biomacromolecules 2020, 21, 5077–5085. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-W.; Liu, X.; Sun, J.; Guo, J.; Tao, L.; Gao, W. Polymerization-Induced Coassembly of Enzyme–Polymer Conjugates into Comicelles with Tunable and Enhanced Cascade Activity. Nano Lett. 2020, 20, 1383–1387. [Google Scholar] [CrossRef]
- Bao, C.; Chen, J.; Li, D.; Zhang, A.; Zhang, Q. Synthesis of lipase–polymer conjugates by Cu(0)-mediated reversible deactivation radical polymerization: Polymerization vs. degradation. Polym. Chem. 2020, 11, 1386–1392. [Google Scholar] [CrossRef]
- Shu, J.Y.; Panganiban, B.; Xu, T. Peptide-Polymer Conjugates: From Fundamental Science to Application. Annu. Rev. Phys. Chem. 2013, 64, 631–657. [Google Scholar] [CrossRef]
- Callmann, C.E.; Thompson, M.P.; Gianneschi, N.C. Poly (peptide): Synthesis, Structure, and Function of Peptide–Polymer Amphiphiles and Protein-like Polymers. Acc. Chem. Res. 2020, 53, 400–413. [Google Scholar] [CrossRef]
- Paik, B.A.; Mane, S.R.; Jia, X.; Kiick, K.L. Responsive hybrid (poly) peptide–polymer conjugates. J. Mater. Chem. B 2017, 5, 8274–8288. [Google Scholar] [CrossRef]
- Li, M.; Zhao, G.; Su, W.-K.; Shuai, Q. Enzyme-Responsive Nanoparticles for Anti-tumor Drug Delivery. Front. Chem. 2020, 8, 647. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ng, D.Y.W.; Weil, T. Polymer bioconjugates: Modern design concepts toward precision hybrid materials. Prog. Polym. Sci. 2020, 105, 101241. [Google Scholar] [CrossRef]
- Nguyen, M.M.; Carlini, A.S.; Chien, M.-P.; Sonnenberg, S.; Luo, C.; Braden, R.L.; Osborn, K.G.; Li, Y.; Gianneschi, N.C.; Christman, K.L. Enzyme-Responsive Nanoparticles for Targeted Accumulation and Prolonged Retention in Heart Tissue after Myocardial Infarction. Adv. Mater. 2015, 27, 5547–5552. [Google Scholar] [CrossRef]
