Photo-Induced Micellization of Block Copolymers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
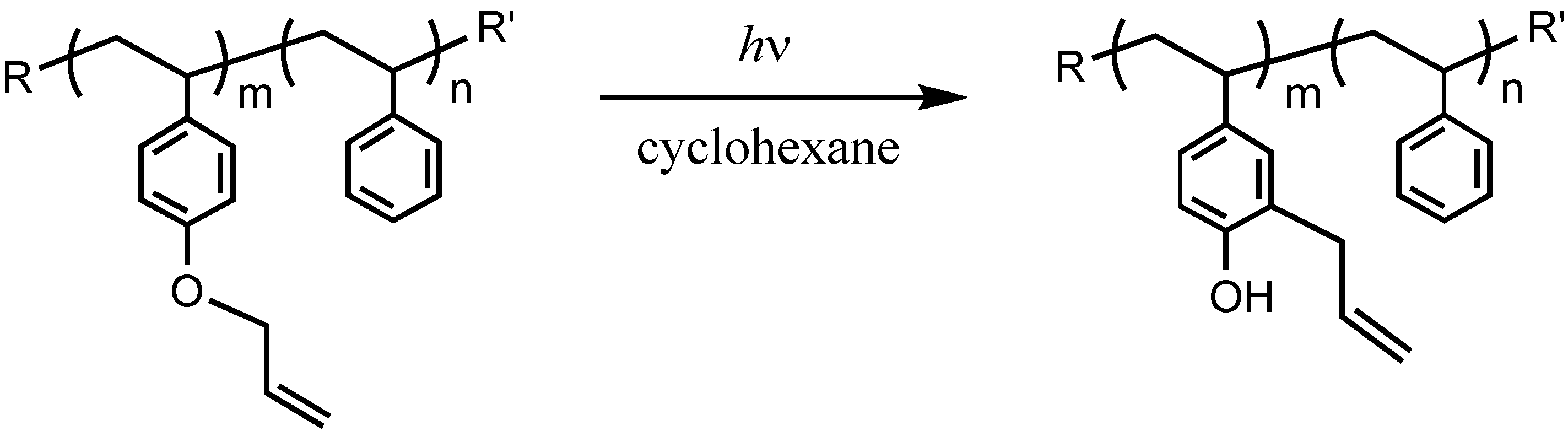{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
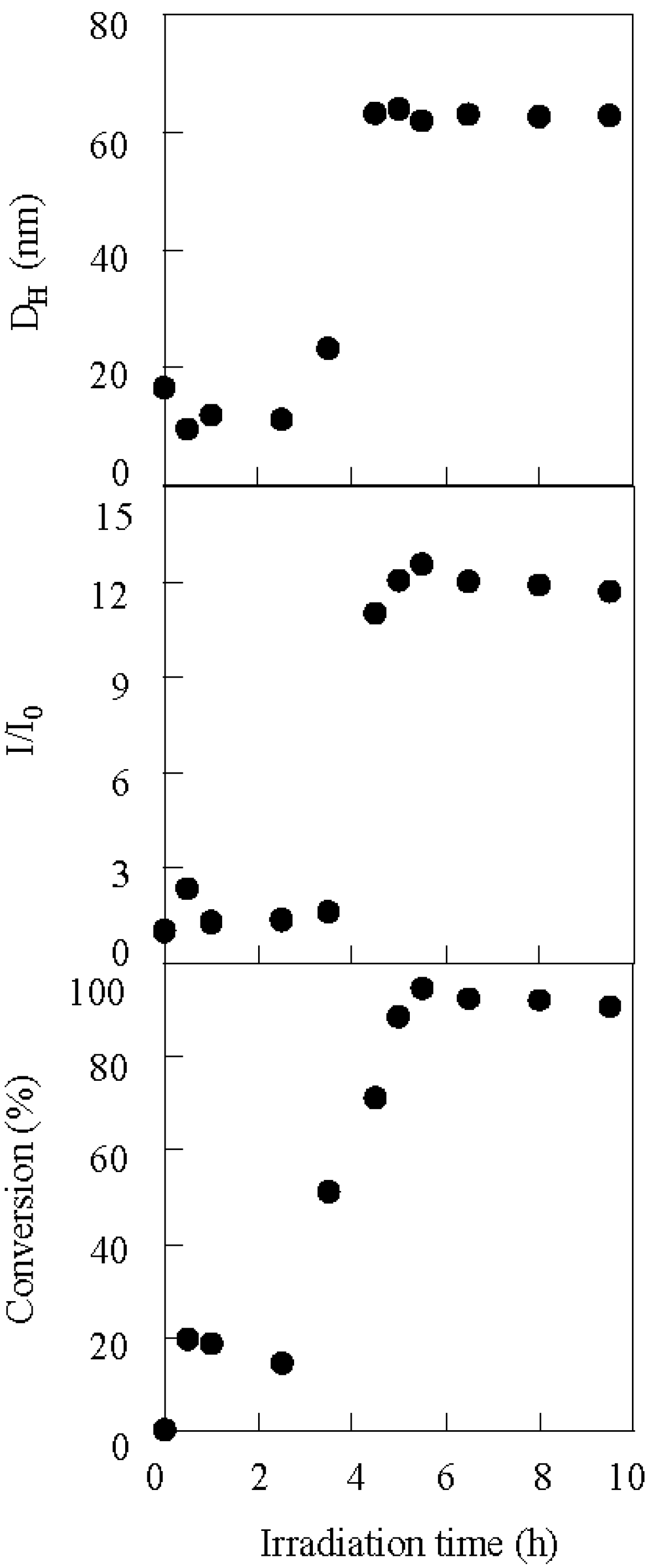
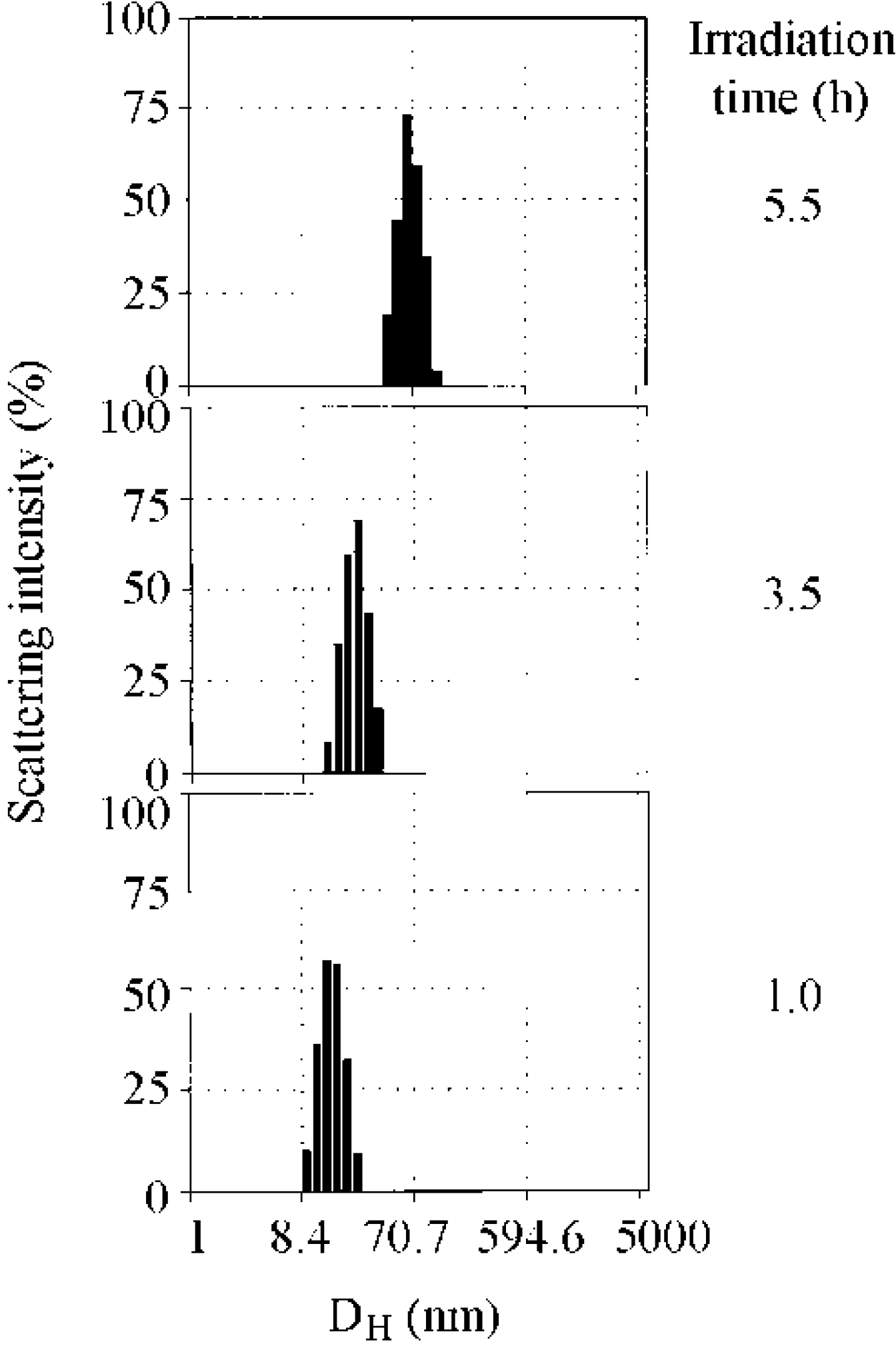
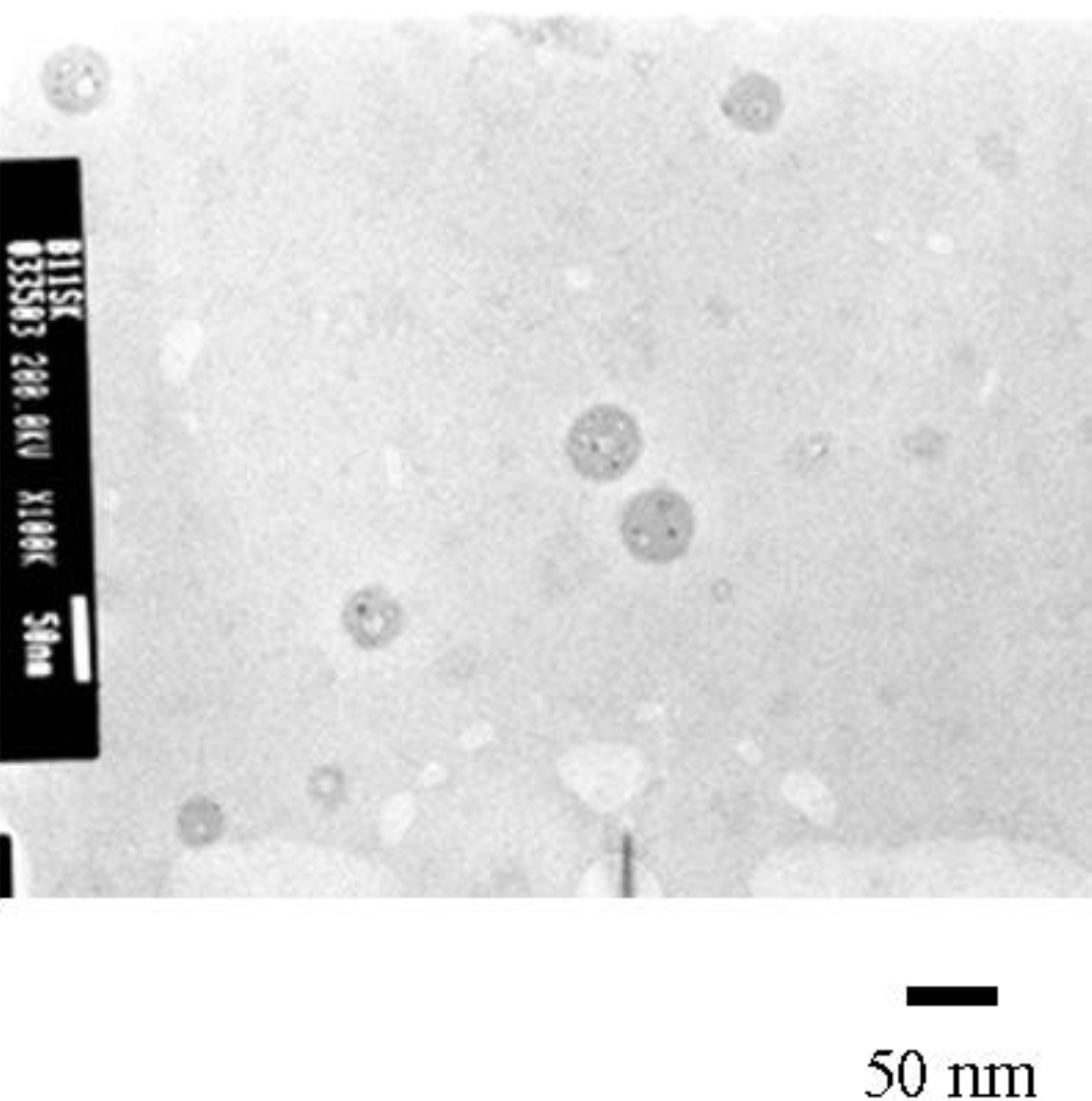
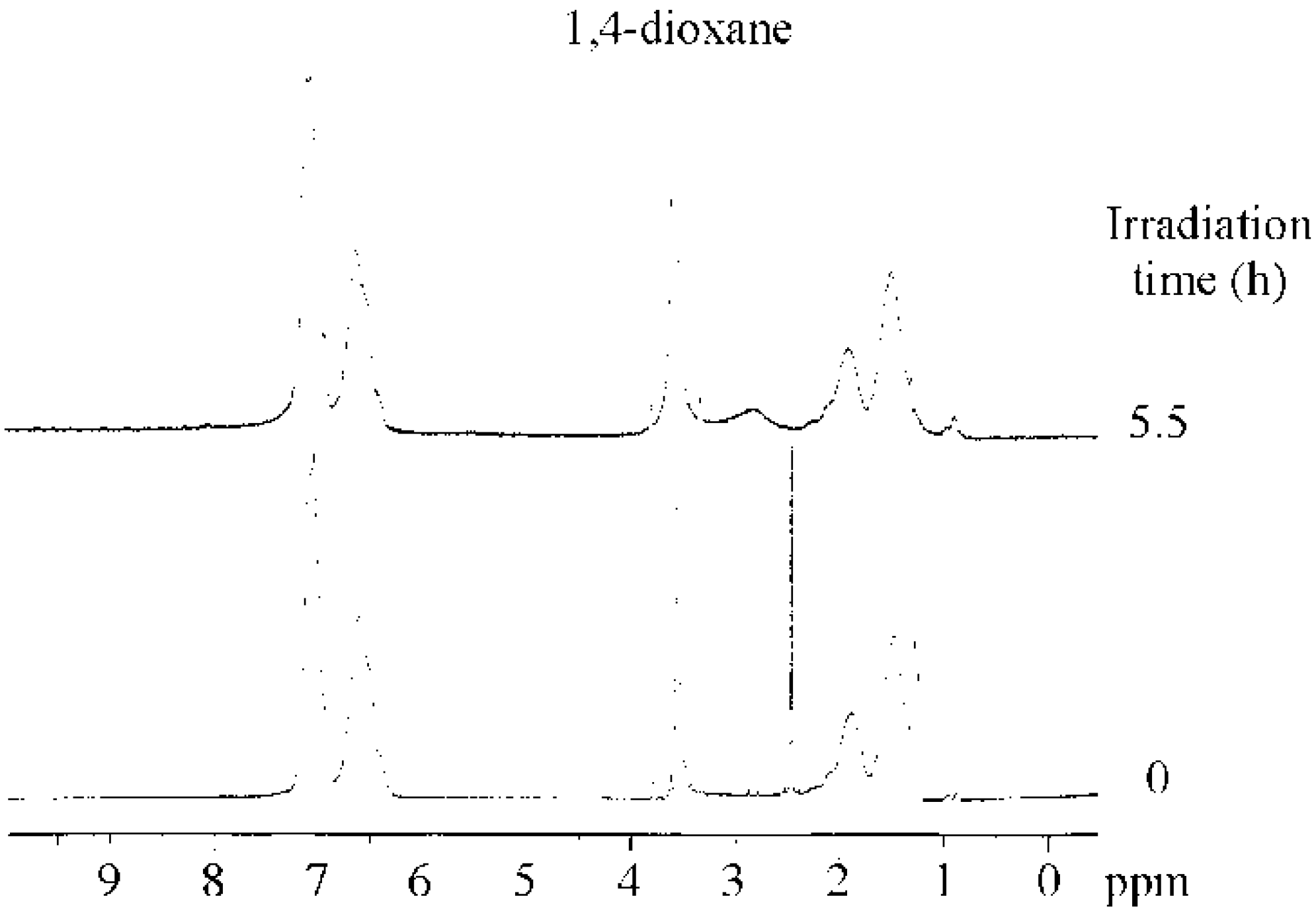
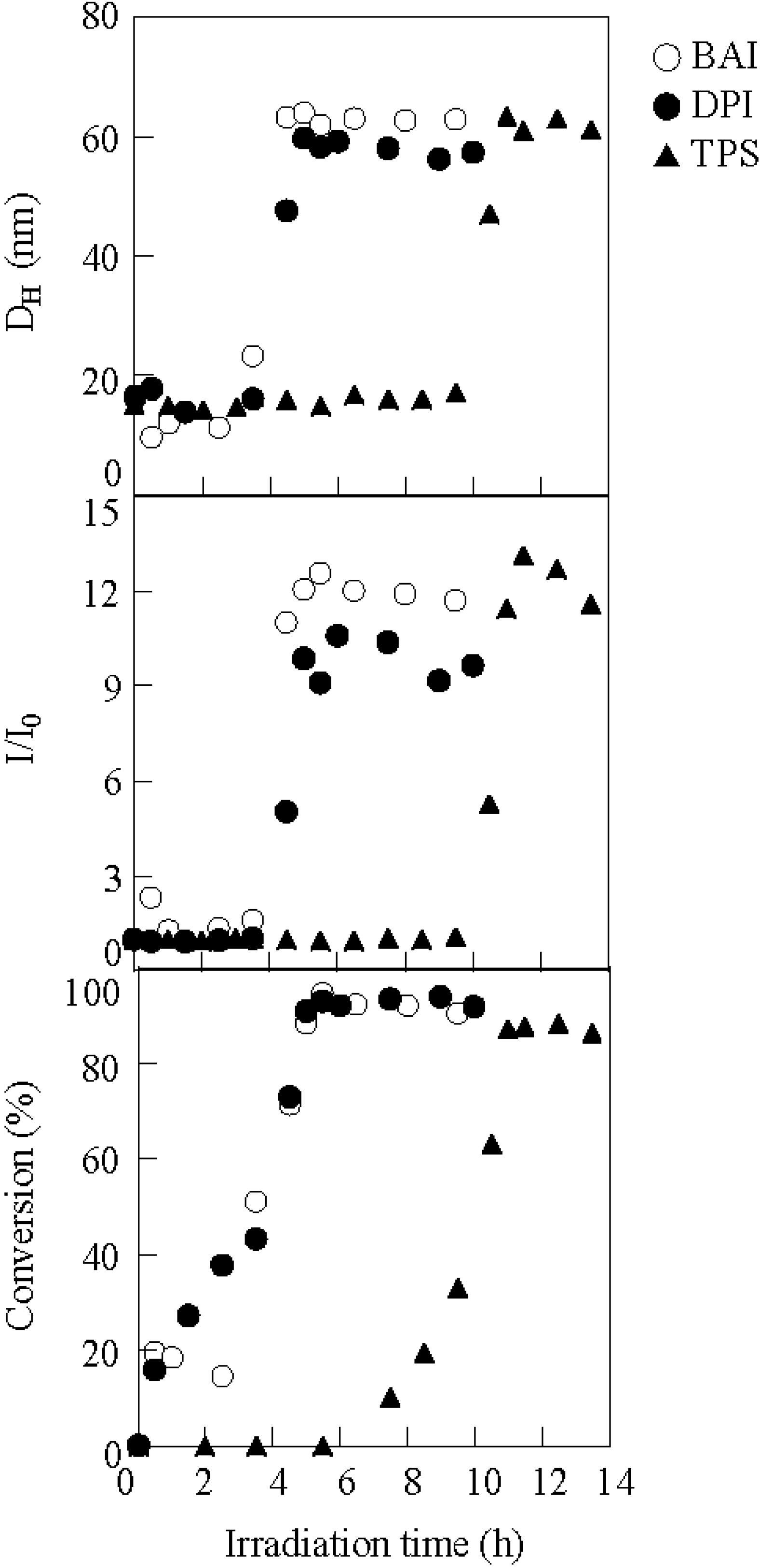
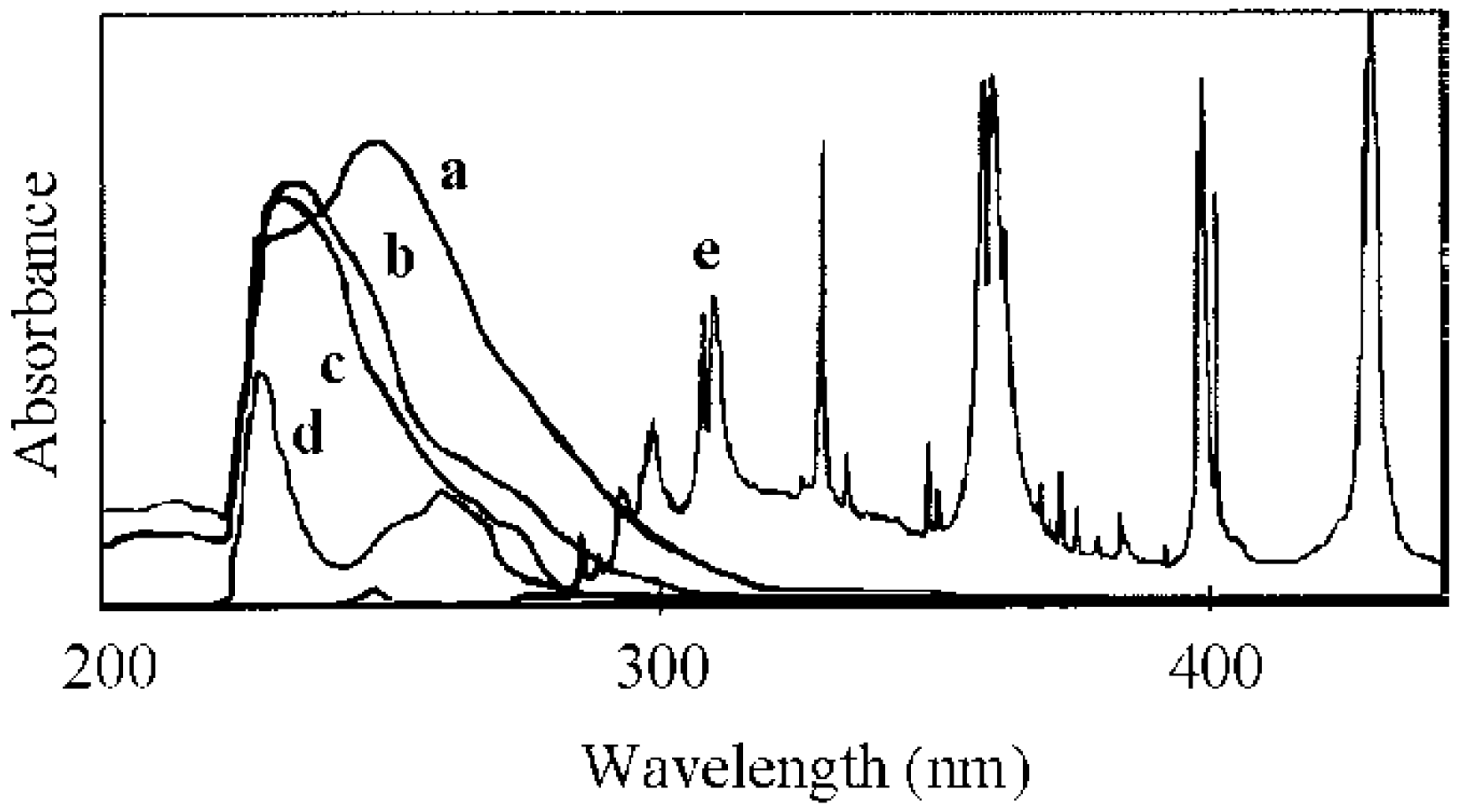
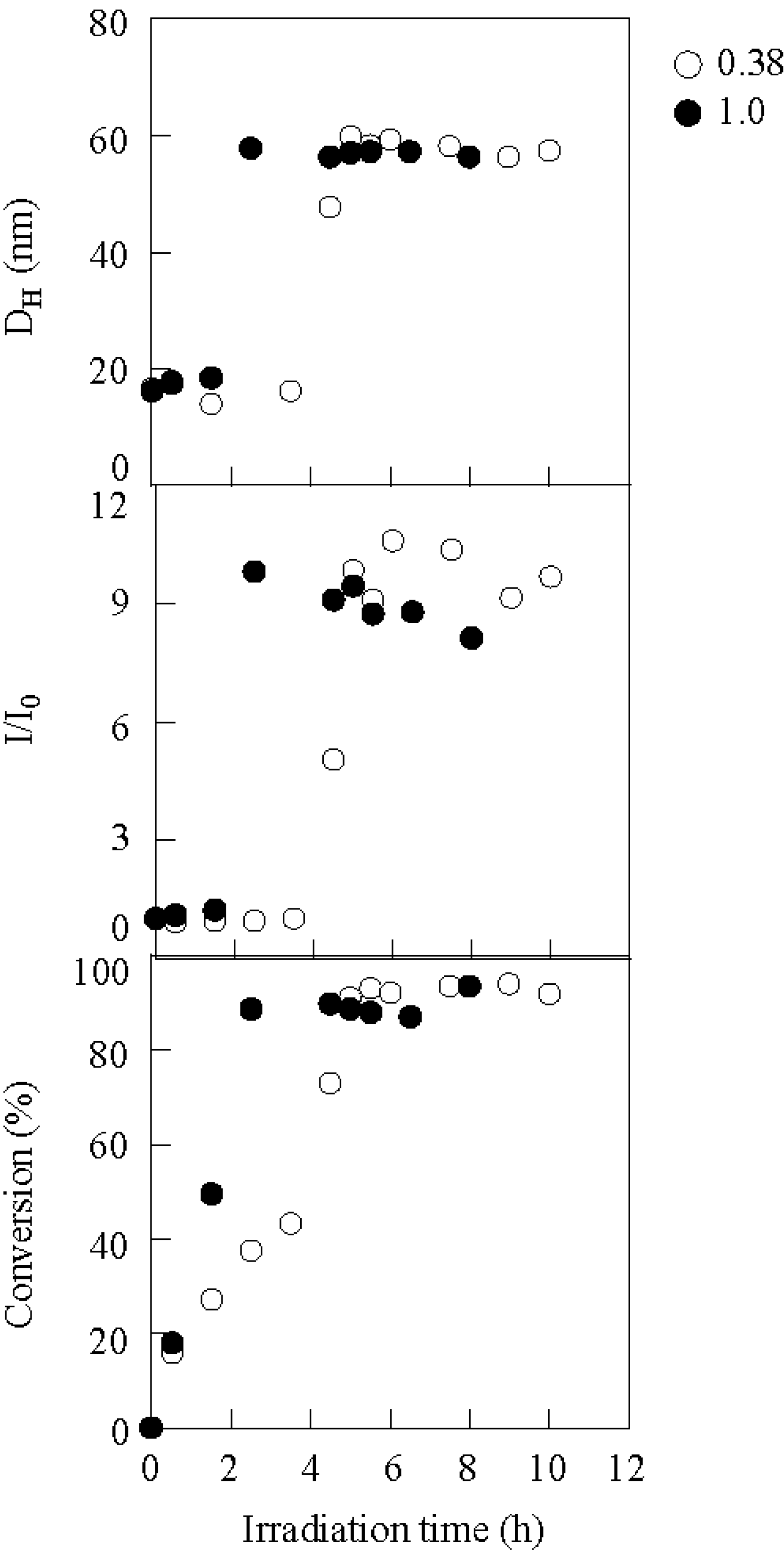
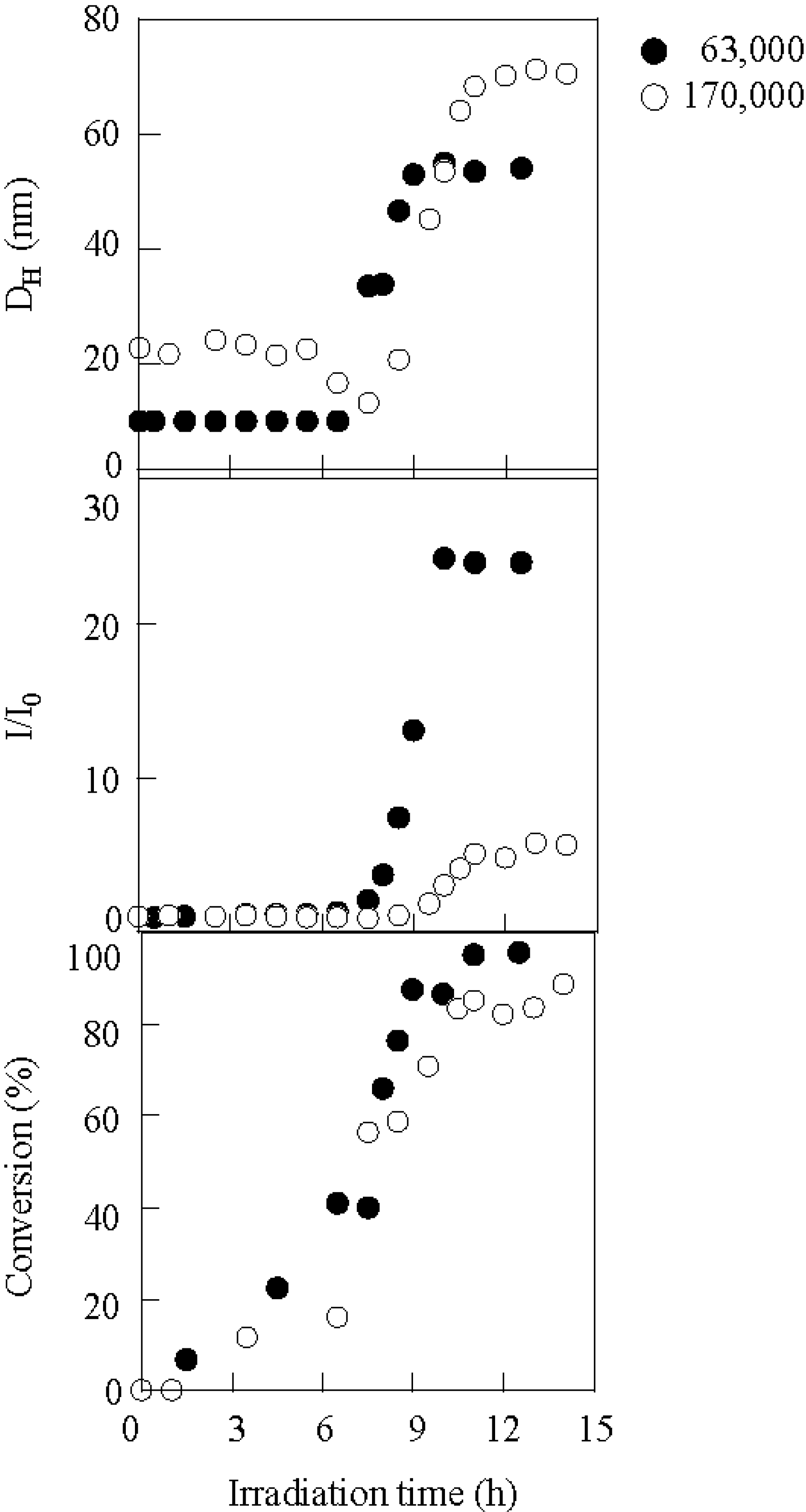
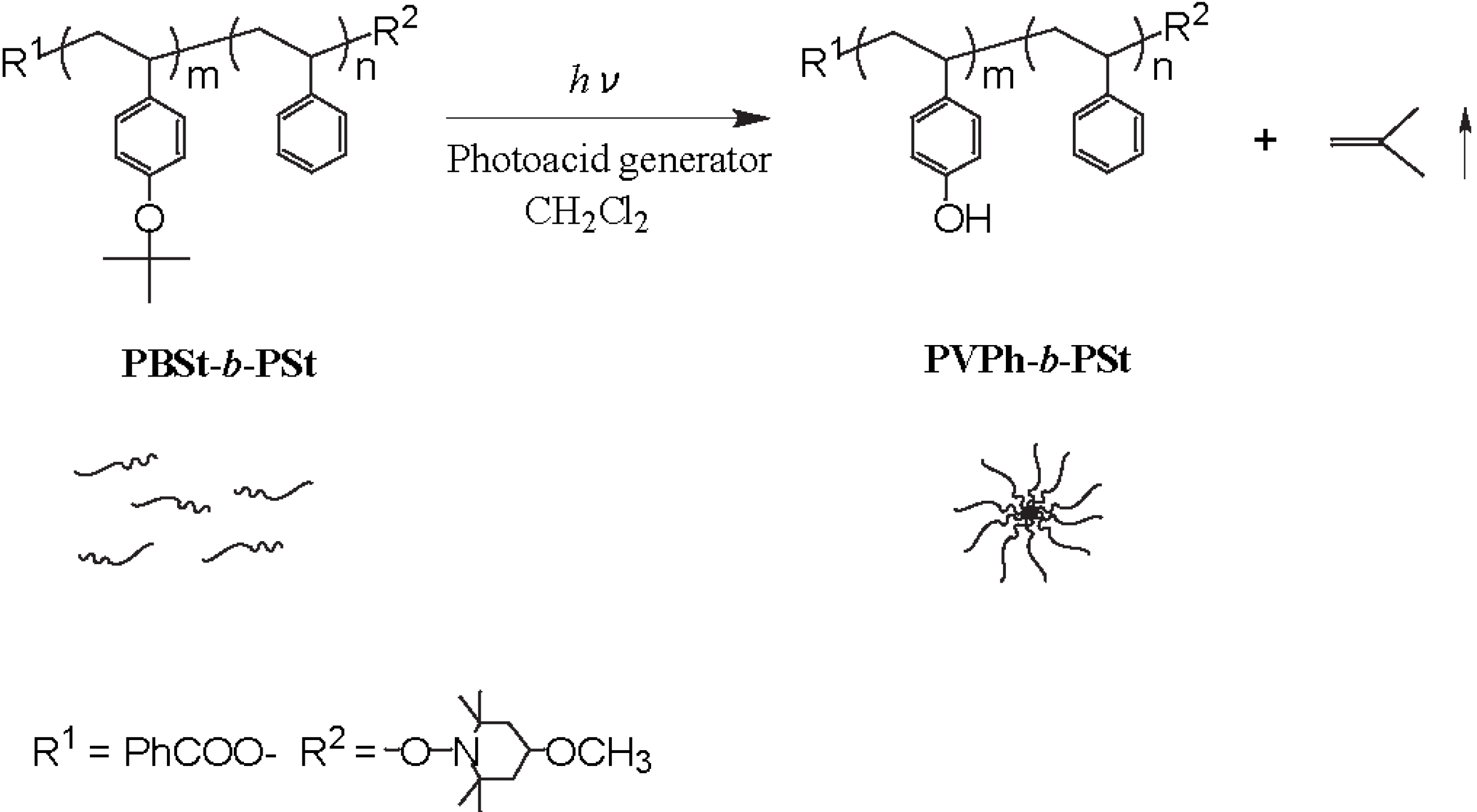
2.1. Photolysis-Induced Micellization [50,51]










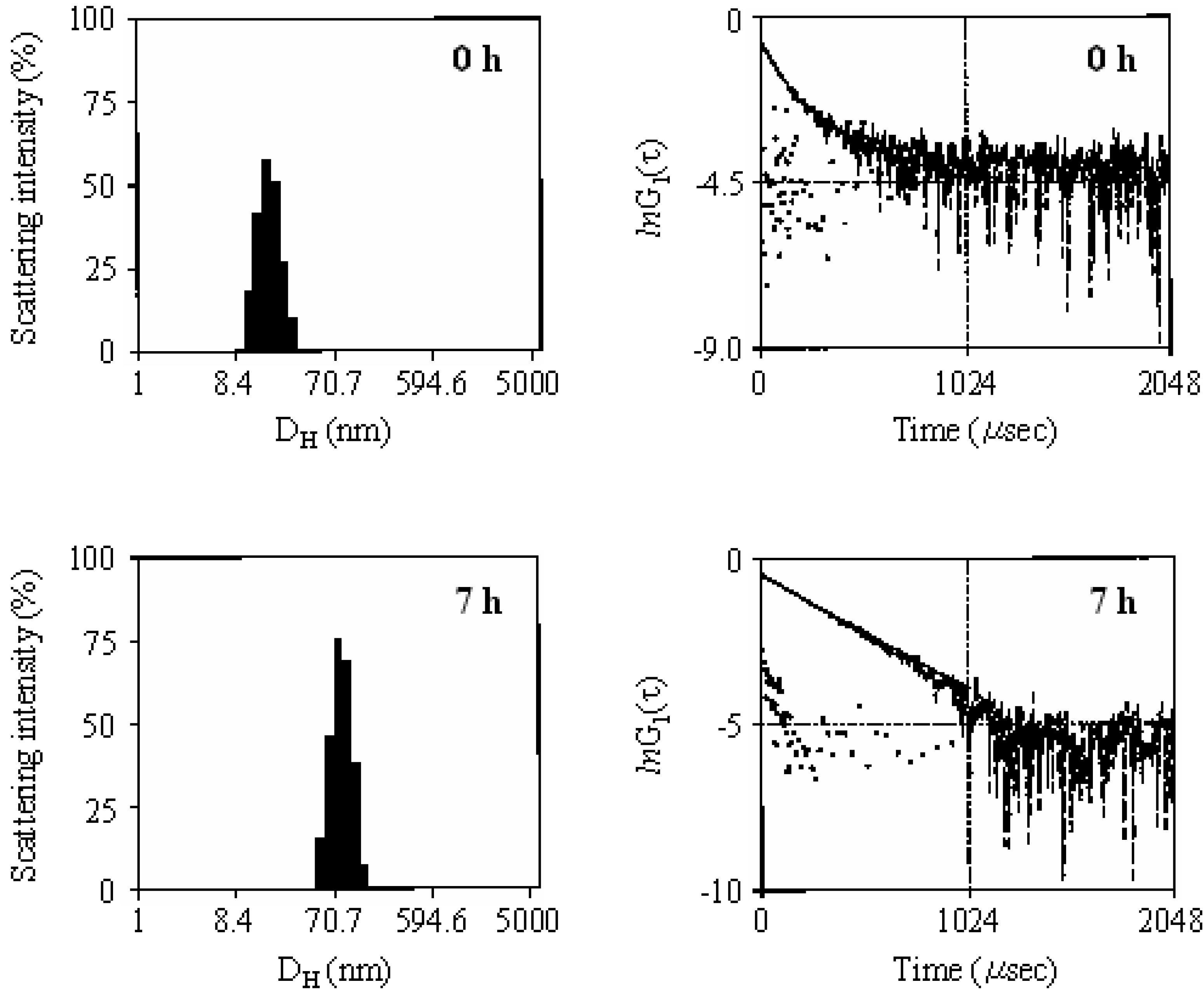
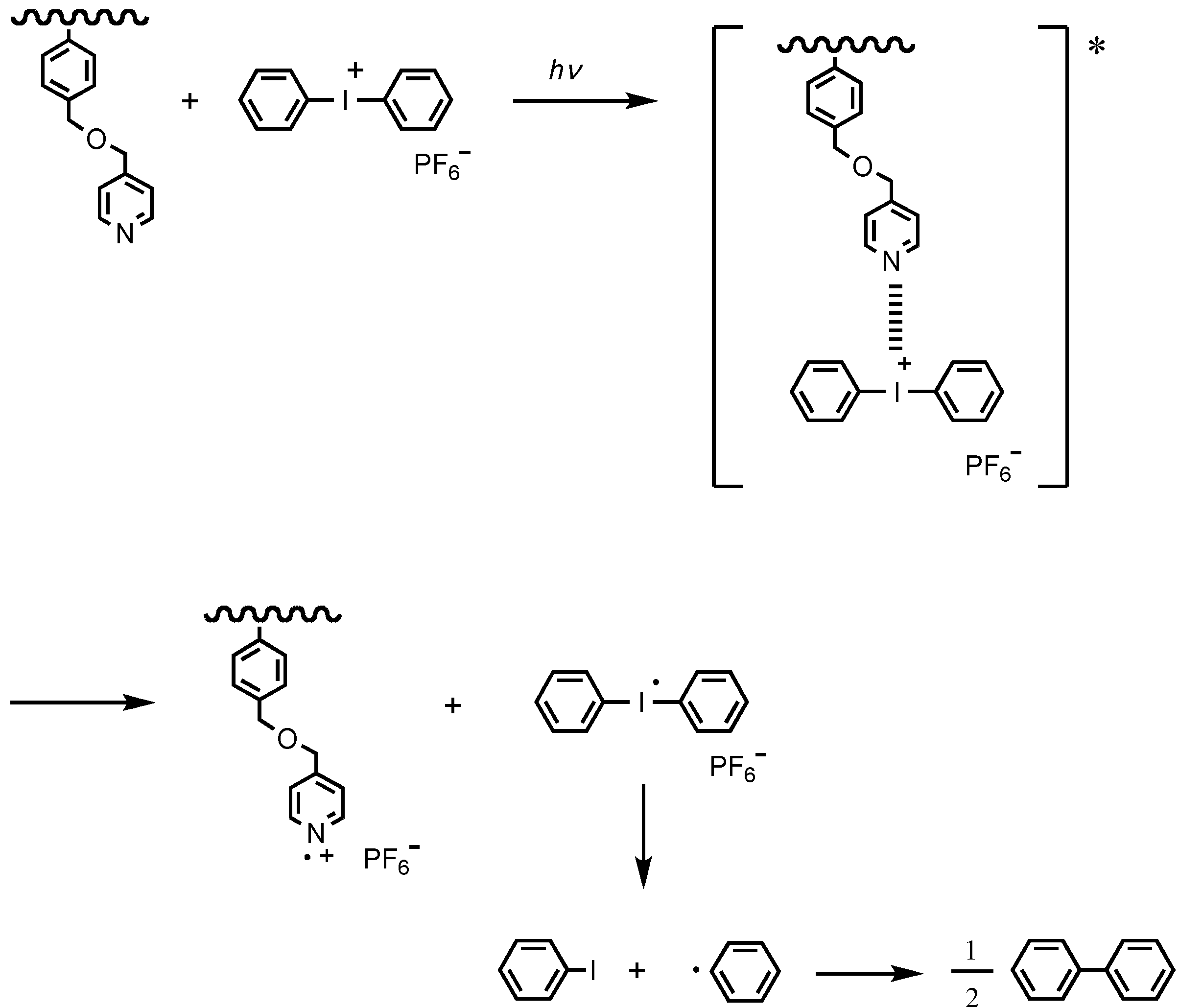
2.2. Photoelectron Transfer-Induced Micellization [54]




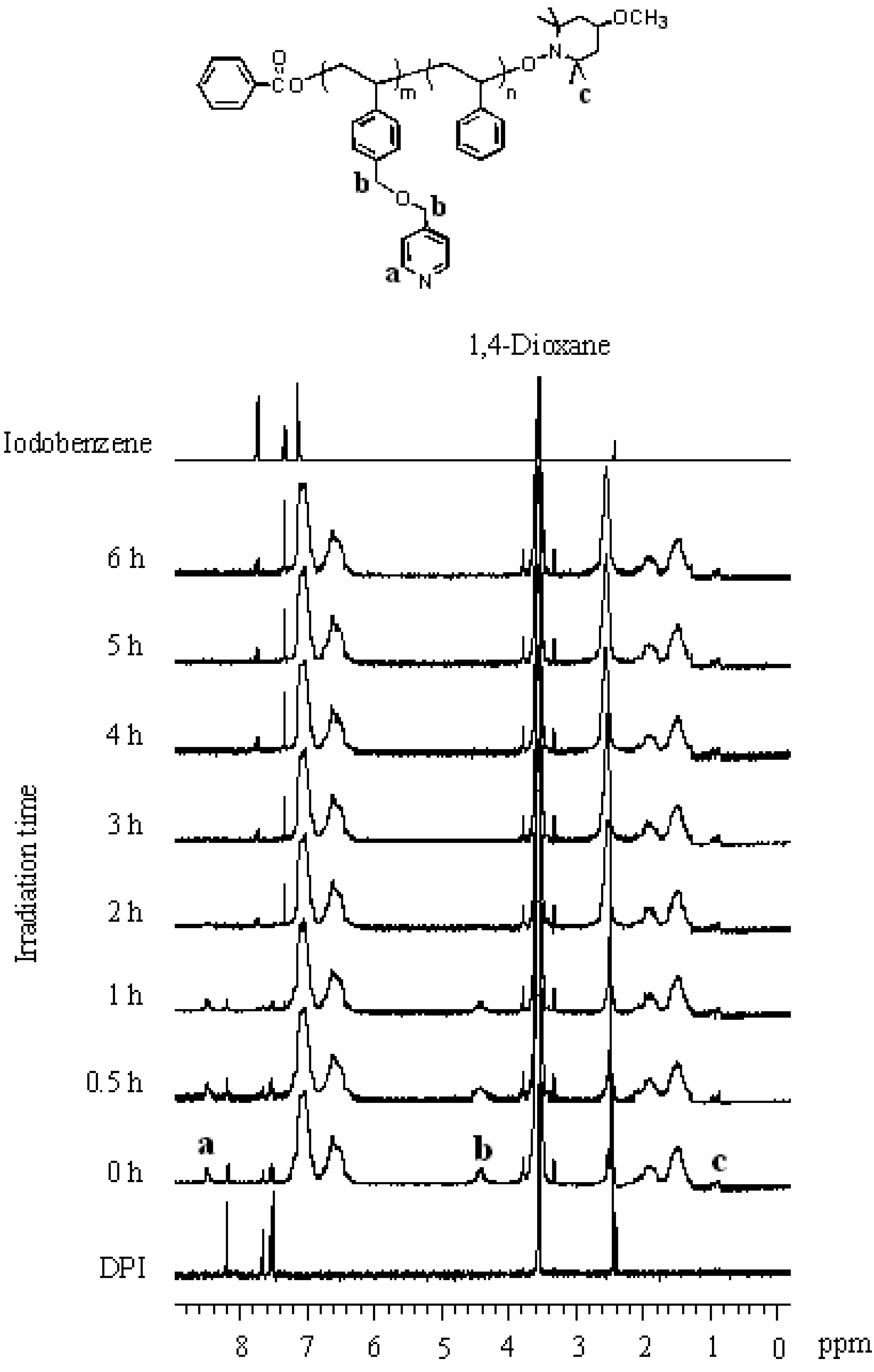
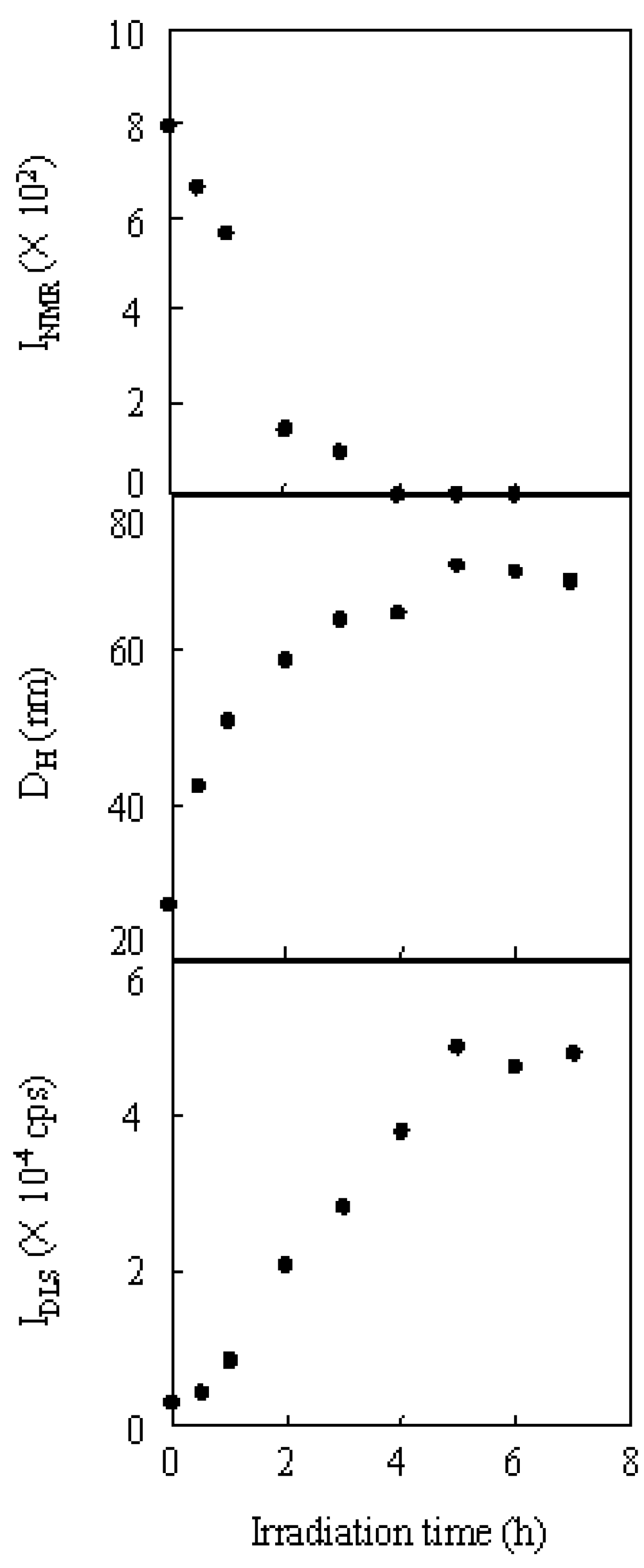
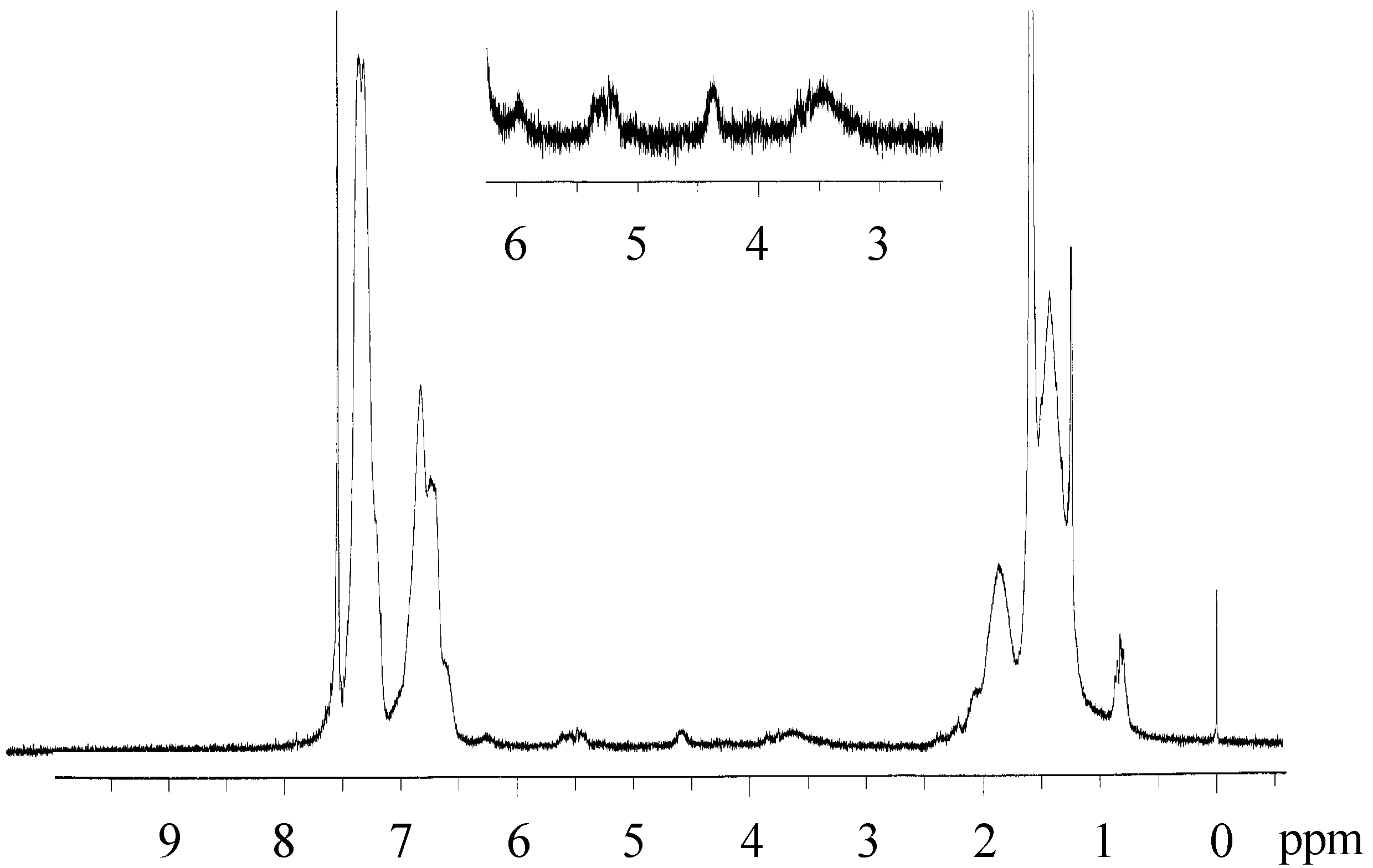
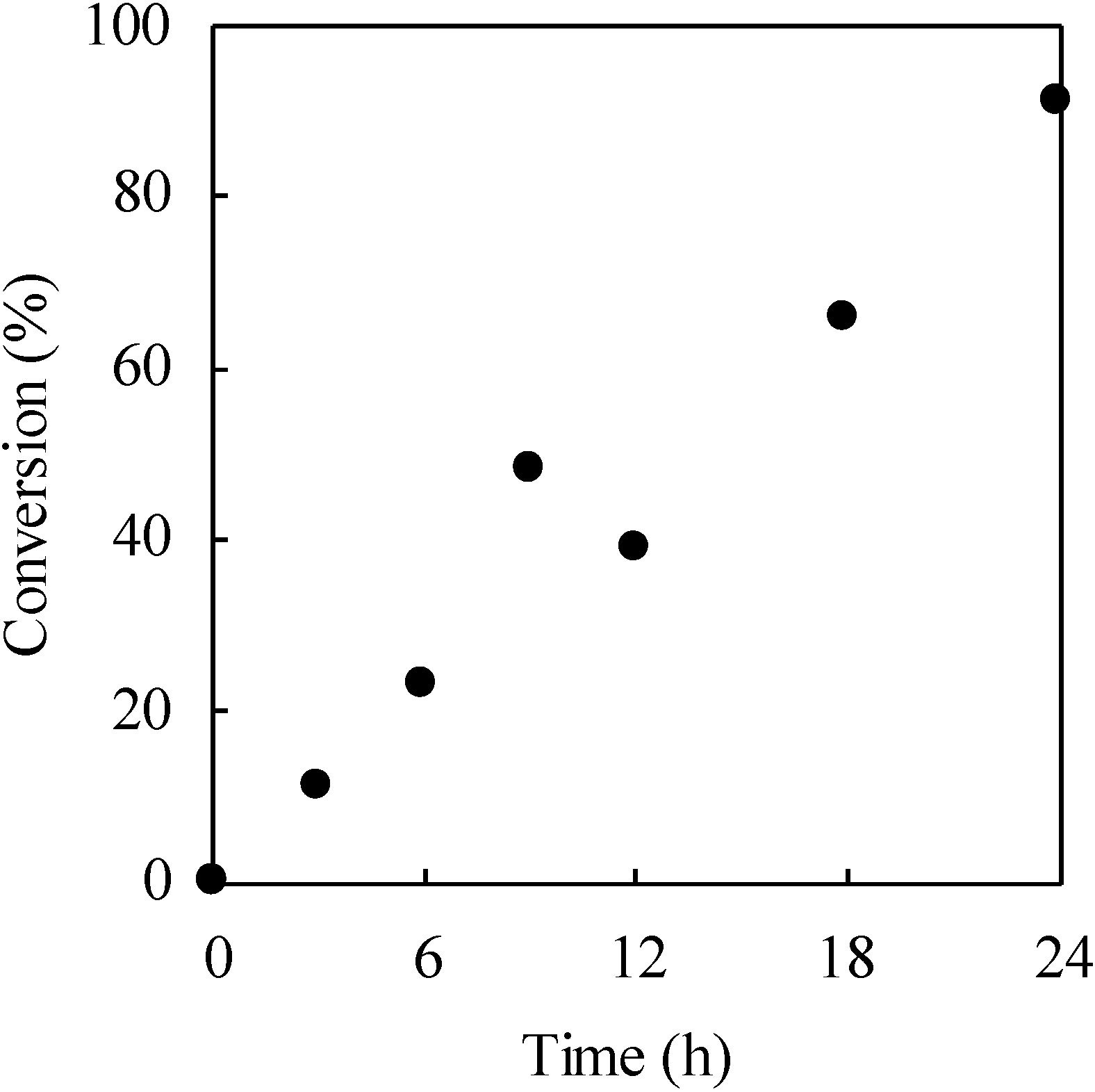
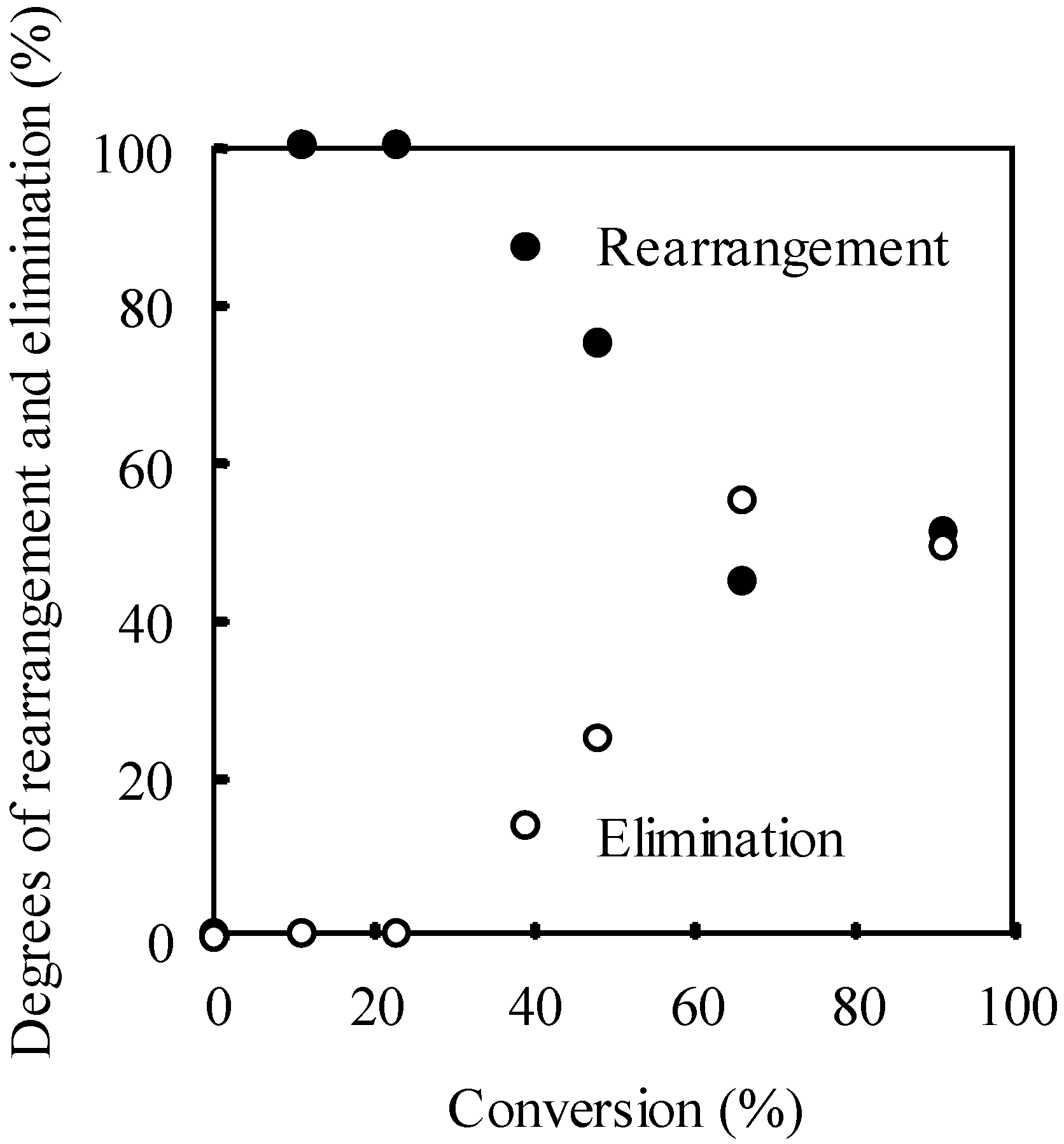
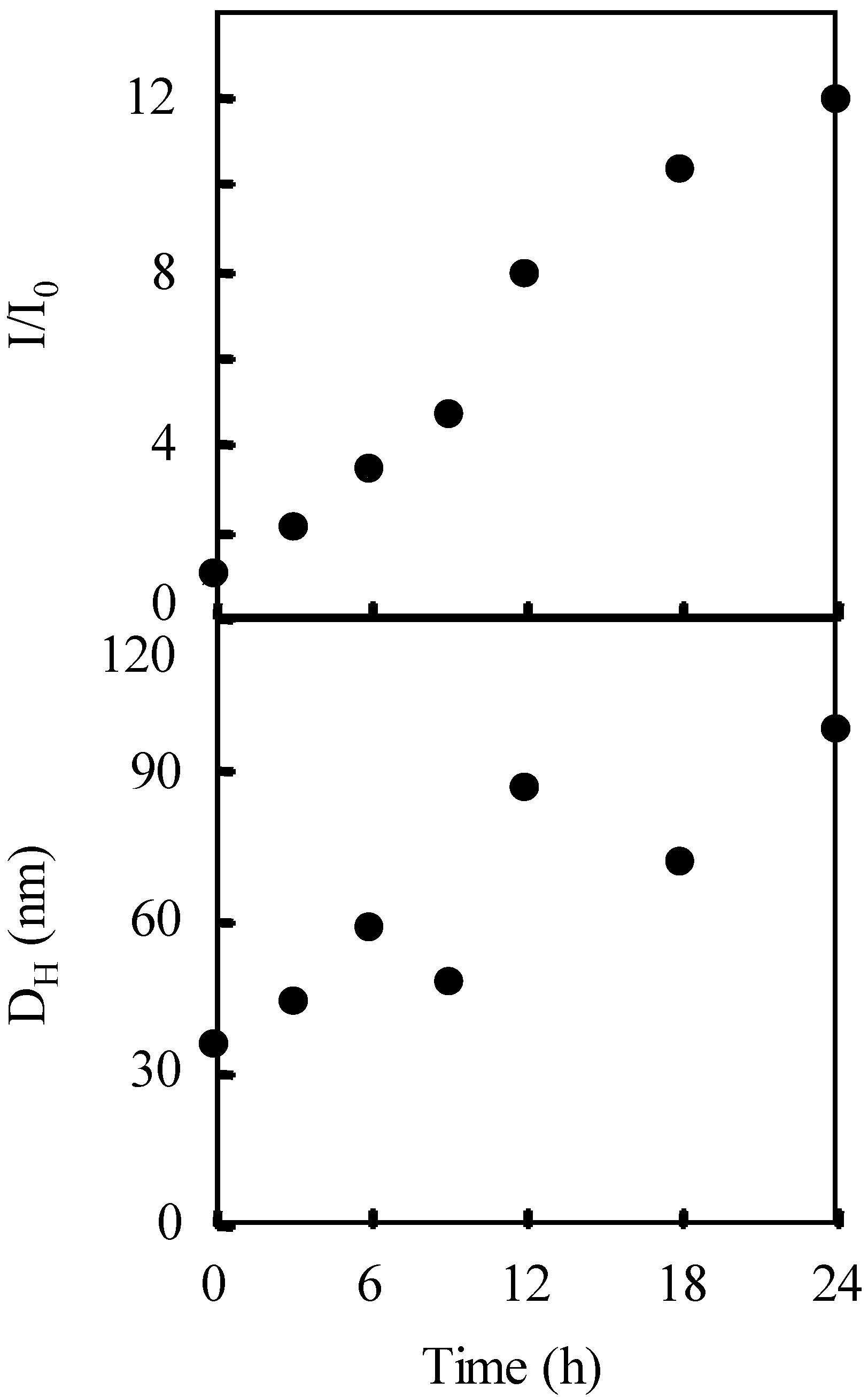
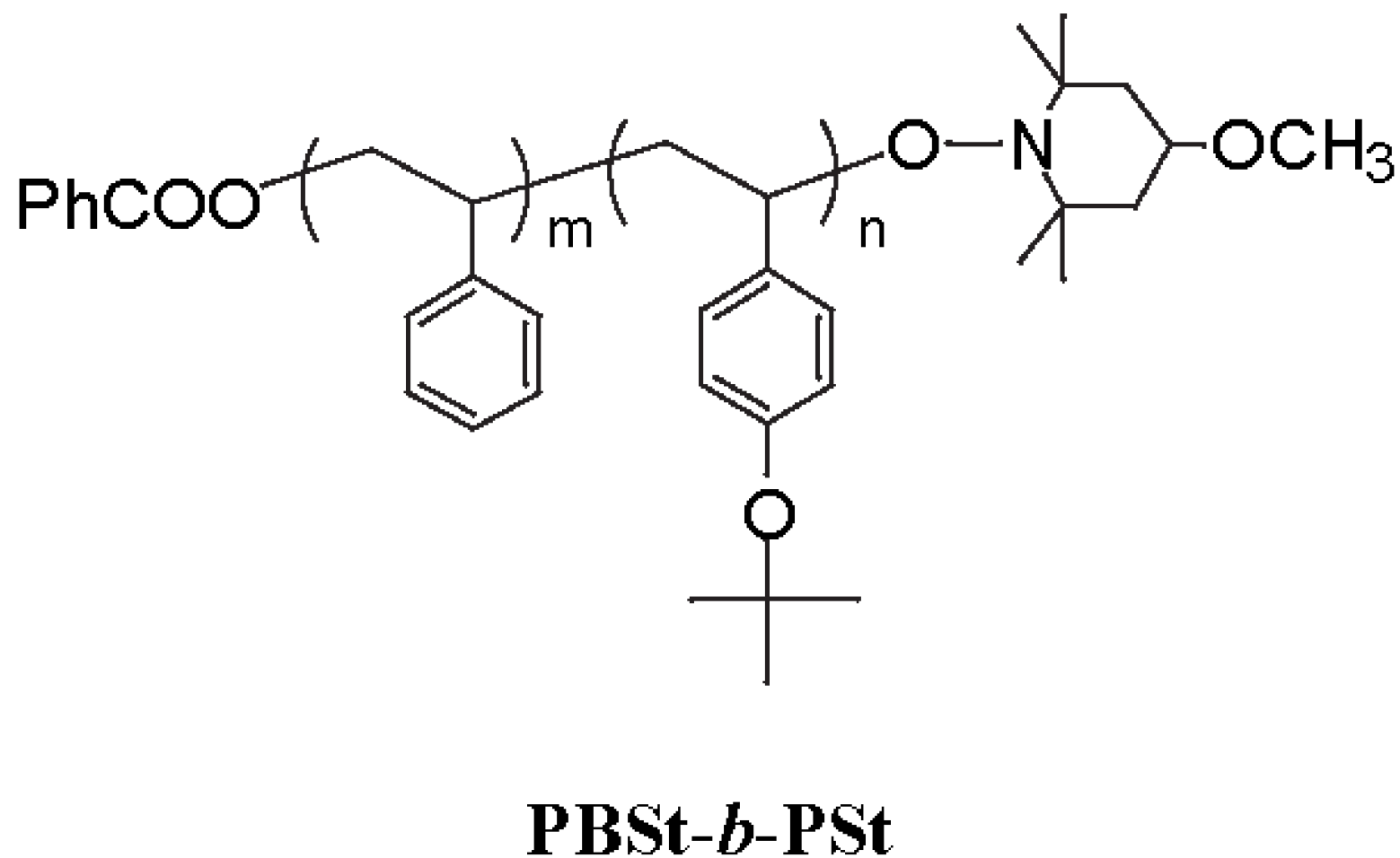
2.3. Photo-Claisen Rearrangement-Induced Micellization [69]






3. Experimental Section
4. Conclusions
References
- Ahlheim, M.; Hellensleben, M.L. Radikalisch polymerisierbare gallens uren in monoschichten, mizellen und vesikeln. Makromol. Chem. 1992, 193, 779–797. [Google Scholar] [CrossRef]
- Isahara, M.; Nakanishi, K.; Ono, K.; Sato, M.; Kikchi, M.; Sito, Y.; Yura, H.; Matsui, T.; Hattori, H.; Uenoyama, M.; Kurita, A. Photocrosslinkable chitosan as a dressing for wound occlusion and accelerator in healing process. Biomaterials 2002, 23, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Hu, Z.; Li, Y. Polymer gel as thermally responsive attenuator for ultrasonic waves. Appl. Phys. Lett. 1999, 74, 2233–2235. [Google Scholar] [CrossRef]
- Sideratou, Z.; Tsiourvas, D.; Paleos, C.M. Quaternized poly(propylene imine) dendrimers as novel pH-sensitive controlled-release systems. Langmuir 2000, 16, 1766–1769. [Google Scholar] [CrossRef]
- Yoon, X.A.; Burgess, D.J. Effect of cationic surfactant on transport of model drugs in emulsion systems. J. Pharm. Pharmacol. 1997, 49, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.J.; Lawrence, S.M.; Barlow, D.J. Aggregation and surface properties of synthetic double-chain non-ionic surfactants in aqueous solution. J. Pharm. Pharmacol. 1997, 49, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Smela, E. Conjugated polymer actuators for biomedical applications. Adv. Mater. 2003, 15, 481–494. [Google Scholar] [CrossRef]
