
Optical Characterization of the Hole Polaron in a Series of Diketopyrrolopyrrole Polymers Used for Organic Photovoltaics


Abstract
:
1. Introduction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Jsc (mA/cm2) | Voc (V) | Fill Factor (FF) | PCE (%) | μsat (cm2·V−1·s−1) * | Ref. |
|---|---|---|---|---|---|---|
| PDPP-FPF | 3.6 | 0.56 | 37 | 0.78 | 0.04 | [30] |
| PDPP-FNF | 8.6 | 0.71 | 42 | 2.6 | 0.11 | [30] |
| PDPP-FAF | 7.8 | 0.69 | 45 | 2.5 | 0.07 | [30] |
| PDPP-TNT | 13 | 0.73 | 47 | 4.5 | 0.65 | [31,32] |
2. Experimental Section
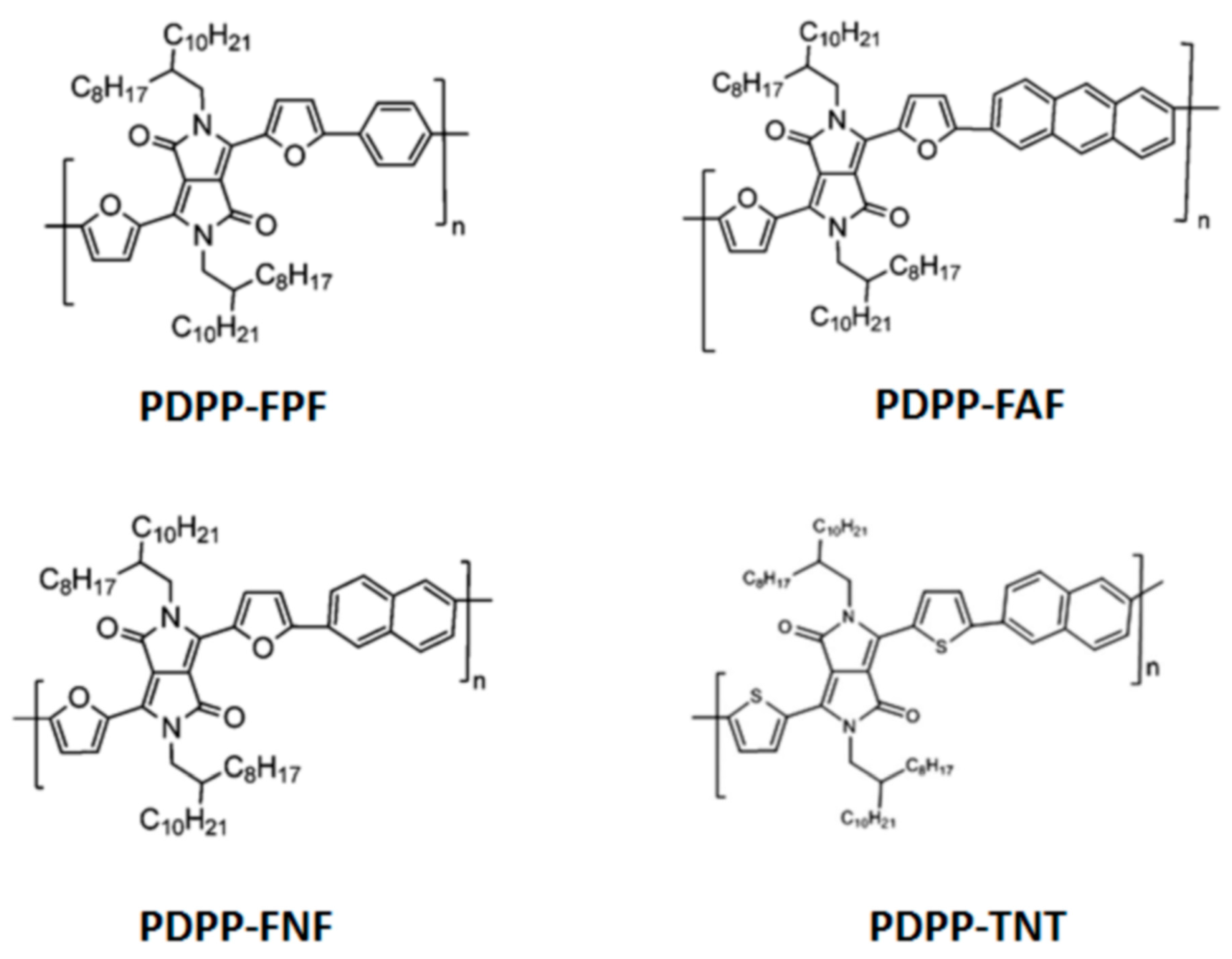
2.1. Polymer Synthesis
2.2. UV-Vis Spectroscopy and Chemical Doping
2.3. Spectroelectrochemical Measurements
2.4. Photoinduced Absorption Measurements
2.5. Photoluminescence Measurements
2.6. Density Functional Theory (DFT) Calculations
3. Results and Discussion
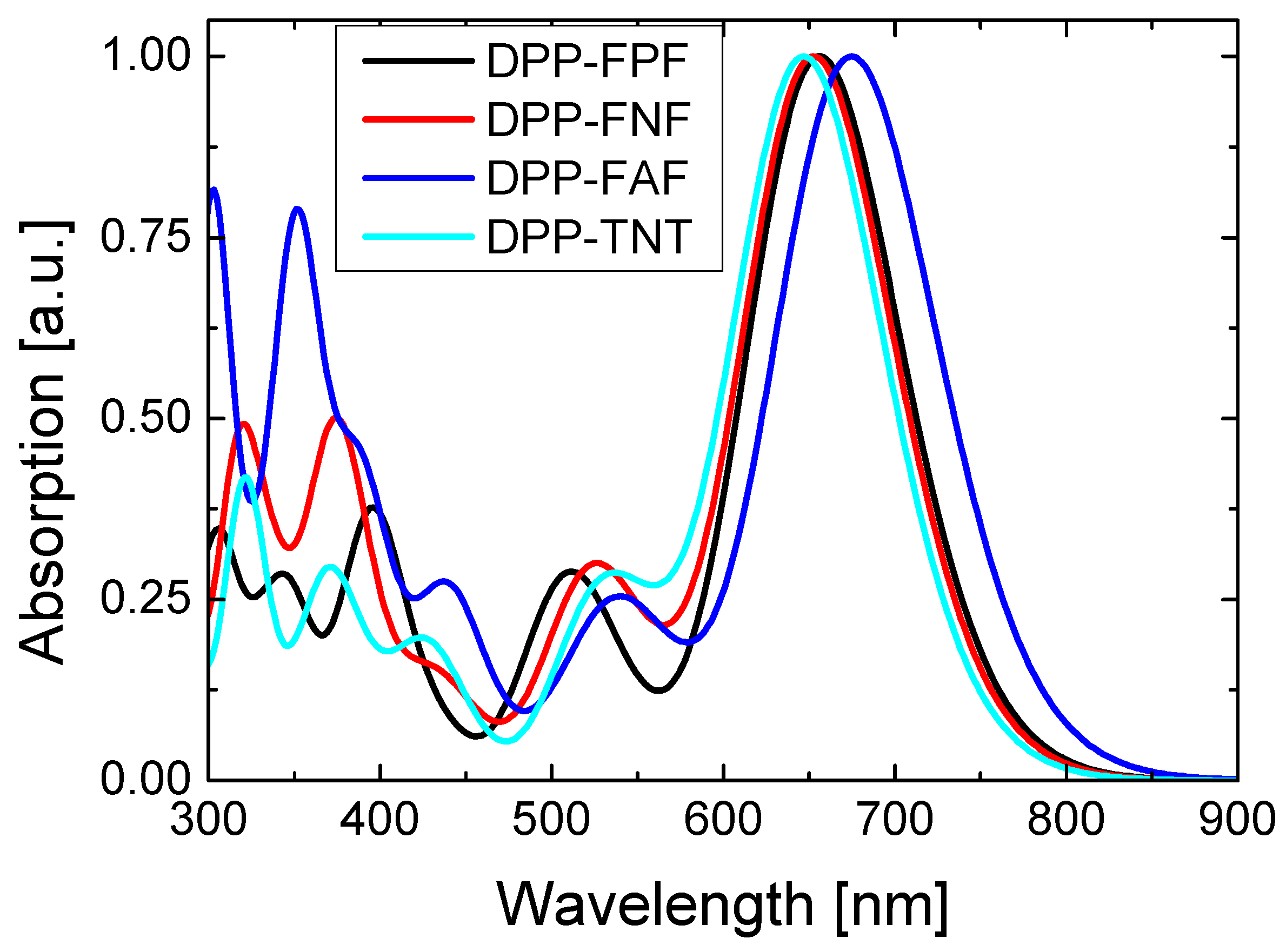
3.1. UV-Vis Absorption

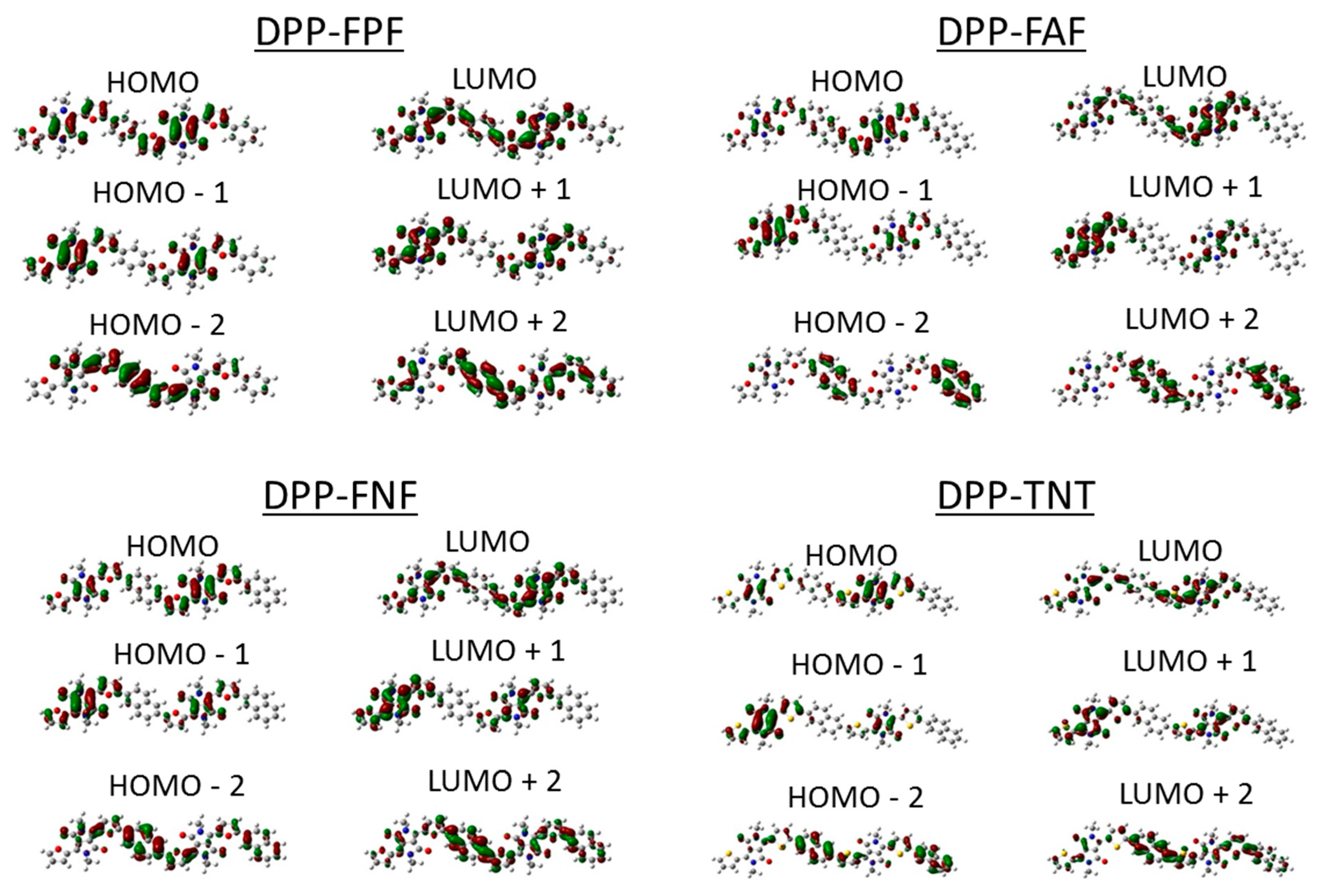
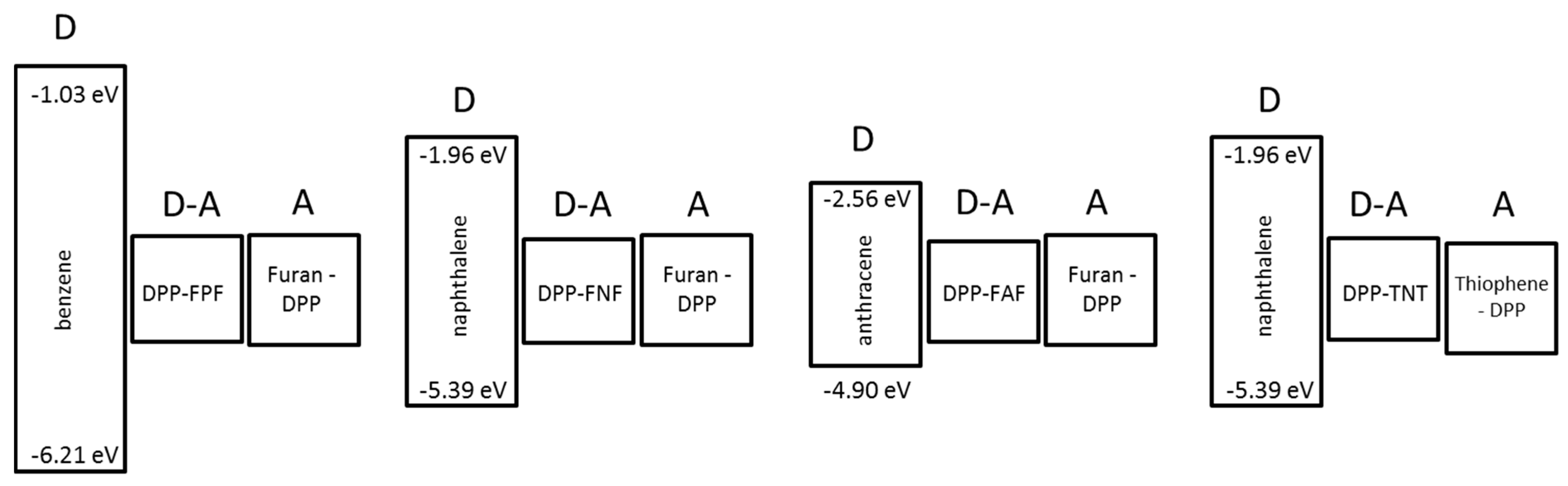
3.2. DFT Modeling

| Monomer unit | HOMO (eV) | LUMO (eV) | Band gap (eV) | HOMO (eV) CV [ref] | HOMO (eV) PESA [ref] | Optical band gap (eV) [ref] |
|---|---|---|---|---|---|---|
| benzene | −6.21 | −1.03 | 5.18 | - | - | - |
| naphthalene | −5.39 | −1.96 | 3.43 | - | - | - |
| anthracene | −4.90 | −2.56 | 2.35 | - | - | - |
| furan-DPP | −4.59 | −3.18 | 1.41 | - | - | - |
| thiophene-DPP | −4.70 | −3.30 | 1.40 | - | - | - |
| DPP-FPF | −4.48 | −3.11 | 1.37 | 5.40 [30] | 5.48 [30] | 1.55 [30] |
| DPP-FNF | −4.48 | −3.13 | 1.35 | 5.34 [30] | 5.50 [30] | 1.63 [30] |
| DPP-FAF | −4.45 | −3.17 | 1.28 | 5.33 [30] | 5.52 [30] | 1.64 [30] |
| DPP-TNT | −4.60 | −3.28 | 1.32 | 5.29 [31] | 5.40 | 1.60 |


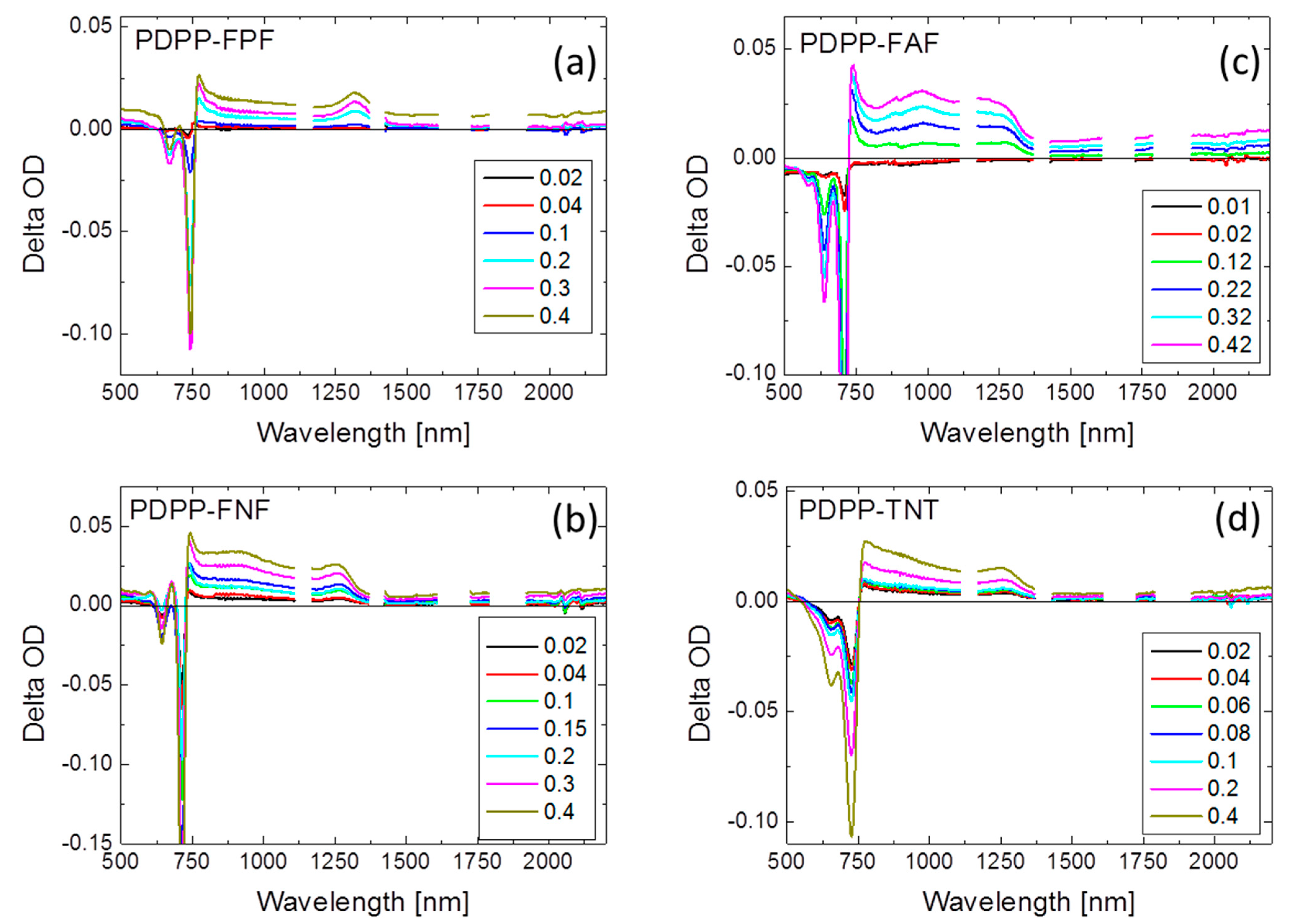