- Chien, M.-P.; Carlini, A.S.; Hu, D.; Barback, C.V.; Rush, A.M.; Hall, D.J.; Orr, G.; Gianneschi, N.C. Enzyme-Directed Assembly of Nanoparticles in Tumors Monitored by in vivo Whole Animal Imaging and ex vivo Super-Resolution Fluorescence Imaging. J. Am. Chem. Soc. 2013, 135, 18710–18713. [Google Scholar] [CrossRef] [Green Version]
- Callmann, C.E.; Barback, C.V.; Thompson, M.P.; Hall, D.J.; Mattrey, R.F.; Gianneschi, N.C. Therapeutic Enzyme-Responsive Nanoparticles for Targeted Delivery and Accumulation in Tumors. Adv. Mater. 2015, 27, 4611–4615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, W.; Li, J.; Zhao, K.; Zha, Z.; Han, Y.; Wang, Y.; Yin, W.; Zhang, P.; Ge, Z. Modular Design and Facile Synthesis of Enzyme-Responsive Peptide-Linked Block Copolymers for Efficient Delivery of Doxorubicin. Biomacromolecules 2016, 17, 3268–3276. [Google Scholar] [CrossRef]
- Das, D.; Gerboth, D.; Postma, A.; Srinivasan, S.; Kern, H.; Chen, J.; Ratner, D.M.; Stayton, P.S.; Convertine, A.J. Synthesis of zwitterionic, hydrophobic, and amphiphilic polymers via RAFT polymerization induced self-assembly (PISA) in acetic acid. Polym. Chem. 2016, 7, 6133–6143. [Google Scholar] [CrossRef]
- Wright, D.B.; Touve, M.A.; Adamiak, L.; Gianneschi, N.C. ROMPISA: Ring-Opening Metathesis Polymerization-Induced Self-Assembly. ACS Macro Lett. 2017, 6, 925–929. [Google Scholar] [CrossRef]
- Wright, D.B.; Thompson, M.P.; Touve, M.A.; Carlini, A.S.; Gianneschi, N.C. Enzyme-Responsive Polymer Nanoparticles via Ring-Opening Metathesis Polymerization-Induced Self-Assembly. Macromol. Rapid Commun. 2019, 40, 1800467. [Google Scholar] [CrossRef]