- Hara, S.; Zama, T.; Takashima, W.; Kaneto, K. Artificial muscles based on polypyrrole actuators with large strain and stress induced electrically. Polym. J. 2004, 36, 151–161. [Google Scholar] [CrossRef]
- Matsuyama, T.; Kawata, Y. Field enhancement by surface plasmon polariton in self-assembling nanopatterned media. Appl. Phys. Lett. 2006, 88, 123113. [Google Scholar] [CrossRef]
- Feringa, B.L.; Jager, W.F.; De Lange, B. Organic materials for reversible optical data storage. Tetrahedron 1993, 49, 8267–8310. [Google Scholar] [CrossRef]
- Liang, L.; Feng, X.D.; Liu, J.; Rieke, P.C.; Fryxell, G.E. Reversible surface properties of glass plate and capillary tube grafted by photopolymerization of N-isopropylacrylamide. Macromolecules 1998, 31, 7845–7850. [Google Scholar] [CrossRef]
- Arotcarena, M.; Heise, B.; Ishaya, S.; Laschewsky, A. Switching the inside and outside of aggregates of water-soluble block copolymers with double thermoresponsivity. J. Am. Chem. Sos. 2002, 124, 3787–3793. [Google Scholar] [CrossRef]
- Leclair, S.; Mathew, L.; Giguere, M.; Motallebi, S.; Zhao, Y. Photoinduced alignment of ferroelectric liquid crystals using azobenzene polymer networks of chiral polyacrylates and polymethacrylates. Macromolecules 2003, 36, 9024–9032. [Google Scholar] [CrossRef]
- Yu, Y.; Nakano, M.; Ikeda, T. Photomechanics: directed bending of a polymer film by light. Nature 2003, 425, 145. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, O.; Fabbri, D.; Fissi, A.; Ciardelli, F. Photomodulated conformational changes of azo-modified poly(L-glutamic acid) in micellar systems. Makromol. Chem. Rapid Commun. 1988, 9, 637–640. [Google Scholar] [CrossRef]
- Fissi, A.; Pieroni, O.; Ciardelli, F.; Ruggeri, G.; Umezawa, K. Photoresponsive polypeptides: photochromism and conformation of poly(L-glutamic acid) containing spiropyran units. Biopolymers 1993, 33, 1505–1517. [Google Scholar] [CrossRef]
- Chen, W.J.; Li, G.Z.; Zhou, G.W.; Zhai, L.M.; Li, Z.M. pH-induced spontaneous vesicle formation from NaDEHP. Chem. Phys. Lett. 2003, 374, 482–486. [Google Scholar] [CrossRef]
- Bergsma, M.; Fielden, M.L.; Engberts, J.B.F.N. pH-Dependent aggregation behavior of a sugar-amine Gemini surfactant in water: Vesicles, micelles, and monolayers of hexane-1,6-bis(hexadecyl-1’-deoxyglucitylamine. J. Colloid Interface Sci. 2001, 243, 491–495. [Google Scholar] [CrossRef]