3.3. Chemical Doping



| Polymer (wavelength) | Molar absorption coefficient (cm2/mol) | Polaron cross section (cm2) |
|---|---|---|
| PDPP-FPF (1312 nm) | 2.06 × 107 | 7.87 × 10−17 |
| PDPP-FNF (1252 nm) | 7.08 × 106 | 2.71 × 10−17 |
| PDPP-FAF (982 nm) | 6.80 × 106 | 2.60 × 10−17 |
| PDPP-TNT (1258 nm) | 1.68 × 107 | 6.44 × 10−17 |
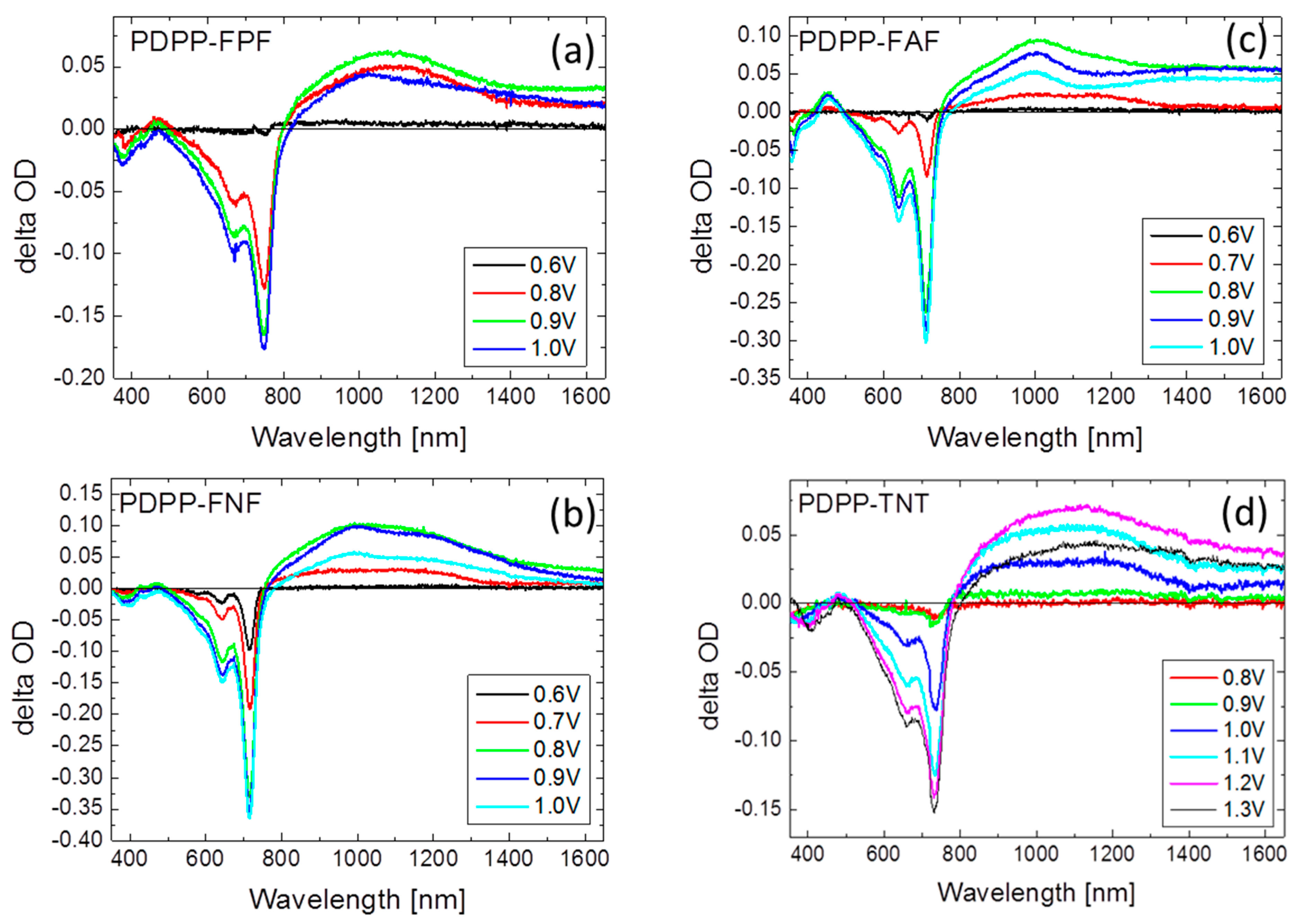
3.4. Spectroeclectrochemistry


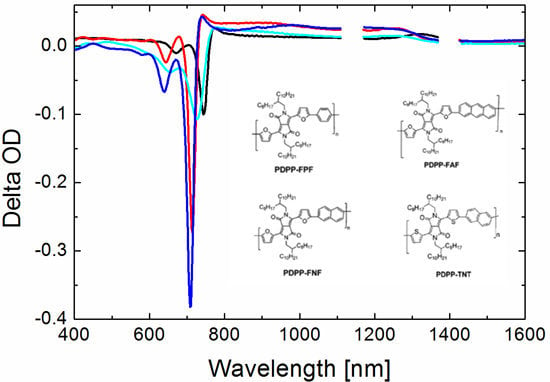
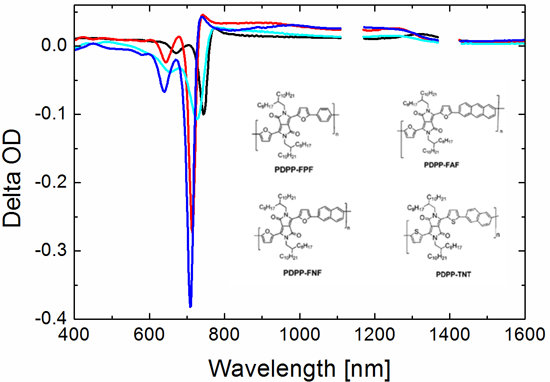
3.5. Photoinduced Absorption Spectroscopy
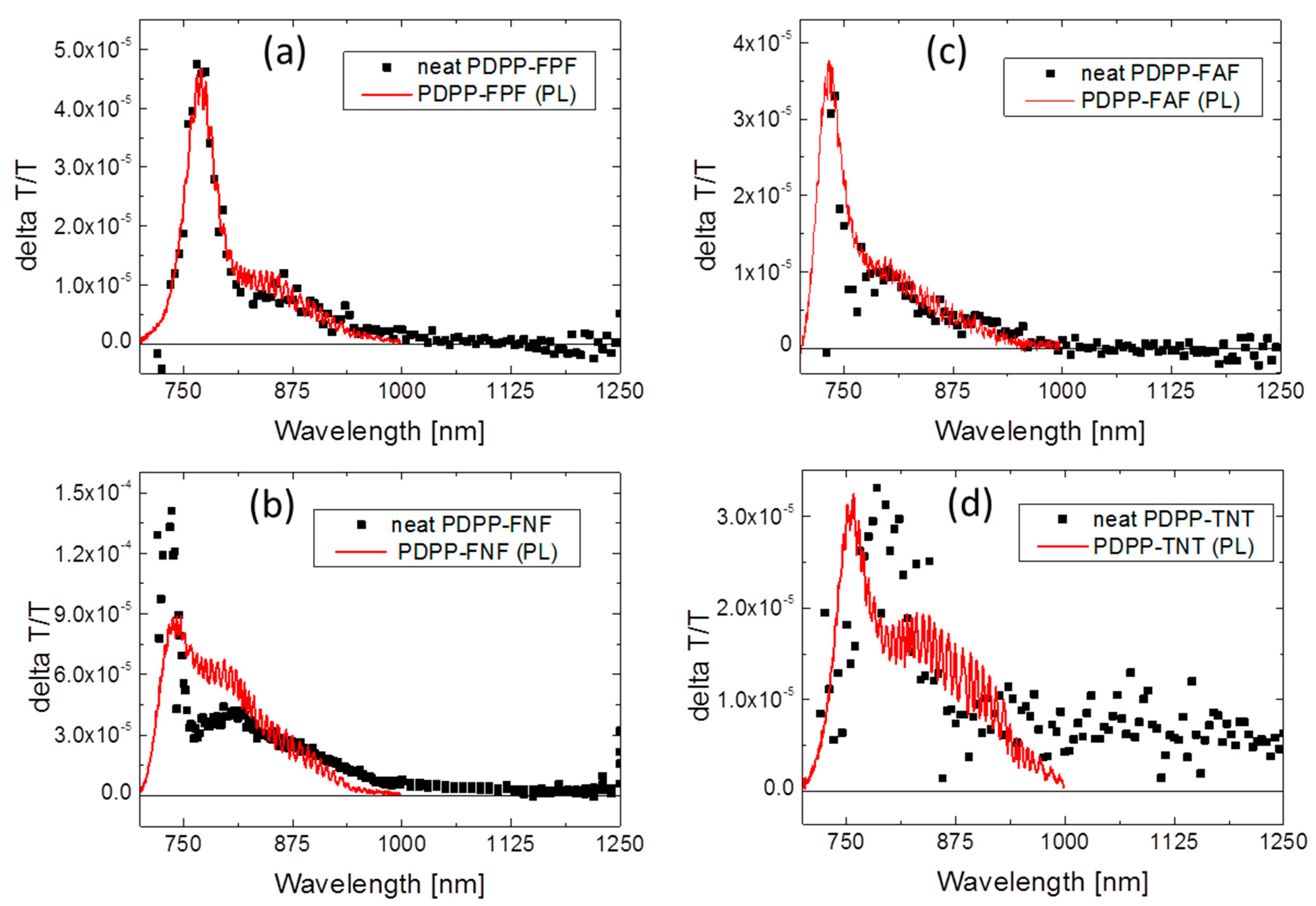
3.5.1. Neat Polymer Films

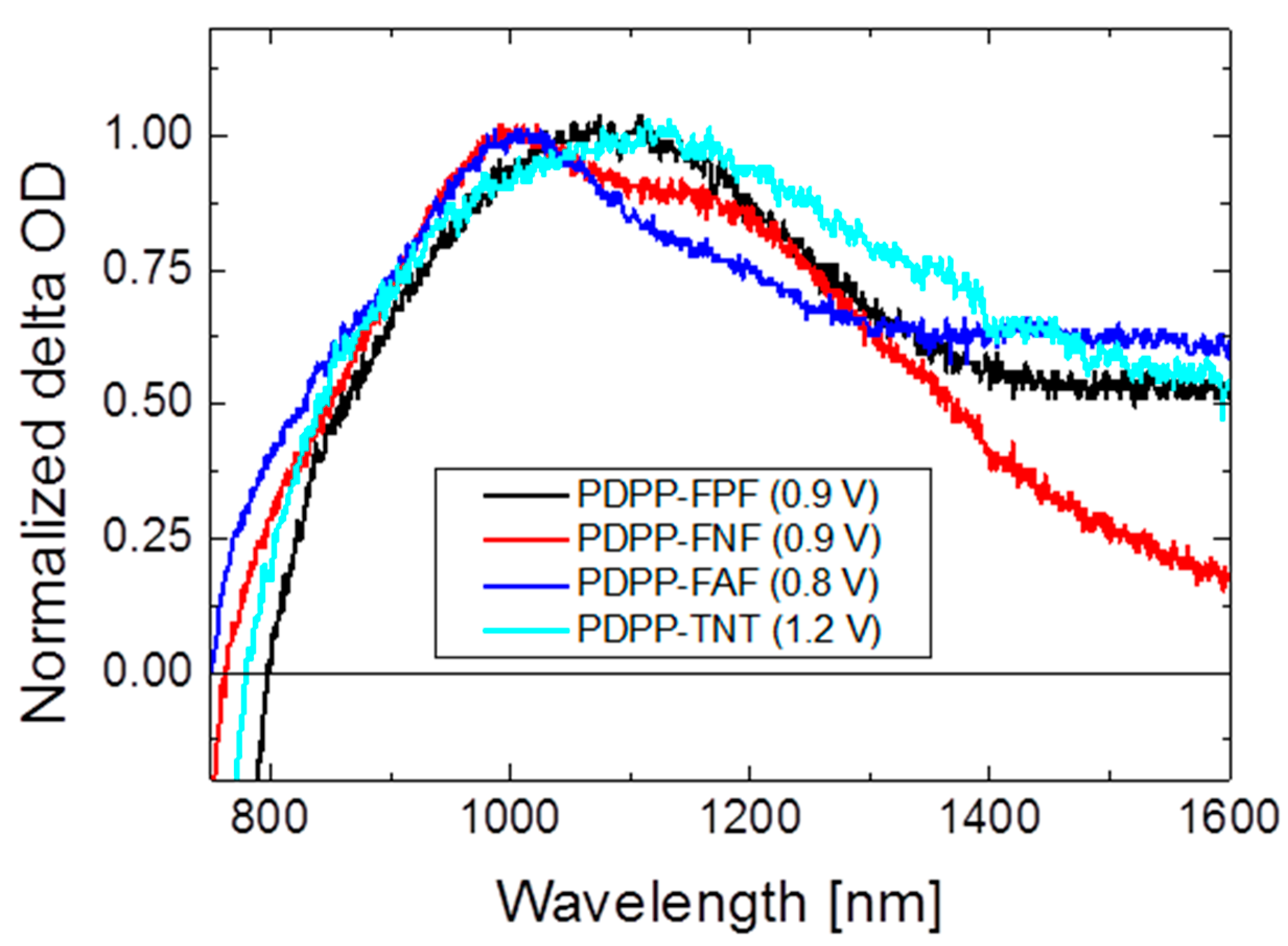
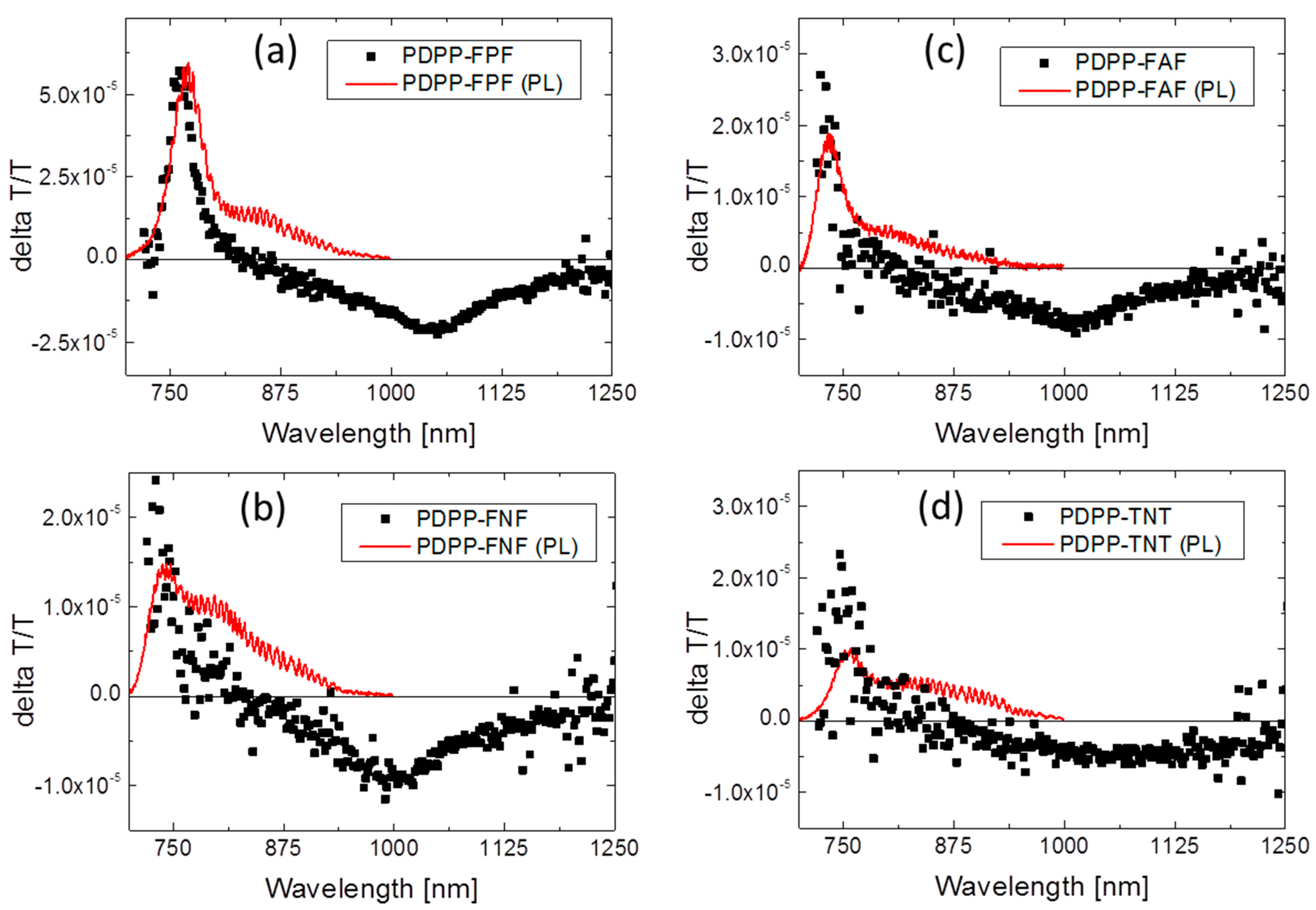
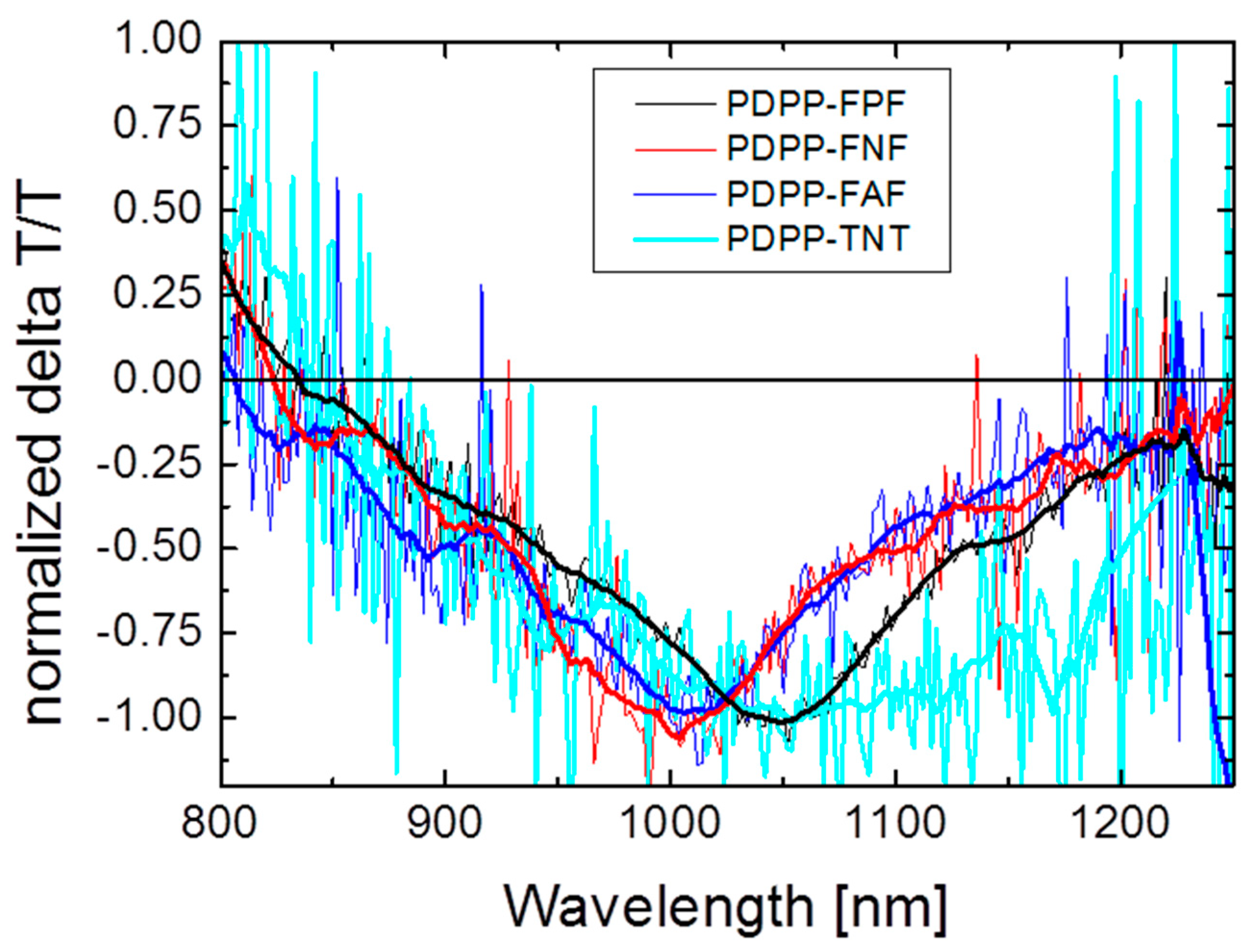
3.5.2. Bulk Heterojunction Films


4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Green, M.A.; Emery, K.; Hishikawa, Y.; Warta, W.; Dunlop, E.D. Solar cell efficiency tables (version 43). Prog. Photovolt. Res. Appl. 2014, 22, 1–9. [Google Scholar] [CrossRef]
- Dennler, G.; Scharber, M.C.; Brabec, C.J. Polymer-fullerene bulk-heterojunction solar cells. Adv. Mater. 2009, 21, 1323–1338. [Google Scholar] [CrossRef]
- Peet, J.; Wen, L.; Byrne, P.; Rodman, S.; Forberich, K.; Shao, Y.; Drolet, N.; Gaudiana, R.; Dennler, G.; Waller, D. Bulk heterojunction solar cells with thick active layers and high fill factors enabled by a bithiophene-co-thiazolothiazole push-pull copolymer. Appl. Phys. Lett. 2011, 98. [Google Scholar] [CrossRef]
- Gregg, B.A. Excitonic solar cells. J. Phys. Chem. B 2003, 107, 4688–4698. [Google Scholar] [CrossRef]
- Brabec, C.J.; Zerza, G.; Cerullo, G.; De Silvestri, S.; Luzzati, S.; Hummelen, J.C.; Sariciftci, S. Tracing photoinduced electron transfer process in conjugated polymer/fullerene bulk heterojunctions in real time. Chem. Phys. Lett. 2001, 340, 232–236. [Google Scholar] [CrossRef]
