
Synthesis and Characterization of Temperature-Responsive N-Cyanomethylacrylamide-Containing Diblock Copolymer Assemblies in Water


 , , , and
, , , and
Abstract
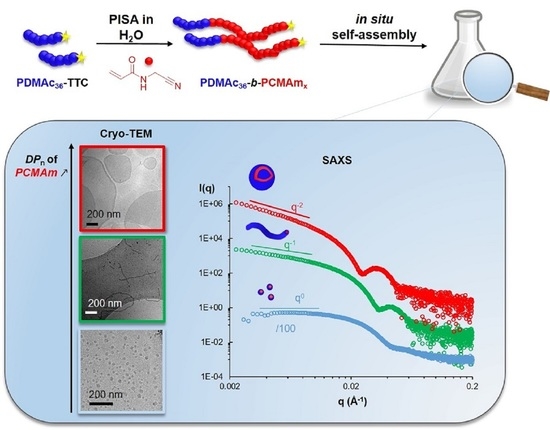
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of the PDMAm MacroRAFT Agents
2.3. Synthesis of PDMAm-b-PCMAm Diblock Copolymers in Water
2.4. Characterization Techniques
2.4.1. Nuclear Magnetic Resonance Spectroscopy (NMR)
2.4.2. Size Exclusion Chromatography (SEC)
2.4.3. Turbidimetry
2.4.4. Dynamic Light Scattering (DLS)
2.4.5. Cryogenic Transmission Electron Microscopy (Cryo-TEM)
2.4.6. Small Angle X-ray Scattering Analyses (SAXS)
3. Results and Discussion
3.1. Synthesis of PDMAm-b-PCMAm Diblock Copolymer Assemblies by RAFT-Mediated Aqueous Dispersion Polymerization
3.2. Impact of DPn of PDMAm and PCMAm on Colloidal Stability and Morphology
3.3. Thermoresponsive Behavior
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, C.; Ma, Z.; Zhu, X.X. Rational Design of Thermoresponsive Polymers in Aqueous Solutions: A Thermodynamics Map. Prog. Polym. Sci. 2019, 90, 269–291. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Matsunaga, Y.T. Thermo-Responsive Polymers and Their Application as Smart Biomaterials. J. Mater. Chem. B 2017, 5, 4307–4321. [Google Scholar] [CrossRef]
- Bansal, K.K.; Upadhyay, P.K.; Saraogi, G.K.; Rosling, A.; Rosenholm, J.M. Advances in Thermo-Responsive Polymers Exhibiting Upper Critical Solution Temperature (UCST). Express Polym. Lett. 2019, 13, 974–992. [Google Scholar] [CrossRef]
- Roy, D.; Brooks, W.L.A.; Sumerlin, B.S. New Directions in Thermoresponsive Polymers. Chem. Soc. Rev. 2013, 42, 7214–7243. [Google Scholar] [CrossRef] [PubMed]
- Seuring, J.; Agarwal, S. Polymers with Upper Critical Solution Temperature in Aqueous Solution. Macromol. Rapid Commun. 2012, 33, 1898–1920. [Google Scholar] [CrossRef] [PubMed]
- Seuring, J.; Agarwal, S. Polymers with Upper Critical Solution Temperature in Aqueous Solution: Unexpected Properties from Known Building Blocks. ACS Macro Lett. 2013, 2, 597–600. [Google Scholar] [CrossRef]
- Seuring, J.; Agarwal, S. First Example of a Universal and Cost-Effective Approach: Polymers with Tunable Upper Critical Solution Temperature in Water and Electrolyte Solution. Macromolecules 2012, 45, 3910–3918. [Google Scholar] [CrossRef]
- Seuring, J.; Agarwal, S. Non-Ionic Homo- and Copolymers with H-Donor and H-Acceptor Units with an UCST in Water. Macromol. Chem. Phys. 2010, 211, 2109–2117. [Google Scholar] [CrossRef]
- Käfer, F.; Pretscher, M.; Agarwal, S. Tuning the Phase Transition from UCST-Type to LCST-Type by Composition Variation of Polymethacrylamide Polymers. Macromol. Rapid Commun. 2018, 39, 1800640. [Google Scholar] [CrossRef]
- Akiyama, Y. Synthesis of Temperature-Responsive Polymers Containing Piperidine Carboxamide and N,N-diethylcarbamoly Piperidine Moiety via RAFT Polymerization. Macromol. Rapid Commun. 2021, 42, 2100208. [Google Scholar] [CrossRef]
- Audureau, N.; Veith, C.; Coumes, F.; Nguyen, T.P.T.; Rieger, J.; Stoffelbach, F. RAFT-Polymerized N-Cyanomethylacrylamide-Based (Co)Polymers Exhibiting Tunable UCST Behavior in Water. Macromol. Rapid Commun. 2021, 42, 2100556. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Zhao, B. Thermoreversible Physically Crosslinked Hydrogels from UCST-Type Thermosensitive ABA Linear Triblock Copolymers. Polym. Chem. 2016, 7, 6980–6991. [Google Scholar] [CrossRef]