- Yin, H.Q.; Zhou, Z.K.; Huang, J.B.; Zheng, R.; Zhang, Y.Y. Temperature-induced micelle to vesicle transition in the sodium dodecylsulfate/dodecyltriethylammonium bromide system. Angew. Chem. Int. Ed. 2003, 42, 2188–2191. [Google Scholar] [CrossRef]
- Majhi, P.R.; Blume, A. Thermodynamic characterization of temperature-induced micellization and demicellization of detergents studied by differential scanning calorimetry. Langmuir 2001, 17, 3844–3851. [Google Scholar] [CrossRef]
- Yoshida, E.; Ohta, M.; Terada, Y. Reversible control of micellization induced by hydrogen bond crosslinking for a nonamphiphilic diblock copolymer with an α,ω-diamine. Polym. Adv. Technol. 2005, 16, 183–188. [Google Scholar] [CrossRef]
- Yoshida, E.; Tanaka, M.; Takata, T. Self-assembly control of a pyridine-containing diblock copolymer by perfluorinated counter anions during salt-induced micellization. Colloid Polym. Sci. 2005, 283, 1100–1107. [Google Scholar] [CrossRef]
- McClain, J.B.; Canelas, D.A.; Samulski, E.T.; DeSimone, J.M.; Londono, J.D.; Cochran, H.D.; Wignall, G.D.; Chillura-Martino, G.D.; Triolo, R. Design of nonionic surfactants for supercritical carbon dioxide. Science 1996, 274, 2049–2052. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Chu, B. Laser light scattering study of pressure-induced micellization of a diblock copolymer of poly(1,1-dihydroperfluorooctylacrylate) and poly(vinyl acetate) in supercritical carbon dioxide. Macromolecules 1998, 31, 5300–5308. [Google Scholar] [CrossRef]
- Celso, L.; Triolo, A.; Triolo, F.; Donato, D.I.; Steinhart, M.; Kriechbaum, M.; Amenitsch, H.; Triolo, R. Synchrotron SAXS investigation on aggregation phenomena in supercritical carbon dioxide. Eur. Phys. J. Soft Matter 2002, 8, 311–314. [Google Scholar] [CrossRef]
- Yoshida, E.; Nagakubo, A. Convenient synthesis of microspheres by self-assembly of random copolymers in supercritical carbon dioxide. Colloid Polym. Sci. 2007, 285, 441–447. [Google Scholar] [CrossRef]
- Yoshida, E.; Tanaka, T. Oxidation-induced micellization of a diblock copolymer containing stable nitroxyl radicals. Colloid Polym. Sci. 2006, 285, 135–144. [Google Scholar] [CrossRef]
- Yoshida, E.; Tanaka, T. Reduction-induced micellization of a diblock copolymer containing stable nitroxyl radicals. Colloid Polym. Sci. 2008, 286, 827–830. [Google Scholar] [CrossRef]
