An SNP-Based High-Density Genetic Linkage Map for Tetraploid Potato Using Specific Length Amplified Fragment Sequencing (SLAF-Seq) Technology


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Extraction
2.3. SLAF Library Preparation and Sequencing
2.4. SLAF Data Analysis and Development of SNP Markers
2.5. Construction of High-Density Linkage Map
3. Results
3.1. SLAF Library Construction and SLAF Labels Development
3.2. SNP Marker Detection
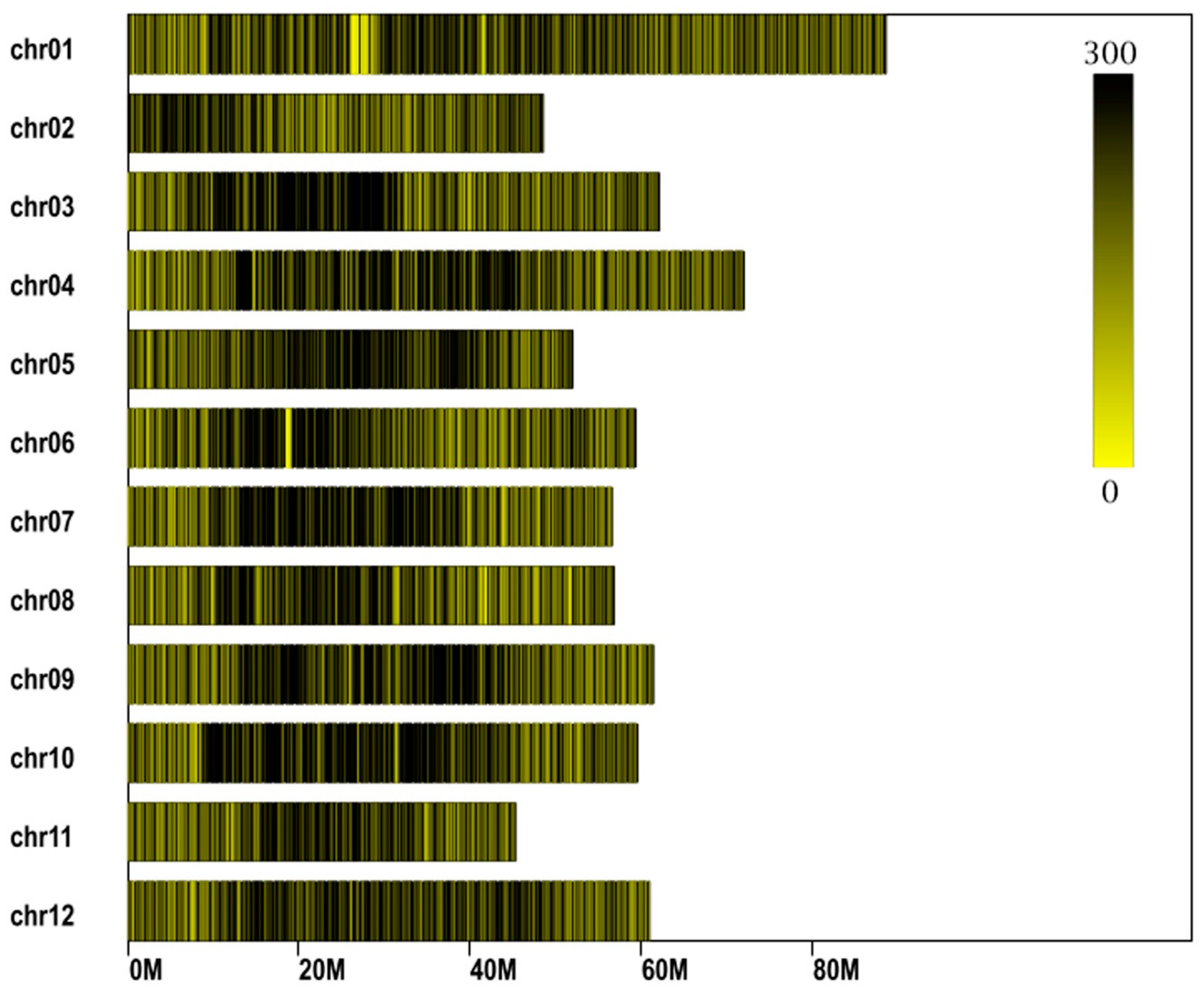
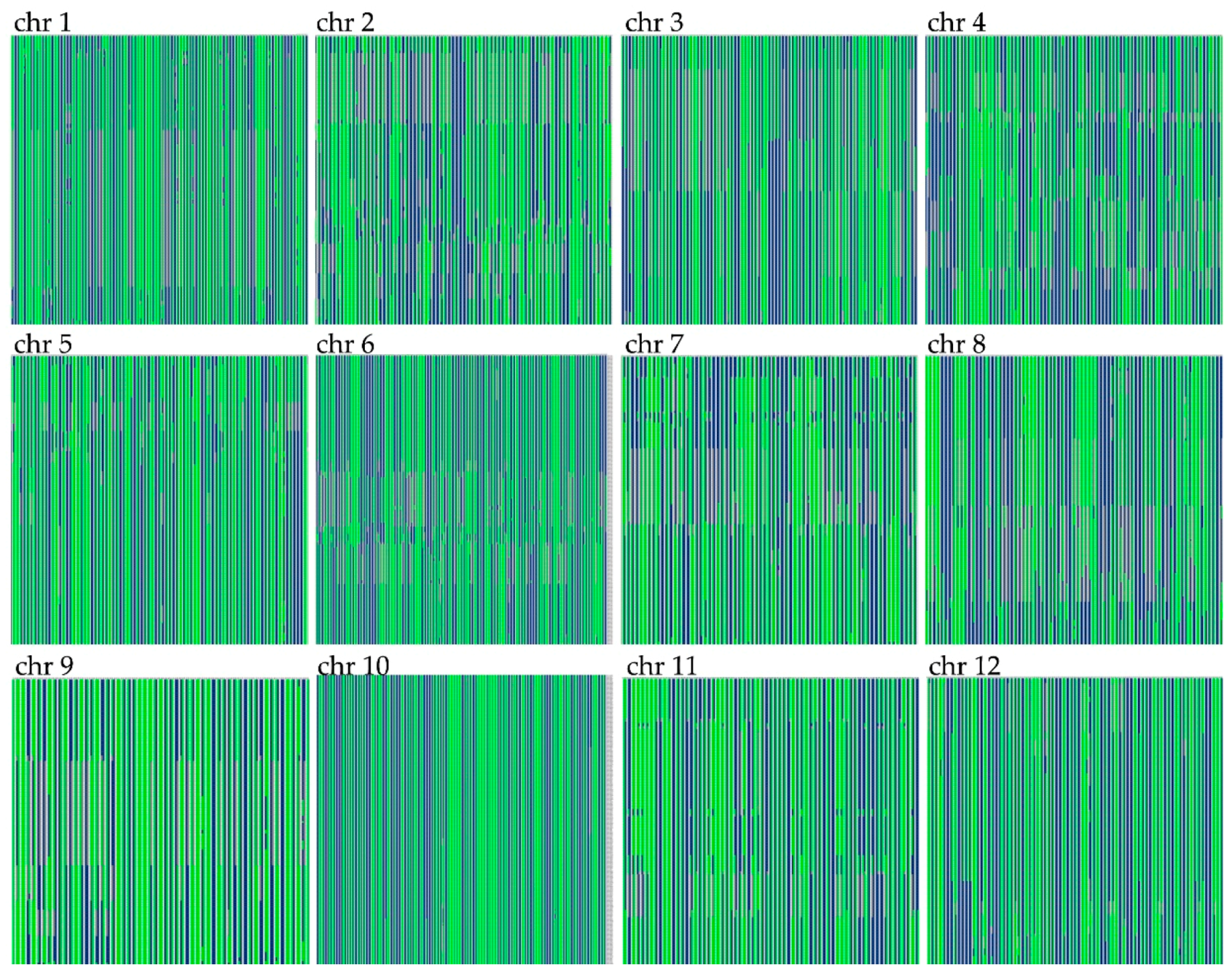
3.3. Construction of the Genetic Linkage Map
3.4. Evaluation of the Genetic Map
4. Discussion
4.1. The Development of SNP Markers Using SLAF-seq Technology
4.2. Mapping Population and Strategies
4.3. The High-Density Genetic Map of Tetraploid Potato