- Bredas, J.-L.; Norton, J.E.; Cornil, J.; Coropceanu, V. Molecular understanding of organic solar cells: The challenges. Acc. Chem. Res. 2009, 42, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Beljonne, D.; Cornil, J.; Muccioli, L.; Zannoni, C.; Brédas, J.L.; Castet, F. Electronic processes at organic−organic interfaces: Insight from modeling and implications for opto-electronic devices. Chem. Mater. 2011, 23, 591–609. [Google Scholar] [CrossRef]
- Gelinas, S.; Rao, A.; Kumar, A.; Smith, S.L.; Chin, A.W.; Clark, J.; van der Poll, T.S.; Bazan, G.C.; Friend, R.H. Ultrafast long-range charge separation in organic semiconductor photovoltaic diodes. Science 2014, 343, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Grancini, G.; Maiuri, M.; Fazzi, D.; Petrozza, A.; Egelhaaf, H.J.; Brida, D.; Cerullo, G.; Lanzani, G. Hot exciton dissociation in polymer solar cells. Nat. Mater. 2013, 12, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.W.; Soci, C.; Moses, D.; Zhu, Z.; Waller, D.; Gaudiana, R.; Brabec, C.J.; Heeger, A.J. Ultrafast electron transfer and decay dynamics in a small band gap bulk heterojunction material. Adv. Mater. 2007, 19, 2307–2312. [Google Scholar] [CrossRef]
- Heremans, P.; Cheyns, D.; Rand, B.P. Strategies for increasing the efficiency of heterojunction organic solar cells: Material selection and device architecture. Acc. Chem. Res. 2009, 42, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Riedel, I.; Dyakonov, V. Influence of electronic transport properties of polymer-fullerene blends on the performance of bulk heterojunction photovoltaic devices. Phys. Status Solidi A 2004, 201, 1332–1341. [Google Scholar] [CrossRef]