- Saji, T.; Hoshino, K.; Aoyagi, S. Reversible formation and disruption of micelles by control of the redox state of the surfactant tail group. J. Chem. Soc. Chem. Commun. 1985, 13, 865–866. [Google Scholar] [CrossRef]
- Saji, T.; Ebata, K.; Sugawara, K.; Liu, S.; Kobayashi, K. Electroless plating of organic thin films by reduction of nonionic surfactants containing an azobenzene group. J. Am. Chem. Soc. 1994, 116, 6053–6054. [Google Scholar] [CrossRef]
- Yoshida, E.; Ogawa, H. Micelle formation induced by disproportionation of stable nitroxyl radicals supported on a diblock copolymer. J. Oleo Sci. 2007, 56, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, E. Control of micellization induced by disproportionation of 2,2,6,6-tetramethylpiperidine-1-oxyl supported on side chains of a block copolymer. Colloid Polym. Sci. 2009, 287, 1365–1368. [Google Scholar] [CrossRef]
- Lovrien, R. The photoviscosity effect. Proc. Natl Acad. Sci. USA 1967, 57, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Matejka, L.; Dusek, K. Photochromic polymers: Photoinduced conformational changes and effect of polymeric matrix on the isomerization of photochromes. Makromol. Chem. 1981, 182, 3223–3236. [Google Scholar] [CrossRef]
- Pieroni, O.; Fissi, A. Synthetic photochromic polypeptides: possible models for photoregulation in biology. J. Photochem. Photobiol. B. Biol. 1992, 12, 125–140. [Google Scholar] [CrossRef]
- Menju, A.; Hayashi, K.; Irie, M. Photoresponsive polymers. 3. Reversible solution viscosity change of poly(methacrylic acid) having spirobenzopyran pendant groups in methanol. Macromolecules 1981, 14, 755–758. [Google Scholar]
- Taguchi, M.; Li, G.; Gu, Z.; Sato, O.; Einaga, Y. Magnetic vesicles of amphiphilic spiropyran containing iron oxide particles on a solid state substrate. Chem. Mater. 2003, 15, 4756–4760. [Google Scholar] [CrossRef]
- Meier, H. The photochemistry of stilbenoid compounds and their role in materials technology. Angew. Chem. Int. Ed. Engl. 1992, 31, 1399–1420. [Google Scholar] [CrossRef]
- Szczubialka, K.; Nowakoaska, M. Response of micelles formed by smart terpolymers to stimuli studied by dynamic light scattering. Polymer 2003, 44, 5269–5274. [Google Scholar] [CrossRef]
- Eastoe, J.; Sanchez, M.; Dominguez, S.; Wyatt, P.; Beeby, A.; Heenan, R. Properties of a stilibene-containing Gemini photosurfactant: Light-triggered changes in surface tension and aggregation. Langmuir 2002, 18, 7837–7844. [Google Scholar] [CrossRef]
