
Modifying Anti-Compression Property and Water-Soluble Ability of Polyglycolic Acid via Melt Blending with Polyvinyl Alcohol



Abstract
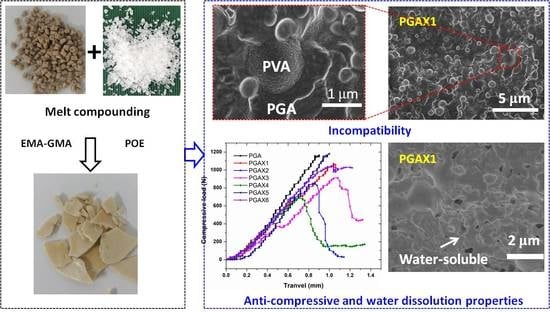
:
1. Introduction
2. Experimental Details
2.1. Materials
2.2. Sample Preparation
2.3. Characterizations
3. Results and Discussion
3.1. Morphological Characterization
3.2. Anti-Compression Properties
3.3. Rheological Behaviors
3.4. Thermal Stability
3.5. Water-Soluble Ability
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lebreton, L.; Andrady, A. Future scenarios of global plastic waste generation and disposal. Palgrave Commun. 2019, 5, 6. [Google Scholar] [CrossRef]
- Gross, R.A.; Kalra, B. Biodegradable Polymers for the Environment. Science 2002, 297, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Van de Velde, K.; Kiekens, P. Biopolymers: Overview of several properties and consequences on their applications. Polym. Test 2002, 21, 433–442. [Google Scholar] [CrossRef]
- Wang, Z.; Ganewatta, M.S.; Tang, C. Sustainable polymers from biomass: Bridging chemistry with materials and processing. Prog. Polym. Sci. 2019, 101, 101197. [Google Scholar] [CrossRef]
- Ji, J.; Pang, Y.; Sutoko, S.; Horimoto, Y.; Sun, W.; Niino, T.; Sakai, Y. Design, Fabrication, and Evaluation of Polyglycolic Acid Modules with Canals as Tissue Elements in Cellular-Assembly Technology. Appl. Sci. 2020, 10, 3478. [Google Scholar] [CrossRef]
- Yeo, T.; Ko, Y.G.; Kim, E.J.; Kwon, O.K.; Chung, H.Y.; Kwon, O.H. Promoting bone regeneration by 3D-printed poly(glycolic acid)/hydroxyapatite composite scaffolds. J. Ind. Eng. Chem. 2021, 94, 343–351. [Google Scholar] [CrossRef]
- Samantaray, P.K.; Farris, S.; McNally, T.; Tan, B.; Sun, Z.; Huang, W.; Ji, Y.; Wan, C. Ellingford, Reactive extrusion of biodegradable PGA/PBAT blends to enhance flexibility and gas barrier properties. J. Appl. Polym. Sci. 2022, 139, 51617. [Google Scholar]
- Samantaray, P.K.; Ellingford, C.; Farris, S.; O’Sullivan, D.; Tan, B.; Sun, Z.; McNally, T.; Wan, C. Electron Beam-Mediated Cross-Linking of Blown Film-Extruded Biodegradable PGA/PBAT Blends toward High Toughness and Low Oxygen Permeation. ACS Sustain. Chem. Eng. 2022, 10, 1267–1276. [Google Scholar] [CrossRef]
- Schultz, R.L.; Watson, B.W.; Ferguson, A.M.; Funkhouser, G.P. Flow Control in Subterranean Wells. Invention Patent US 9567825 B2, 14 February 2014. assigned to THRU TUBING SOLUTIONS, INC., 2017. [Google Scholar]
- Echols, R.H. Fluid Flow Control Device and Method for Use of Same. Invention Patent US 2004/0035591 A1, 26 February 2004. [Google Scholar]
- Funkhouser, G.P.; Watson, B.W.; Ferguson, A.M.; Robertson, J.N.; Schultz, R.L. Plugging Devices and Deployment in Subterranean Wells. Invention Patent US 9816 341 B2, 14 November 2017. assigned to THRU TUBING SOLUTIONS, INC., 2017. [Google Scholar]
- Niu, D.; Xu, P.; Sun, Z.; Yang, W.; Dong, W.; Ji, Y.; Liu, T.; Du, M.; Lemstra, P.J.; Ma, P. Superior toughened bio-compostable Poly(glycolic acid)-based blends with enhanced melt strength via selective interfacial localization of in-situ grafted copolymers. Polymer 2021, 235, 124269. [Google Scholar] [CrossRef]
- Samantaray, P.K.; Little, A.; Haddleton, D.M.; McNally, T.; Tan, B.; Sun, Z.; Huang, W.; Ji, Y.; Wan, C. Poly (glycolic acid)(PGA): A versatile building block expanding high performance and sustainable bioplastic applications. Green Chem. 2020, 22, 4055–4081. [Google Scholar] [CrossRef]