- Augé, A.; Fortin, D.; Tong, X.; Zhao, Y. Nanogel-like UCST Triblock Copolymer Micelles Showing Large Volume Expansion before Abrupt Dissolution. Polym. Chem. 2018, 9, 4660–4673. [Google Scholar] [CrossRef]
- Wu, L.; Zong, L.; Ni, H.; Liu, X.; Wen, W.; Feng, L.; Cao, J.; Qi, X.; Ge, Y.; Shen, S. Magnetic Thermosensitive Micelles with Upper Critical Solution Temperature for NIR Triggered Drug Release. Biomater. Sci. 2019, 7, 2134–2143. [Google Scholar] [CrossRef]
- Chen, L.; Yang, T.; Niu, Y.; Mu, X.; Gong, Y.; Feng, Y.; de Rooij, N.F.; Wang, Y.; Li, H.; Zhou, G. Building a Smart Surface with Converse Temperature-Dependent Wettability Based on Poly(Acrylamide-co-Acrylonitrile). Chem. Commun. 2020, 56, 2837–2840. [Google Scholar] [CrossRef]
- Zhou, C.; Chen, Y.; Huang, M.; Ling, Y.; Yang, L.; Zhao, G.; Chen, J. A PH and UCST Thermo-Responsive Tri-Block Copolymer (PAA-b-PDMA-b-P(AM-Co-AN)) with Micellization and Gelatinization in Aqueous Media for Drug Release. New J. Chem. 2020, 44, 14551–14559. [Google Scholar] [CrossRef]
- Lertturongchai, P.; Ibrahim, M.I.A.; Durand, A.; Sunintaboon, P.; Ferji, K. Synthesis of Thermoresponsive Copolymers with Tunable UCST-Type Phase Transition Using Aqueous Photo-RAFT Polymerization. Macromol. Rapid Commun. 2020, 41, 2000058. [Google Scholar] [CrossRef]
- Audureau, N.; Coumes, F.; Guigner, J.-M.; Nguyen, T.P.T.; Ménager, C.; Stoffelbach, F.; Rieger, J. Thermoresponsive Properties of Poly(Acrylamide- co -Acrylonitrile)-Based Diblock Copolymers Synthesized (by PISA) in Water. Polym. Chem. 2020, 11, 5998–6008. [Google Scholar] [CrossRef]
- Chapiro, A.; Perec-Spritzer, L. Influence des solvants sur la copolymérisation de l’acrylamide avec l’acrylonitrile. Eur. Polym. J. 1975, 11, 59–69. [Google Scholar] [CrossRef]
- FUJIFILM Wako Pure Chemical Corporation. 2,2’-Azobis[2-(2-Imidazolin-2-Yl)Propane]Dihydrochloride (VA-044). Available online: https://specchem-wako-jp.fujifilm.com/en/waterazo/VA-044.htm# (accessed on 17 June 2021).
- Grazon, C.; Rieger, J.; Sanson, N.; Charleux, B. Study of Poly(N,N-Diethylacrylamide) Nanogel Formation by Aqueous Dispersion Polymerization of N,N-Diethylacrylamide in the Presence of Poly(Ethylene Oxide)-b-Poly(N,N-Dimethylacrylamide) Amphiphilic Macromolecular RAFT Agents. Soft Matter 2011, 7, 3482–3490. [Google Scholar] [CrossRef]
- Rieger, J.; Osterwinter, G.; Bui, C.; Stoffelbach, F.; Charleux, B. Surfactant-Free Controlled/Living Radical Emulsion (Co)Polymerization of n-Butyl Acrylate and Methyl Methacrylate via RAFT Using Amphiphilic Poly(Ethylene Oxide)-Based Trithiocarbonate Chain Transfer Agents. Macromolecules 2009, 42, 5518–5525. [Google Scholar] [CrossRef]
- Zhang, W.; Charleux, B.; Cassagnau, P. Viscoelastic Properties of Water Suspensions of Polymer Nanofibers Synthesized via RAFT-Mediated Emulsion Polymerization. Macromolecules 2012, 45, 5273–5280. [Google Scholar] [CrossRef]
- Magnet, S.; Inoubli, R.; Couvreur, L.; Charleux, B.; Brusseau, S. Filamentous Polymer Particles and Use Thereof as Rheology Modifieres. Patent WO 2012085473, 28 June 2012. [Google Scholar]