- Wright, D.B.; Touve, M.A.; Thompson, M.P.; Gianneschi, N.C. Aqueous-Phase Ring-Opening Metathesis Polymerization-Induced Self-Assembly. ACS Macro Lett. 2018, 7, 401–405. [Google Scholar] [CrossRef]
- D’Agosto, F.; Rieger, J.; Lansalot, M. RAFT-Mediated Polymerization-Induced Self-Assembly. Angew. Chem. Int. Ed. 2020, 59, 8368–8392. [Google Scholar] [CrossRef]
- Luppi, L.; Babut, T.; Petit, E.; Rolland, M.; Quemener, D.; Soussan, L.; Moradi, M.A.; Semsarilar, M. Antimicrobial polylysine decorated nano-structures prepared through polymerization induced self-assembly (PISA). Polym. Chem. 2019, 10, 336–344. [Google Scholar] [CrossRef]
- Dao, T.P.T.; Vezenkov, L.; Subra, G.; Amblard, M.; In, M.; Le Meins, J.-F.; Aubrit, F.; Moradi, M.-A.; Ladmiral, V.; Semsarilar, M. Self-Assembling Peptide—Polymer Nano-Objects via Polymerization-Induced Self-Assembly. Macromolecules 2020, 53, 7034–7043. [Google Scholar] [CrossRef]
- Habibi, N.; Kamaly, N.; Memic, A.; Shafiee, H. Self-assembled peptide-based nanostructures: Smart nanomaterials toward targeted drug delivery. Nano Today 2016, 11, 41–60. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Cao, W.; Zang, N.; Clemons, T.D.; Scheutz, G.M.; Hu, Z.; Thompson, M.P.; Liang, Y.; Vratsanos, M.; Zhou, X.; et al. Proapoptotic Peptide Brush Polymer Nanoparticles via Photoinitiated Polymerization-Induced Self-Assembly. Angew. Chem. Int. Ed. 2020, 59, 19136–19142. [Google Scholar] [CrossRef]
- Dao, T.P.T.; Vezenkov, L.; Subra, G.; Ladmiral, V.; Semsarilar, M. Nano-assemblies with core-forming hydrophobic polypeptide via polymerization-induced self-assembly (PISA). Polym. Chem. 2021, 12, 113–121. [Google Scholar] [CrossRef]