- Su, Y.W.; Lan, S.C.; Wei, K.H. Organic photovoltaics. Mater. Today 2012, 15, 554–562. [Google Scholar] [CrossRef]
- Scharber, M.C.; Mühlbacher, D.; Koppe, M.; Denk, P.; Waldauf, C.; Heeger, A.J.; Brabec, C.J. Design rules for donors in bulk-heterojunction solar cells—Towards 10 % energy-conversion efficiency. Adv. Mater. 2006, 18, 789–794. [Google Scholar] [CrossRef]
- Brabec, C.J.; Cravino, A.; Meissner, D.; Sariciftci, N.S.; Fromherz, T.; Rispens, M.T.; Sanchez, L.; Hummelen, J.C. Origin of the open circuit voltage of plastic solar cells. Adv. Funct. Mater. 2001, 11, 374–380. [Google Scholar] [CrossRef]
- Chen, H.Y.; Hou, J.; Zhang, S.; Liang, Y.; Yang, G.; Yang, Y.; Yu, L.; Wu, Y.; Li, G. Polymer solar cells with enhanced open-circuit voltage and efficiency. Nat. Photon. 2009, 3, 649–653. [Google Scholar] [CrossRef]
- Gadisa, A.; Svensson, M.; Andersson, M.R.; Inganas, O. Correlation between oxidation potential and open-circuit voltage of composite solar cells based on blends of polythiophenes/fullerene derivative. Appl. Phys. Lett. 2004, 84, 1609–1611. [Google Scholar] [CrossRef]
- Roncali, J. Molecular engineering of the band gap of π-conjugated systems: Facing technological applications. Macromol. Rapid Commun. 2007, 28, 1761–1775. [Google Scholar] [CrossRef]
- Bérubé, N.; Gaudreau, J.; Côté, M. Low band gap polymers design approach based on a mix of aromatic and quinoid structures. Macromolecules 2013, 46, 6873–6880. [Google Scholar] [CrossRef]
- Pandey, L.; Risko, C.; Norton, J.E.; Brédas, J.L. Donor–acceptor copolymers of relevance for organic photovoltaics: A theoretical investigation of the impact of chemical structure modifications on the electronic and optical properties. Macromolecules 2012, 45, 6405–6414. [Google Scholar] [CrossRef]