- Irie, M.; Hosoda, M. Photoresponsive polymers. Reversible solution viscosity change of poly(N,N-dimethylacrylamide) with pendant triphenylmethane leucohydroxide residues in methanol. Makromol. Chem. Rapid Commun. 1985, 6, 533–536. [Google Scholar]
- Dunkin, I.R.; Gittinger, A.; Sherrington, D.C.; Whittaker, P. A photodestructible surfactant. J. Chem. Soc. Chem. Commun. 1994. [CrossRef]
- Mezger, T.; Nuyken, O.; Meindl, K.; Wokaun, A. Light decomposable emulsifiers: application of alkyl-substituted aromatic azosulfonates in emulsion polymerization. Prog. Org. Coatings 1996, 29, 147–157. [Google Scholar] [CrossRef]
- Nuyken, O.; Voit, B. The photoactive diazosulfonate group and its role in polymer chemistry. Macromol. Chem. Phys. 1997, 198, 2337–2372. [Google Scholar] [CrossRef]
- Haubs, M.; Ringsdorf, H. Photosensitive monolayers, bilayer membranes and polymers. New J. Chem. 1987, 11, 151–156. [Google Scholar]
- Okamoto, Y.; Yoshida, H.; Takamuku, S. Photochemical reaction of [4(4’-alkoxybenzoyl)phenylmethylphosphonic acids. Application to a photo-degradable surfactant. Chem. Lett. 1988, 17, 569–572. [Google Scholar]
- Veronese, A.; Berclaz, N.; Luisi, P.L. Photoinduced formation of bilayer vesicles. J. Phys. Chem. B 1998, 102, 7078–7080. [Google Scholar] [CrossRef]
- Cohen, S.M.; Young, R.H.; Markhart, A.H. Transparent ultraviolet-barrier coatings. J. Polym. Sci. A 1971, 9, 3263–3299. [Google Scholar] [CrossRef]
- Tessier, T.G.; Frechet, J.M.J. The Photo-fries rearrangement and its use in polymeric imaging systems. ACS Sympo. Ser. 1985, 266, 269–292. [Google Scholar]
- Yoshida, E.; Kuwayama, S. Micelle formation induced by photolysis of a poly(tert-butoxystyrene)-block-polystyrene diblock copolymer. Colloid Polym. Sci. 2007, 285, 1287–1291. [Google Scholar] [CrossRef]
- Yoshida, E.; Kuwayama, S. Photolysis-induced micellization of a poly(4-tert-butoxystyrene)-block-polystyrene diblock copolymer. Colloid Polym. Sci. 2008, 286, 1621–1627. [Google Scholar] [CrossRef]
- Marquardt, D.W. An algorithm for least-squares estimation of nonlinear parameters. J. Soc. Indust. Appl. Math. 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Conlon, D.A.; Crivello, J.V.; Lee, J.L.; O’Brien, M.J. The synthesis, characterization, and deblocking of poly(4-tert-butoxystyrene) and poly(4-tert-butoxy-.alpha.-methylstyrene). Macromolecules 1989, 22, 509–516. [Google Scholar] [CrossRef]