- Panda, J.J.; Chauhan, V.S. Short peptide based self-assembled nanostructures: Implications in drug delivery and tissue engineering. Polym. Chem. 2014, 5, 4418–4436. [Google Scholar] [CrossRef]
- Amblard, M.; Fehrentz, J.-A.; Martinez, J.; Subra, G. Methods and protocols of modern solid phase peptide synthesis. Mol. Biotechnol. 2006, 33, 239–254. [Google Scholar] [CrossRef]
- Deming, T.J. Synthetic polypeptides for biomedical applications. Prog. Polym. Sci. 2007, 32, 858–875. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Sakellariou, G. Synthesis of Well-Defined Polypeptide-Based Materials via the Ring-Opening Polymerization of α-Amino Acid N-Carboxyanhydrides. Chem. Rev. 2009, 109, 5528–5578. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.D.; Saha, N.; Zandraa, O.; Zuckermann, R.N.; Sáha, P. Peptoids and polypeptoids: Biomimetic and bioinspired materials for biomedical applications. Polym. Bull. 2017, 74, 3455–3466. [Google Scholar] [CrossRef]
- Huang, J.; Heise, A. Stimuli responsive synthetic polypeptides derived from N-carboxyanhydride (NCA) polymerisation. Chem. Soc. Rev. 2013, 42, 7373–7390. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wang, J.; Song, Z.; Yin, L.; Zhang, Y.; Tang, H.; Tu, C.; Lin, Y.; Cheng, J. Recent advances in amino acid N-carboxyanhydrides and synthetic polypeptides: Chemistry, self-assembly and biological applications. Chem. Commun. 2014, 50, 139–155. [Google Scholar] [CrossRef]
- Liarou, E.; Varlas, S.; Skoulas, D.; Tsimblouli, C.; Sereti, E.; Dimas, K.; Iatrou, H. Smart polymersomes and hydrogels from polypeptide-based polymer systems through α-amino acid N-carboxyanhydride ring-opening polymerization. From chemistry to biomedical applications. Prog. Polym. Sci. 2018, 83, 28–78. [Google Scholar] [CrossRef]