- Havinga, E.E.; Tenhoeve, W.; Wynberg, H. A new class of small band-gap organic polymer conductors. Polym. Bull. 1992, 29, 119–126. [Google Scholar] [CrossRef]
- Park, S.H.; Roy, A.; Beaupré, S.; Cho, S.; Coates, N.; Moon, J.S.; Moses, D.; Leclerc, M.; Lee, K.; Heeger, A.J. Bulk heterojunction solar cells with internal quantum efficiency approaching 100%. Nat. Photon. 2009, 3, 297–302. [Google Scholar] [CrossRef]
- Li, W.; Hendriks, K.H.; Furlan, A.; Roelofs, W.S.C.; Wienk, M.M.; Janssen, R.A.J. Universal correlation between fibril width and quantum efficiency in diketopyrrolopyrrole-based polymer solar cells. J. Am. Chem. Soc. 2013, 135, 18942–18948. [Google Scholar] [CrossRef] [PubMed]
- Wienk, M.M.; Turbiez, M.; Gilot, J.; Janssen, R.A.J. Narrow-bandgap diketo-pyrrolo-pyrrole polymer solar cells: The effect of processing on the performance. Adv. Mater. 2008, 20, 2556–2560. [Google Scholar] [CrossRef]
- Tamayo, A.B.; Walker, B.; Nguyen, T.Q. A low band gap, solution processable oligothiophene with a diketopyrrolopyrrole core for use in organic solar cells. J. Phys. Chem. C 2008, 112, 11545–11551. [Google Scholar] [CrossRef]
- Chen, G.Y.; Chiang, C.M.; Kekuda, D.; Lan, S.C.; Chu, C.W.; Wei, K.H. Synthesis and characterization of a narrow-bandgap polymer containing alternating cyclopentadithiophene and diketo-pyrrolo-pyrrole units for solar cell applications. J. Polym. Sci. A Polym. Chem. 2010, 48, 1669–1675. [Google Scholar] [CrossRef]
- Biniek, L.; Schroeder, B.C.; Nielsen, C.B.; McCulloch, I. Recent advances in high mobility donor–acceptor semiconducting polymers. J. Mater. Chem. 2012, 22, 14803–14813. [Google Scholar] [CrossRef]