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linkage Group (ID) | Total Marker | Total Distance (cM) | Average Distance (cM) | Max Gap (Cm) | Gap ≥ 5 cM (%) |
|---|---|---|---|---|---|
| chr1 | 43 | 54.06 | 1.29 | 26.6 | 95.24 |
| chr2 | 70 | 199.96 | 2.9 | 66.56 | 86.96 |
| chr3 | 138 | 58.29 | 0.43 | 48.73 | 99.27 |
| chr4 | 107 | 149.13 | 1.41 | 30.15 | 93.4 |
| chr5 | 167 | 45.02 | 0.27 | 21.7 | 98.8 |
| chr6 | 235 | 172.13 | 0.74 | 31.9 | 95.73 |
| chr7 | 153 | 282.89 | 1.86 | 88.65 | 94.74 |
| chr8 | 153 | 116.85 | 0.77 | 15.34 | 96.71 |
| chr9 | 80 | 91.37 | 1.16 | 55.92 | 98.73 |
| chr10 | 341 | 32.82 | 0.1 | 7.32 | 99.85 |
| chr11 | 57 | 123.36 | 2.2 | 31.91 | 91.07 |
| chr12 | 94 | 57.8 | 0.62 | 16.63. | 96.77 |
| Total | 1638 | 1383.68 | 0.84 | 88.65 | 95.62 |