- Varlas, S.; Georgiou, P.G.; Bilalis, P.; Jones, J.R.; Hadjichristidis, N.; O’Reilly, R.K. Poly(sarcosine)-Based Nano-Objects with Multi-Protease Resistance by Aqueous Photoinitiated Polymerization-Induced Self-Assembly (Photo-PISA). Biomacromolecules 2018, 19, 4453–4462. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Ingram, N.; Rowley, J.V.; Parkinson, S.; Green, D.C.; Warren, N.J.; Thornton, P.D. Thermoresponsive polysarcosine-based nanoparticles. J. Mater. Chem. B 2019, 7, 4217–4223. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, X.; Fan, Z.; Du, J. Ring-Opening Polymerization of N-Carboxyanhydride-Induced Self-Assembly for Fabricating Biodegradable Polymer Vesicles. ACS Macro Lett. 2019, 8, 1216–1221. [Google Scholar] [CrossRef]
- Grazon, C.; Salas-Ambrosio, P.; Ibarboure, E.; Buol, A.; Garanger, E.; Grinstaff, M.W.; Lecommandoux, S.; Bonduelle, C. Aqueous Ring-Opening Polymerization-Induced Self-Assembly (ROPISA) of N-Carboxyanhydrides. Angew. Chem. Int. Ed. 2020, 59, 622–626. [Google Scholar] [CrossRef]
- van Hest, J.C.M. Biosynthetic-Synthetic Polymer Conjugates. Polym. Rev. 2007, 47, 63–92. [Google Scholar] [CrossRef]
- Brisson, E.R.L.; Xiao, Z.; Connal, L.A. Amino Acid Functional Polymers: Biomimetic Polymer Design Enabling Catalysis, Chiral Materials, and Drug Delivery. Aust. J. Chem. 2016, 69, 705–716. [Google Scholar] [CrossRef]
- Chung, I.-D.; Britt, P.; Xie, D.; Harth, E.; Mays, J. Synthesis of amino acid-based polymers via atom transfer radical polymerization in aqueous media at ambient temperature. Chem. Commun. 2005, 1046–1048. [Google Scholar] [CrossRef]