- Zhang, X.; Richter, L.J.; DeLongchamp, D.M.; Kline, R.J.; Hammond, M.R.; McCulloch, I.; Heeney, M.; Ashraf, R.S.; Smith, J.N.; Anthopoulos, T.D.; et al. Molecular packing of high-mobility diketo pyrrolo-pyrrole polymer semiconductors with branched alkyl side chains. J. Am. Chem. Soc. 2011, 133, 15073–15084. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sonar, P.; Murphy, L.; Hong, W. High mobility diketopyrrolopyrrole (dpp)-based organic semiconductor materials for organic thin film transistors and photovoltaics. Energy Environ. Sci. 2013, 6, 1684–1710. [Google Scholar] [CrossRef]
- Sonar, P.; Singh, S.P.; Williams, E.L.; Li, Y.; Soh, M.S.; Dodabalapur, A. Furan containing diketopyrrolopyrrole copolymers: Synthesis, characterization, organic field effect transistor performance and photovoltaic properties. J. Mater. Chem. 2012, 22, 4425–4435. [Google Scholar] [CrossRef]
- Sonar, P.; Singh, S.P.; Li, Y.; Ooi, Z.E.; Ha, T.J.; Wong, I.; Soh, M.S.; Dodabalapur, A. High mobility organic thin film transistor and efficient photovoltaic devices using versatile donor–acceptor polymer semiconductor by molecular design. Energy Environ. Sci. 2011, 4, 2288–2296. [Google Scholar] [CrossRef]
- Williams, E.L.; Gorelik, S.; Phang, I.; Bosman, M.; Vijila, C.; Subramanian, G.S.; Sonar, P.; Hobley, J.; Singh, S.P.; Matsuzaki, H.; et al. Nanoscale phase domain structure and associated device performance of organic solar cells based on a diketopyrrolopyrrole polymer. RSC Adv. 2013, 3, 20113–20124. [Google Scholar] [CrossRef]