- Yoshida, E.; Kuwayama, S.; Kawaguchi, S. Photo-induced micellization of poly(4-pyridinemethoxymethylstyrene)-block-polystyrene using a photo-acid generator. Colloid Polym. Sci. 2010, 288, 91–95. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. New photoinitiators for cationic polymerization. J. Polym. Sci. Symp. 1976, 56, 383–395. [Google Scholar]
- Crivello, J.V.; Lam, J.H.W.; Volante, C.N. Photoinitiated cationic polymerization using diaryliodonium salts. J. Radiat. Curing 1977, 4, 2–16. [Google Scholar]
- Crivello, J.V.; Sangermano, M. Visible and long-wavelength photoinitiated cationic polymerization. J. Polym. Sci. A Polym. Chem. 2001, 39, 343–356. [Google Scholar] [CrossRef]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate by a nitroxide mediator. Colloid Polym. Sci. 2008, 286, 1663–1666. [Google Scholar] [CrossRef]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate by 2,2,6,6-tetramethylpiperidine-1-oxyl in the presence of a photo-acid generator. Colloid Polym. Sci. 2009, 287, 767–772. [Google Scholar] [CrossRef]
- Yoshida, E. Synthesis of poly(methyl methacrylate)-block-poly(tetrahydrofuran) by photo-living radical polymerization using a 2,2,6,6-tetramethylpiperidine-1-oxyl macromediator. Colloid Polym. Sci. 2009, 287, 1417–1424. [Google Scholar] [CrossRef]
- Yoshida, E. Photo-living radical polymerization of methyl methacrylate using alkoxyamine as an initiator. Colloid Polym. Sci. 2010, 288, 7–13. [Google Scholar] [CrossRef]
- Yoshida, E. Nitroxide-mediated photo-living radical polymerization of vinyl acetate. Colloid Polym. Sci. 2010, 288, 73–78. [Google Scholar] [CrossRef]
- Yoshida, E. Effect of azoinitiators on nitroxide-mediated photo-living radical polymerization of methyl methacrylate. Colloid Polym. Sci. 2010, 288, 341–345. [Google Scholar] [CrossRef]
- Crivello, J.V. The discovery and development of onium salt cationic photoinitiators. J. Polym. Sci. A Polym. Chem. 1999, 37, 4241–4254. [Google Scholar] [CrossRef]
- Pappas, S.P.; Gatechair, L.; Jilek, J.H. Photoinitiation of cationic polymerization. III. Photosensitization of diphenyliodonium and triphenylsulfonium salts. J. Polym. Sci. Polym. Chem. Ed. 1984, 22, 77–84. [Google Scholar] [CrossRef]