- Higashi, N.; Matsubara, S.; Nishimura, S.-n.; Koga, T. Stepwise Thermo-Responsive Amino Acid-Derived Triblock Vinyl Polymers: ATRP Synthesis of Polymers, Aggregation, and Gelation Properties via Flower-Like Micelle Formation. Materials 2018, 11, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skey, J.; Hansell, C.F.; O’Reilly, R.K. Stabilization of Amino Acid Derived Diblock Copolymer Micelles through Favorable D: L side chain interactions. Macromolecules 2010, 43, 1309–1318. [Google Scholar] [CrossRef]
- Skey, J.; Willcock, H.; Lammens, M.; Du Prez, F.; O’Reilly, R.K. Synthesis and Self-Assembly of Amphiphilic Chiral Poly (amino acid) Star Polymers. Macromolecules 2010, 43, 5949–5955. [Google Scholar] [CrossRef]
- Roy, S.G.; De, P. pH responsive polymers with amino acids in the side chains and their potential applications. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Bauri, K.; Nandi, M.; De, P. Amino acid-derived stimuli-responsive polymers and their applications. Polym. Chem. 2018, 9, 1257–1287. [Google Scholar] [CrossRef]
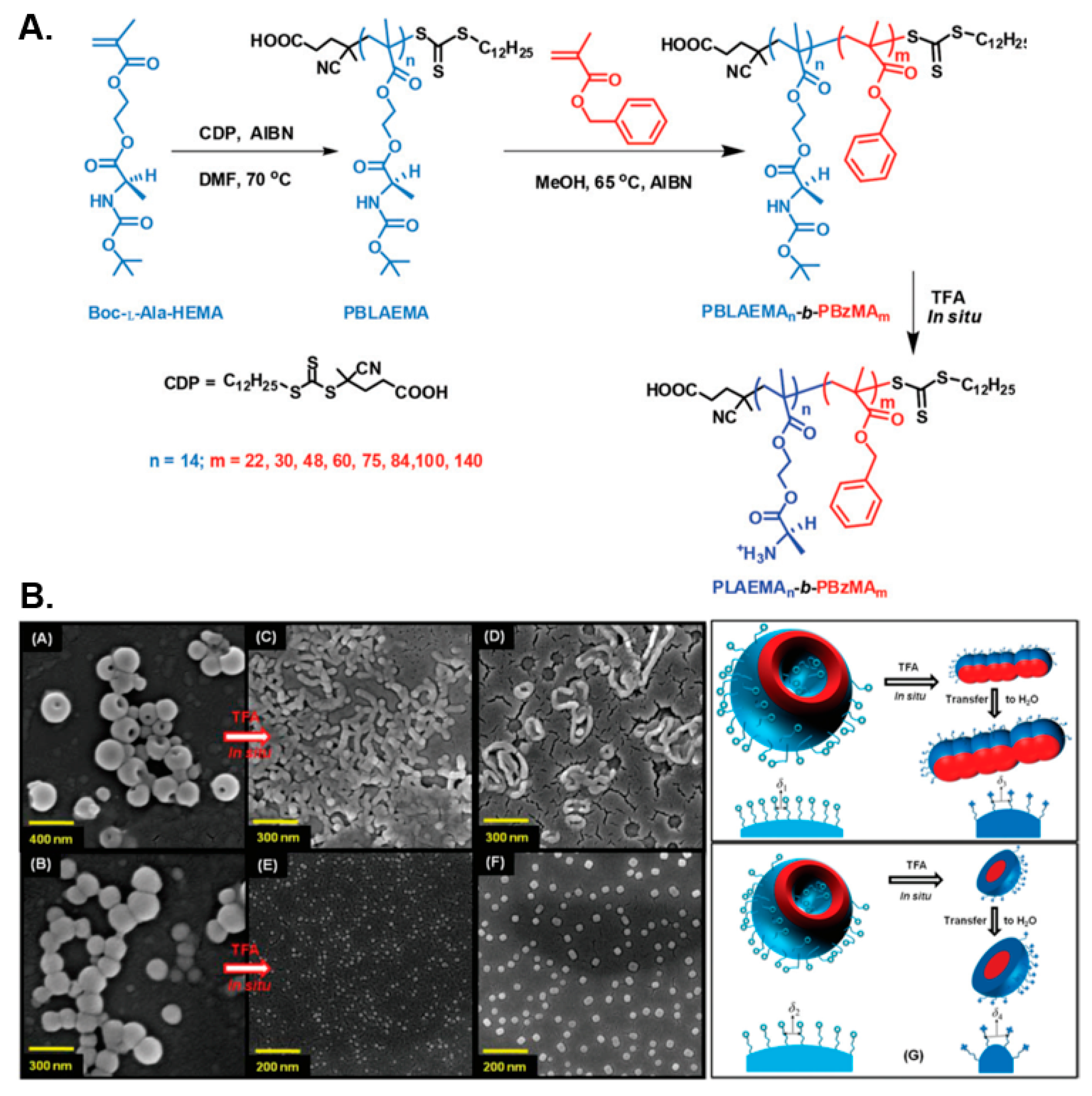
- Ladmiral, V.; Charlot, A.; Semsarilar, M.; Armes, S.P. Synthesis and characterization of poly (amino acid methacrylate)-stabilized diblock copolymer nano-objects. Polym. Chem. 2015, 6, 1805–1816. [Google Scholar] [CrossRef]
- Bauri, K.; Narayanan, A.; Haldar, U.; De, P. Polymerization-induced self-assembly driving chiral nanostructured materials. Polym. Chem. 2015, 6, 6152–6162. [Google Scholar] [CrossRef]
- Bauri, K.; Maiti, B.; De, P. Leucine-Based Block Copolymer Nano-Objects via Polymerization-Induced Self-Assembly (PISA). Macromol. Symp. 2016, 369, 101–107. [Google Scholar] [CrossRef]