- Tautz, R.; da Como, E.; Limmer, T.; Feldmann, J.; Egelhaaf, H.J.; von Hauff, E.; Lemaur, V.; Beljonne, D.; Yilmaz, S.; Dumsch, I.; et al. Structural correlations in the generation of polaron pairs in low-bandgap polymers for photovoltaics. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Guo, J.; Liang, Y.; Xiao, S.; Szarko, J.M.; Sprung, M.; Mukhopadhyay, M.K.; Wang, J.; Yu, L.; Chen, L.X. Structure and dynamics correlations of photoinduced charge separation in rigid conjugated linear donor–acceptor dyads towards photovoltaic applications. New J. Chem. 2009, 33, 1497–1507. [Google Scholar] [CrossRef]
- Deussen, M.; Bassler, H. Anion and cation absorption-spectra of conjugated oligomers and polymers. Chem. Phys. 1992, 164, 247–257. [Google Scholar] [CrossRef]
- Çelikbilek, Ö.; İçli-Özkut, M.; Algi, F.; Önal, A.M.; Cihaner, A. Donor–acceptor polymer electrochromes with cyan color: Effect of alkyl chain length on doping processes. Org. Electron. 2012, 13, 206–213. [Google Scholar] [CrossRef]
- Comoretto, D.; Moggio, I.; Cuniberti, C.; Musso, G.F.; Dellepiane, G.; Borghesi, A.; Kajzar, F.; Lorin, A. Long-lived photoexcited states in polydiacetylenes: The photoinduceda-absorption spectra of PDA-4BCMU. Phys. Rev. B 1998, 57, 7071–7078. [Google Scholar] [CrossRef]
- Roncali, J. Synthetic principles for bandgap control in linear pi-conjugated systems. Chem. Rev. 1997, 97, 173–205. [Google Scholar] [CrossRef] [PubMed]
- Berson, S.; de Bettignies, R.; Bailly, S.; Guillerez, S. Poly(3-hexylthiophene) fibers for photovoltaic applications. Adv. Funct. Mater. 2007, 17, 1377–1384. [Google Scholar] [CrossRef]
- Peng, Q.; Park, K.; Lin, T.; Durstock, M.; Dai, L. Donor–π–acceptor conjugated copolymers for photovoltaic applications: Tuning the open-circuit voltage by adjusting the donor/acceptor ratio. J. Phys. Chem. B 2008, 112, 2801–2808. [Google Scholar] [CrossRef] [PubMed]