| Linkage Group (ID) | Total Marker | Total Distance (cM) | Average Distance (cM) | Max Gap (cM) | Gap ≥ 5 cM (%) |
|---|---|---|---|---|---|
| chr1 | 243 | 117.29 | 0.48 | 12.87 | 99.17 |
| chr2 | 156 | 167.27 | 1.08 | 28.45 | 96.77 |
| chr3 | 125 | 51.51 | 0.42 | 15.34 | 96.77 |
| chr4 | 132 | 151.04 | 1.15 | 51.29 | 95.42 |
| chr5 | 76 | 83.47 | 1.11 | 10.46 | 93.33 |
| chr6 | 83 | 77.56 | 0.95 | 37.57 | 96.34 |
| chr7 | 52 | 101.5 | 1.99 | 71.52 | 96.08 |
| chr8 | 71 | 88.68 | 1.27 | 70.27 | 98.57 |
| chr9 | 82 | 108.18 | 1.34 | 16.63 | 93.83 |
| chr10 | 102 | 52.42 | 0.52 | 10.46 | 98.02 |
| chr11 | 138 | 178.97 | 1.31 | 41.74 | 96.35 |
| chr12 | 142 | 26.05 | 0.18 | 3.92 | 100 |
| Total | 1402 | 1203.94 | 0.87 | 71.52 | 96.72 |



References
- FAOSTAT. 2017. Available online: http://faostat.fao.org/site/339/default.aspx (accessed on 2 September 2019).