- Chen, S.; Chang, X.; Sun, P.; Zhang, W. Versatile multicompartment nanoparticles constructed with two thermo-responsive, pH-responsive and hydrolytic diblock copolymers. Polym. Chem. 2017, 8, 5593–5602. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, Q.; Li, C.; Cao, L.; Ma, L.; Wang, X.; Cai, Y. Noncovalent structural locking of thermoresponsive polyion complex micelles, nanowires, and vesicles via polymerization-induced electrostatic self-assembly using an arginine-like monomer. Chem. Commun. 2020, 56, 4954–4957. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Corona-Forming Block | Core-Forming Block | PISA Solvent | Morphologies 1 | Application | Ref. |
|---|---|---|---|---|---|
| BSA, HSA, reduced human calcitonin | PS | PBS (w. DMSO in some cases) | S | Nanoreactors | [54] |
| BSA, HSA, GOx, β-galactosidase | PS, PDMAEMA, substituted styrenes | PBS, water | S | Nanoreactors | [55] |
| BSA | MA, MMA, tBA, tBMA | Water | S | - | [56] |
| BSA | PHPMA | Water | S | Drug delivery | [57] |
| HSA | PHPMA | PBS | S/W/V | Protein delivery | [58] |
| HSA | PDPA | PBS | S/W/A/V | Bioimaging | [46] |
| IFN-g-POEGMA | PHPMA | PBS | S | Antitumor agents | [47] |
| sfGFP | PHPMA | PBS | S | - | [59] |
| GOx/HRP | PHPMA | PBS | S | Glucose detection | [60] |
| CALB | NIPAAm, tBAm, MA | Water/methanol | S | - | [61] |
| Corona-Forming Block | Core-Forming Block | PISA Solvent | Morphologies 1 | Application | Ref. |
|---|---|---|---|---|---|
| P(HEMA-stat-O300) | P(DMAPS-stat-(MAm-AhxWSGPGVWGASVK)) | Acetic acid | S | - | [71] |
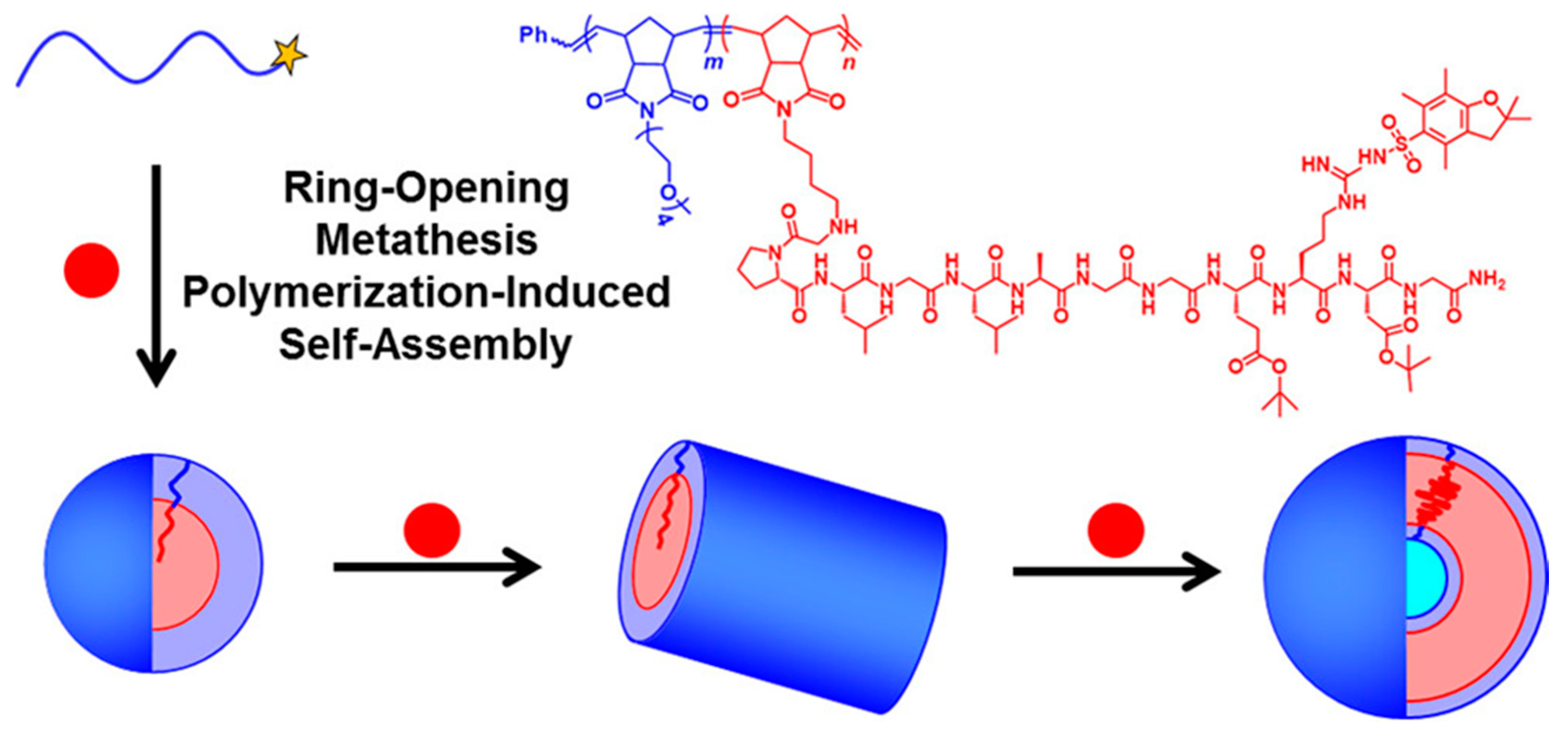
| PNB-OEG | P(NB-GPLGLAGGERDG) | DMF/methanol | S/W/V | - | [72] |
| P(NB-GPLGLAGGWGERDGS) | PNB-APh | Water | S/V | Biomimicry | [73] |
| KKK | PHPMA | Water | S/W/V | Antibacterial membranes | [76] |
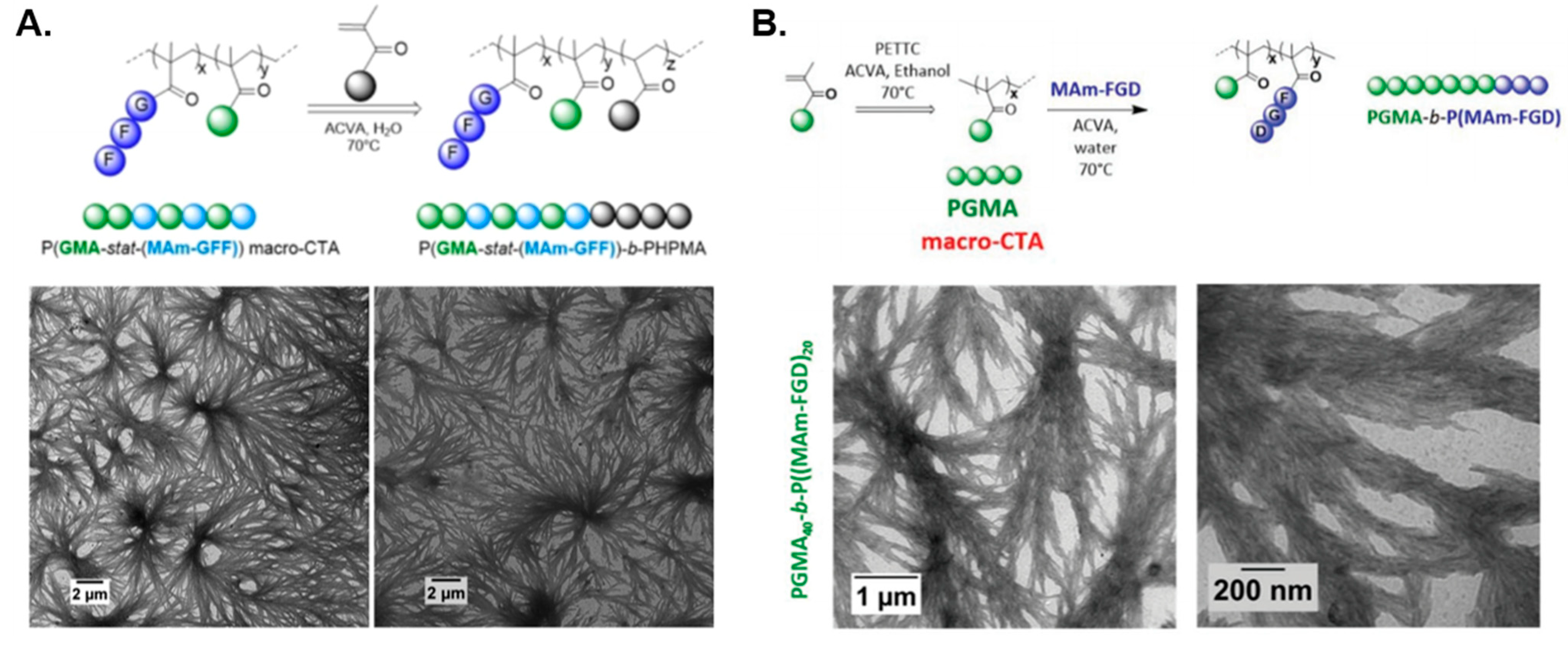