- Atkins, P.W. Physical Chemistry; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Berlman, I.B. Handbook of Fluorescence Spectra of Aromatic Molecules; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Goh, W.P.; Ooi, Z.E.; Williams, E.L.; Yang, R.B.; Koh, W.S.; Mhaisalkar, S.; Institute of Materials Research and Engineering, 3 Research Link, Singapore. Unpublished work. 2014.
- Ohkita, H.; Ito, S. Transient absorption spectroscopy of polymer-based thin-film solar cells. Polymer 2011, 52, 4397–4417. [Google Scholar] [CrossRef]
- Guldi, D.M.; Prato, M. Excited-state properties of C-60 fullerene derivatives. Acc. Chem. Res. 2000, 33, 695–703. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, E.L.; Ang, T.S.; Ooi, Z.; Sonar, P.; Lin, T.T.; Neo, W.T.; Song, J.; Hobley, J. Optical Characterization of the Hole Polaron in a Series of Diketopyrrolopyrrole Polymers Used for Organic Photovoltaics. Polymers 2015, 7, 69-90. https://doi.org/10.3390/polym7010069
Williams EL, Ang TS, Ooi Z, Sonar P, Lin TT, Neo WT, Song J, Hobley J. Optical Characterization of the Hole Polaron in a Series of Diketopyrrolopyrrole Polymers Used for Organic Photovoltaics. Polymers. 2015; 7(1):69-90. https://doi.org/10.3390/polym7010069
Chicago/Turabian StyleWilliams, Evan L., Ter Shien Ang, Zien Ooi, Prashant Sonar, Ting Ting Lin, Wei Teng Neo, Jing Song, and Jonathan Hobley. 2015. "Optical Characterization of the Hole Polaron in a Series of Diketopyrrolopyrrole Polymers Used for Organic Photovoltaics" Polymers 7, no. 1: 69-90. https://doi.org/10.3390/polym7010069
APA StyleWilliams, E. L., Ang, T. S., Ooi, Z., Sonar, P., Lin, T. T., Neo, W. T., Song, J., & Hobley, J. (2015). Optical Characterization of the Hole Polaron in a Series of Diketopyrrolopyrrole Polymers Used for Organic Photovoltaics. Polymers, 7(1), 69-90. https://doi.org/10.3390/polym7010069