- Kunze, A.; Muller, U.; Tittes, K.; Fouassier, J.P.; Morlet-Savary, F. Triplet quenching by onium salts in polar and nonpolar solvents. J. Photochem. Photobiol. A 1997, 110, 115–122. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. Dye-sensitized photoinitiated cationic polymerization. J. Polym. Sci. Polym. Chem. Ed. 1976, 16, 2441–2451. [Google Scholar] [CrossRef]
- Brown, W. Light Scattering Principles and Development; Clarendon Press Oxford: Gloucestershire, UK, 1996; pp. 439–442. [Google Scholar]
- Kellmann, A. Primary photochemical processes of cationic acridine orange in aqueous solution studied by flash photolysis. Photochem. Photobiol. 1974, 20, 103–108. [Google Scholar] [CrossRef]
- Devoe, R.J.; Sahyun, M.R.V.; Serpone, N.; Sharma, D.K. Transient intermediates in the photolysis of iodonium cations. Can. J. Chem. 1987, 65, 2342–2349. [Google Scholar] [CrossRef]
- Yoshida, E.; Kuwayama, S. Micelle formation induced by photo-Claisen rearrangement of poly(4-allyloxystyrene)-block-polystyrene. Colloid Polym. Sci. 2009, 287, 789–793. [Google Scholar] [CrossRef]
- Fischer, M. Photochemische reaktionen, V: Photochemische synthese mittlerer und grosser stickstoffhaltiger ringe. Tetrahedron Lett. 1968, 9, 4295–4298. [Google Scholar] [CrossRef]
- Fischer, M.; Mattheus, A. Photochemische reaktionen, VI. Photoumlagerungen von N-phenyl-lactamen. Chem. Ber. 1969, 102, 342–350. [Google Scholar]
- Bellus, D.; Schaffner, K. Photochemische reaktionen. 43. Mitteilung [1]. UV.-bestrahlung von N-phenylurethan und N-phenylthiourethan. Helv. Chim. Acta 1968, 51, 221–224. [Google Scholar]
- Kan, R.O.; Furey, R.L. Photochemical generation and decomposition of dibenzoylaniline. Tetrahedron Lett. 1966, 7, 2573–2578. [Google Scholar] [CrossRef]
- Finnegan, R.A.; Hagen, A.W. An analogue of the photo-fries rearrangement; the photolysis of vinyl benzoate. Tetrahedron Lett. 1963, 4, 365–368. [Google Scholar] [CrossRef]
- Gorodetsky, M.; Mazur, Y. Photochemistry of enolic systems. II. Irradiation of dienol acetates. J. Am. Chem. Soc. 1964, 86, 5213–5218. [Google Scholar]
- Yogev, A.; Mazur, Y. Irradiation of enol lactones. J. Am. Chem. Soc. 1965, 87, 3520–3521. [Google Scholar] [CrossRef]
- Bertele, E.; Boos, H.; Dunitz, J.D.; Elsinger, F.; Eschenmoser, A.; Felner, I.; Gribi, H.P.; Gschwend, H.; Meyer, E.F.; Pesaro, M.; Scheffold, R. Ein synthetischer Zugang zum Corrinsystem. Angew. Chem. 1964, 76, 393–399. [Google Scholar] [CrossRef]
- Nozaki, H.; Okada, T.; Noyori, R.; Kawanishi, M. Photochemical rearrangement of arenesulphonanilides to p-aminodiarylsulphones. Tetrahedron 1966, 22, 2177–2180. [Google Scholar] [CrossRef]