| P(GMA-stat-(MAm-GFF)) | PHPMA | Water | DF/S 2/W 3 | - | [77] |
| P(DMA-stat-(Am-KLAKLAKKLAKLAK)) | P(DMA-stat-DAAm) | Acetate buffer | S | Therapeutics/biomimicry | [79] |
| PGMA | P((MAm-GFF)-stat-HPMA) | Ethanol | W/V | - | [80] |
| PGMA | P(MAm-GFF) | Water/acetonitrile | S/V | - | [80] |
| PGMA | P(MAm-FGD) | Water | DF | - | [80] |
| Corona-Forming Block | Core-Forming Block | PISA Solvent | Morphologies 1 | Application | Ref. |
|---|---|---|---|---|---|
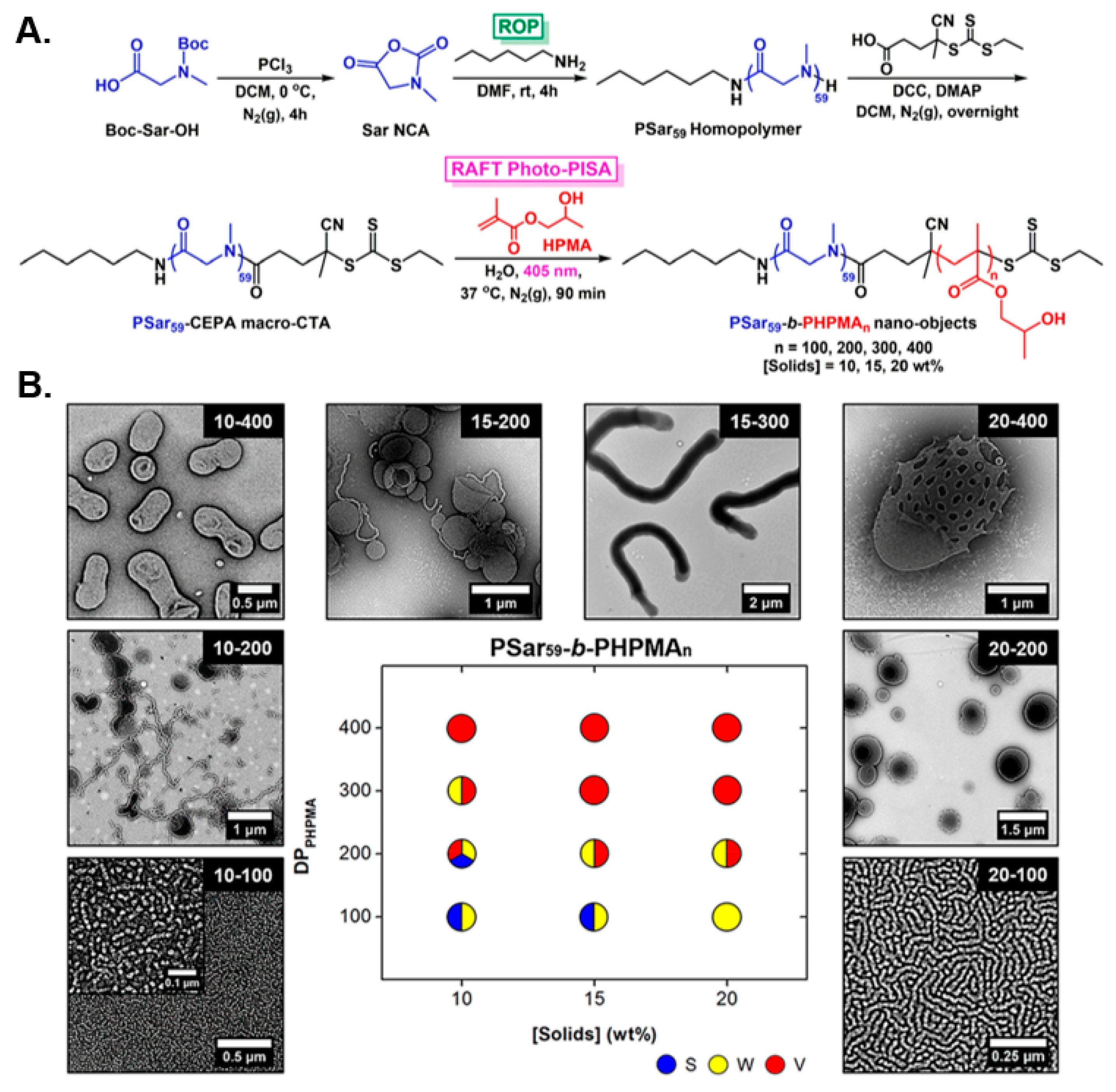
| PSar | PHPMA | Water | S/W/V | Nanoreactors/biomimicry | [89] |
| PSar | PHPMA | Ethanol/water | S | Drug delivery | [90] |
| PEG | PPhe, PBLA | THF | S/V | - | [91] |
| PEG | PBLG, P(NBocLys) | aq. NaHCO3 | W | - | [92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varlas, S.; Maitland, G.L.; Derry, M.J. Protein-, (Poly)peptide-, and Amino Acid-Based Nanostructures Prepared via Polymerization-Induced Self-Assembly. Polymers 2021, 13, 2603. https://doi.org/10.3390/polym13162603
Varlas S, Maitland GL, Derry MJ. Protein-, (Poly)peptide-, and Amino Acid-Based Nanostructures Prepared via Polymerization-Induced Self-Assembly. Polymers. 2021; 13(16):2603. https://doi.org/10.3390/polym13162603
Chicago/Turabian StyleVarlas, Spyridon, Georgia L. Maitland, and Matthew J. Derry. 2021. "Protein-, (Poly)peptide-, and Amino Acid-Based Nanostructures Prepared via Polymerization-Induced Self-Assembly" Polymers 13, no. 16: 2603. https://doi.org/10.3390/polym13162603
APA StyleVarlas, S., Maitland, G. L., & Derry, M. J. (2021). Protein-, (Poly)peptide-, and Amino Acid-Based Nanostructures Prepared via Polymerization-Induced Self-Assembly. Polymers, 13(16), 2603. https://doi.org/10.3390/polym13162603