- Schmitt, E.A.; Flanagan, D.R.; Linhardt, R.J. Importance of distinct water environments in the hydrolysis of poly(DL-lactide-co-glycolide). Macromolecules 1994, 27, 743–748. [Google Scholar] [CrossRef]
- Panda, P.K.; Yang, J.M.; Chang, Y.H. Water-induced shape memory behavior of poly (vinyl alcohol) and p-coumaric acid-modified water-soluble chitosan blended membrane. Carbohyd. Polym. 2021, 257, 117633. [Google Scholar] [CrossRef]
- Bolto, B.; Tran, T.; Hoang, M.; Xie, Z. Crosslinked poly(vinyl alcohol) membranes. Prog. Polym. Sci. 2009, 34, 969–981. [Google Scholar] [CrossRef]
- Panda, P.K.; Dash, P.; Biswal, A.K.; Chang, Y.-H.; Misra, P.K.; Yang, J.-M. Synthesis and Characterization of Modified Poly(vinyl alcohol) Membrane and Study of Its Enhanced Water-Induced Shape-Memory Behavior. J. Polym. Environ. 2022, 30, 3409–3419. [Google Scholar] [CrossRef]
- Yang, J.; Panda, P.K.; Jie, C.J.; Dash, P.; Chang, Y. Poly (vinyl alcohol)/chitosan/sodium alginate composite blended membrane: Preparation, characterization, and water-induced shape memory phenomenon. Polym. Eng. Sci. 2022, 62, 1526. [Google Scholar] [CrossRef]
- Panda, P.K.; Sadeghi, K.; Seo, J. Recent advances in poly (vinyl alcohol)/natural polymer based films for food packaging applications: A review. Food Packag. Shelf 2022, 33, 100904. [Google Scholar] [CrossRef]
- Chiellini, E.; Corti, A.; D′Antone, S.; Solaro, R. Biodegradation of poly (vinyl alcohol) based materials. Prog. Polym. Sci. 2003, 28, 963–1014. [Google Scholar] [CrossRef]
- Matsumura, S.; Tomizawa, N.; Toki, A.; Nishikawa, A.K.; Toshima, K. Novel Poly(vinyl alcohol)-Degrading Enzyme and the Degradation Mechanism. Macromolecules 1999, 32, 7753–7761. [Google Scholar] [CrossRef]
- Corti, A.; Solaro, R.; Chiellini, E. Biodegradation of poly(vinyl alcohol) in selected mixed microbial culture and relevant culture filtrate. Polym. Degrad. Stab. 2002, 75, 447–458. [Google Scholar] [CrossRef]
- Thomas, P.S.; Guerbois, J.-P.; Russell, G.F.; Briscoe, B.J. FTIR Study of the Thermal Degradation of Poly(vinyl Alcohol). J. Therm. Anal. Calorim. 2001, 64, 501–508. [Google Scholar] [CrossRef]
- Cao, Y.; Xiong, D.; Niu, Y.; Mei, Y.; Yin, Z.; Gui, J. Compressive properties and creep resistance of a novel, porous, semidegradable poly(vinyl alcohol)/poly(lactic-co-glycolic acid) scaffold for articular cartilage repair. J. Appl. Polym. Sci. 2014, 131, 40311. [Google Scholar] [CrossRef]
- Shen, J.; Wang, K.; Ma, Z.; Xu, N.; Pang, S.; Pan, L. Biodegradable blends of poly(butylene adipate-co- terephthalate) and polyglycolic acid with enhanced mechanical, rheological and barrier performances. J. Appl. Polym. Sci. 2021, 138, e51285. [Google Scholar] [CrossRef]
- Zhang, Z.; He, F.; Wang, B.; Zhao, Y.; Wei, Z.; Zhang, H.; Sang, L. Biodegradable PGA/PBAT Blends for 3D Printing: Material Performance and Periodic Minimal Surface Structures. Polymers 2021, 13, 3757. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L. Poly(l-Lactic Acid)/Poly(Butylene Succinate) Biobased Biodegradable Blends. Polym. Rev. 2020, 61, 457–492. [Google Scholar] [CrossRef]
- Hivechi, A.; Bahrami, S.H.; Siegel, R.A. Drug release and biodegradability of electrospun cellulose nanocrystal reinforced polycaprolactone. Mater. Sci. Eng. C 2018, 94, 929–937. [Google Scholar] [CrossRef]