- Lovett, J.R.; Derry, M.J.; Yang, P.; Hatton, F.L.; Warren, N.J.; Fowler, P.W.; Armes, S.P. Can Percolation Theory Explain the Gelation Behavior of Diblock Copolymer Worms? Chem. Sci. 2018, 9, 7138–7144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maji, T.; Banerjee, S.; Biswas, Y.; Mandal, T.K. Dual-Stimuli-Responsive l -Serine-Based Zwitterionic UCST-Type Polymer with Tunable Thermosensitivity. Macromolecules 2015, 48, 4957–4966. [Google Scholar] [CrossRef]
- Mellot, G.; Guigner, J.-M.; Jestin, J.; Bouteiller, L.; Stoffelbach, F.; Rieger, J. Unexpected Thermo-Responsiveness of Bisurea-Functionalized Hydrophilic Polymers in Water. J. Colloid Interface Sci. 2021, 581, 874–883. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | [CMAm]0/ [TTC]0/[A]0 a | Time (h) | Conv. b (%) | DPn,thc | Mn,thc (kg mol−1) | SEC DMF | Visual Aspect ≤45 °C e | |
|---|---|---|---|---|---|---|---|---|
| Mnd (kg mol−1) | Đd | |||||||
| PDMAm-macroRAFT DPn = 13 | ||||||||
| 1 | 100/1/0.23 | 2.8 | 87 | 87 | 11.1 | 22.1 | 1.15 | Turbid, phase separation during polymerization |
| PDMAm-macroRAFT DPn = 23 | ||||||||
| 2 | 50/1/0.24 | 2.8 | 83 | 42 | 7.2 | 12.8 | 1.14 | Transparent |
| 3 | 100/1/0.15 | 6 | 93 | 93 | 12.8 | 24.4 | 1.15 | Slightly turbid |
| 4 | 225/1/0.32 | 4 | 86 | 194 | 20.8 | 34.9 | 1.54 | Phase separation during polymerization |
| PDMAm-macroRAFT DPn = 36 | ||||||||
| 5 | 100/1/0.45 | 8.3 | 91 | 91 | 13.8 | 23.4 | 1.16 | Slightly turbid |
| 6 | 200/1/0.35 | 5 | 93 | 186 | 24.3 | 42.4 | 1.23 | Slightly turbid |
| 7 | 400/1/0.35 | 8 | 90 | 360 | 43.4 | 83.5 | 1.63 | Milky solution |
| Sample Name | DPn PCMAm | Morphology | Dza DLS @25 °C (PDI) b | Dnc Cryo-TEM @RT (σ) d | Dnc Cryo-TEM @70 °C (σ) d | D e SAXS @25 °C (σ’) f | Vesicle Membrane Thickness Cryo-TEM @RT (σ) d | Vesicle Membrane Thickness SAXS @ 25 °C (σ’) f |
|---|---|---|---|---|---|---|---|---|
| 5 | 91 | Sphere | 29 nm (0.23) | 14 nm (1.9 nm) | 15 nm (1.2 nm) | 18 nm (0.15) | - | - |
| 6 | 186 | Worm | 153 nm * (0.23) | 21 nm (2.4 nm) | 19 nm (1.5 nm) | 22 nm (0.10) [>300 nm] g | - | - |
| 7 | 360 | Vesicle | 551 nm (0.16) | 592 nm (192 nm) | 391 nm (110 nm) | >300 nm g | 22 nm (2.5 nm) | 26 nm (0.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Audureau, N.; Coumes, F.; Veith, C.; Guibert, C.; Guigner, J.-M.; Stoffelbach, F.; Rieger, J. Synthesis and Characterization of Temperature-Responsive N-Cyanomethylacrylamide-Containing Diblock Copolymer Assemblies in Water. Polymers 2021, 13, 4424. https://doi.org/10.3390/polym13244424
Audureau N, Coumes F, Veith C, Guibert C, Guigner J-M, Stoffelbach F, Rieger J. Synthesis and Characterization of Temperature-Responsive N-Cyanomethylacrylamide-Containing Diblock Copolymer Assemblies in Water. Polymers. 2021; 13(24):4424. https://doi.org/10.3390/polym13244424
Chicago/Turabian StyleAudureau, Nicolas, Fanny Coumes, Clémence Veith, Clément Guibert, Jean-Michel Guigner, François Stoffelbach, and Jutta Rieger. 2021. "Synthesis and Characterization of Temperature-Responsive N-Cyanomethylacrylamide-Containing Diblock Copolymer Assemblies in Water" Polymers 13, no. 24: 4424. https://doi.org/10.3390/polym13244424
APA StyleAudureau, N., Coumes, F., Veith, C., Guibert, C., Guigner, J.-M., Stoffelbach, F., & Rieger, J. (2021). Synthesis and Characterization of Temperature-Responsive N-Cyanomethylacrylamide-Containing Diblock Copolymer Assemblies in Water. Polymers, 13(24), 4424. https://doi.org/10.3390/polym13244424