- Pitchumani, K.; Warri, M.; Ramamurthy, V. Remarkable product selectivity during photo-Fries and photo-Claisen rearrangements within zeolites. J. Am. Chem. Soc. 1996, 118, 9428–9429. [Google Scholar] [CrossRef]
- Pincock, A.L.; Pincock, J.A.; Stefanova, R. Substituent effects on the rate constants for the photo-Claisen rearrangement of allyl aryl ethers. J. Am. Chem. Soc. 2002, 124, 9768–9778. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Veglia, A.V.; Rossi, R.H. β-Cyclodextrin effects on photo-Claisen rearrangement of allyl phenyl ether. Can. J. Chem. 1997, 75, 1151–1155. [Google Scholar] [CrossRef]
- Anderson, J.C.; Reese, C.B. Photo-induced Fries rearrangements. Proc. Chem. Soc. 1960, 217. [Google Scholar]
- Arai, T.; Tobita, S.; Shizuka, H. Direct measurements of the rates of 1,3- and 1,5-sigmatropic hydrogen shifts in the photo-Fries rearrangements of phenyl acetate. Chem. Phys. Lett. 1994, 223, 521–526. [Google Scholar] [CrossRef]
- Magdy, M.; Malik, A.; Mayo, P. Surface photochemistry: The amide photo-Fries rearrangement. Can. J. Chem. 1983, 62, 1275–1278. [Google Scholar]
- Pitchumani, K.; Warrier, M.; Ramamurthy, V. Utility of zeolitic medium in photo-Fries and photo-Claisen rearrangements. Res. Chem. Intermed. 1999, 25, 623–631. [Google Scholar] [CrossRef]
- Miyazawa, T.; Endo, T.; Shiihashi, S.; Ogawara, M. Selective oxidation of alcohols by oxoaminium salts (R2N:O+ X-). J. Org. Chem. 1985, 50, 1332–1334. [Google Scholar] [CrossRef]
- Yoshida, E.; Kunugi, S. Micelle formation of poly(vinyl phenol)-block-polystyrene through hydrogen bond crosslinking by α, ω-diamine. J. Polym. Sci. A Polym. Chem. Ed. 2002, 40, 3063–3067. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yoshida, E.; Kuwayama, S. Photo-Induced Micellization of Block Copolymers. Polymers 2010, 2, 623-648. https://doi.org/10.3390/polym2040623
Yoshida E, Kuwayama S. Photo-Induced Micellization of Block Copolymers. Polymers. 2010; 2(4):623-648. https://doi.org/10.3390/polym2040623
Chicago/Turabian StyleYoshida, Eri, and Satoshi Kuwayama. 2010. "Photo-Induced Micellization of Block Copolymers" Polymers 2, no. 4: 623-648. https://doi.org/10.3390/polym2040623
APA StyleYoshida, E., & Kuwayama, S. (2010). Photo-Induced Micellization of Block Copolymers. Polymers, 2(4), 623-648. https://doi.org/10.3390/polym2040623