- Su, S.-K.; Wu, C.-S.; Siao, J.-W.; Yen, F.-S.; Wu, J.-Y.; Huang, C.M. Biodegradable Blends Prepared from Polycaprolactone and Poly(glutamic acid): Structure, Thermal Properties, and Biodegradability. Polym.-Plast. Technol. 2010, 49, 1361–1370. [Google Scholar] [CrossRef]
- Al-Itry, R.; Lamnawar, K.; Maazouz, A. Improvement of thermal stability, rheological and mechanical properties of PLA, PBAT and their blends by reactive extrusion with functionalized epoxy. Polym. Degrad. Stab. 2012, 97, 1898–1914. [Google Scholar] [CrossRef]
- Kumar, M.; Mohanty, S.; Nayak, S.; Parvaiz, M.R. Effect of glycidyl methacrylate (GMA) on the thermal, mechanical and morphological property of biodegradable PLA/PBAT blend and its nanocomposites. Bioresour. Technol. 2010, 101, 8406–8415. [Google Scholar] [CrossRef]
- Wang, R.; Sun, X.J.; Chen, L.L.; Liang, W.B. Morphological and mechanical properties of biodegradable poly(glycolic acid)/poly(butylene adipate-co-terephthalate) blends with in situ compatibilization. RSC Adv. 2021, 11, 1241. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Sun, X.; Ren, Y.; Wang, R.; Sun, M.; Liang, W. Enhancing melt strength of polyglycolic acid by reactive extrusion with chain extenders. J. Appl. Polym. Sci. 2021, 139, 51796. [Google Scholar] [CrossRef]
- Jem, K.J.; Tan, B. The development and challenges of poly (lactic acid) and poly (glycolic acid). Adv. Ind. Eng. Polym. Res. 2020, 3, 60–70. [Google Scholar] [CrossRef]
- Gautier, E.; Fuertes, P.; Cassagnau, P.; Pascault, J.-P.; Fleury, E. Synthesis and rheology of biodegradable poly(glycolic acid) prepared by melt ring-opening polymerization of glycolide. J. Polym. Sci. Part A: Polym. Chem. 2009, 47, 1440–1449. [Google Scholar] [CrossRef]
- Holland, B.J.; Hay, J.N. The thermal degradation of poly (vinyl alcohol). Polymer 2001, 42, 6775–6783. [Google Scholar] [CrossRef]
- Holland, B.J.; Hay, J.N. The thermal degradation of poly (vinyl acetate) measured by thermal analysis-Fourier transform infrared spectroscopy. Polymer 2002, 43, 2207–2211. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Components | |||
|---|---|---|---|---|
| PGA (wt%) | PVA (wt%) | POE (wt%) | EMA-GMA (wt%) | |
| PGAX1 | 72.7 | 18.2 | 0 | 9.1 |
| PGAX2 | 61.5 | 15.4 | 15.4 | 7.7 |
| PGAX3 | 54.5 | 36.4 | 0 | 9.1 |
| PGAX4 | 46.2 | 30.7 | 15.4 | 7.7 |
| PGAX5 | 36.4 | 54.5 | 0 | 9.1 |
| PGAX6 | 30.7 | 46.2 | 15.4 | 7.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, L.; Ma, S.; Hao, M.; Ma, L.; Lin, X. Modifying Anti-Compression Property and Water-Soluble Ability of Polyglycolic Acid via Melt Blending with Polyvinyl Alcohol. Polymers 2022, 14, 3375. https://doi.org/10.3390/polym14163375
Wei L, Ma S, Hao M, Ma L, Lin X. Modifying Anti-Compression Property and Water-Soluble Ability of Polyglycolic Acid via Melt Blending with Polyvinyl Alcohol. Polymers. 2022; 14(16):3375. https://doi.org/10.3390/polym14163375
Chicago/Turabian StyleWei, Liao, Shuyue Ma, Mengyuan Hao, Lanrong Ma, and Xiang Lin. 2022. "Modifying Anti-Compression Property and Water-Soluble Ability of Polyglycolic Acid via Melt Blending with Polyvinyl Alcohol" Polymers 14, no. 16: 3375. https://doi.org/10.3390/polym14163375
APA StyleWei, L., Ma, S., Hao, M., Ma, L., & Lin, X. (2022). Modifying Anti-Compression Property and Water-Soluble Ability of Polyglycolic Acid via Melt Blending with Polyvinyl Alcohol. Polymers, 14(16), 3375. https://doi.org/10.3390/polym14163375