- Hermsen, J.G.T. Nature, evolution and breeding of polyploids. Iowa State J. Res. 1984, 58, 411–420. [Google Scholar]
- Sybenga, J. Chromosome pairing affinity and quadrivalent formation in polyploids: Do segmental allopolyploids exist? Genome 1996, 39, 1176–1184. [Google Scholar] [CrossRef]
- Cervantes-Flores, J.C.; Yencho, G.C.; Kriegner, A.; Pecota, K.V.; Faulk, M.A.; Mwanga, R.O.M.; Sosinski, B.R. Development of a genetic linkage map and identification of homologous linkage groups in sweetpotato using multiple-dose AFLP markers. Mol. Breed. 2008, 21, 511–532. [Google Scholar] [CrossRef]
- Zhao, N.; Yu, X.X.; Jie, Q.; Li, H.; Li, H.; Hu, J.; Zhai, H.; He, S.Z.; Liu, Q.C. A genetic linkage map based on AFLP and SSR markers and mapping of QTL for dry-matter content in sweetpotato. Mol. Breed. 2013, 32, 807–820. [Google Scholar] [CrossRef]
- Gebhardt, C.; Ritter, E.; Barone, A.; Debener, T.; Walkemeier, B.; Schachtschabel, U.; Kaufmann, H.; Thompson, R.D.; Bonierbale, M.W.; Ganal, M.W. RFLP maps of potato and their alignment with the homoeologous tomato genome. Theor. Appl. Genet. 1991, 83, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Tanksley, S.D.; Ganal, M.W.; Prince, J.P.; De Vicente, M.C.; Bonierbale, M.W.; Broun, P.; Fulton, T.M.; Giovannoni, J.J.; Grandillo, S.; Martin, G.B. High density molecular linkage maps of the tomato and potato genomes. Genetics 1992, 132, 1141–1160. [Google Scholar] [PubMed]
- Jacobs, J.M.E.; Van Eck, H.J.; Arens, P.; Verkerk-Bakker, B.; Te Lintel Hekkert, B.; Bastiaanssen, H.J.M.; El-Kharbotly, A.; Pereira, A.; Jacobsen, E.; Stiekema, W.J. A genetic map of potato (Solanum tuberosum) integrating molecular markers, including transposons, and classical markers. Theor. Appl. Genet. 1995, 91, 289–300. [Google Scholar] [CrossRef]
- Caromel, B.; Mugniéry, D.; Lefebvre, V.; Andrzejewski, S.; Ellissèche, D.; Kerlan, M.C.; Rousselle, P.; Roussellebourgeois, F. Mapping QTLs for resistance against Globodera pallida (Stone) Pa2/3 in a diploid potato progeny originating from Solanum spegazzinii. Theor. Appl. Genet. 2003, 106, 1517–1523. [Google Scholar] [CrossRef]
- Yamanaka, S.; Ikeda, S.; Imai, A.; Luan, Y.; Watanabe, J.A.; Watanabe, K.N. Construction of integrated genetic map between various existing DNA markers and newly developed P450-related PBA markers in diploid potato (Solanum tuberosum). Breed. Sci. 2005, 55, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Van Os, H.; Andrzejewski, S.; Bakker, E.; Barrena, I.; Glenn, J.B.; Caromel, B.; Ghareeb, B.; Isidore, E.; De Jong, W.; Van Koert, P. Construction of a 10,000-marker ultradense genetic recombination map of potato: Providing a framework for accelerated gene isolation and a genomewide physical map. Genetics 2006, 173, 1075–1087. [Google Scholar] [CrossRef] [Green Version]
- Sa, K.J.; Park, J.Y.; Park, K.C.; Ju, K.L. Analysis of genetic mapping in a waxy/dent maize RIL population using SSR and SNP markers. Genes. Genom. 2012, 34, 157–164. [Google Scholar] [CrossRef]
- Su, C.F.; Wang, W.; Gong, S.L.; Zuo, J.H.; Li, S.J.; Xu, S.Z. High density linkage map construction and mapping of yield trait QTLs in maize (Zea mays) using the genotyping-by-sequencing (GBS) technology. Front. Plant Sci. 2017, 8, 706–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, H.J.; Feng, Z.Y.; Liu, X.Y.; Cheng, X.J.; Peng, H.R.; Yao, Y.Y.; Sun, Q.X.; Ni, Z.F. A genetic linkage map with 178 SSR and 1 901 SNP markers constructed using a RIL population in wheat (Triticum aestivum L.). J. Integr. Agric. 2015, 14, 1697–1705. [Google Scholar] [CrossRef]
- Leon, T.B.D.; Linscombe, S.; Subudhi, P.K. Molecular dissection of seedling salinity tolerance in rice (Oryza sativa L.) using a high-density GBS-based SNP linkage map. Rice 2016, 9, 52–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Yang, Z.J.; Tang, H.F.; Yu, Y.; Chen, Z.Y.; Wei, S.H.; Sun, Q.X.; Peng, Z.S. High-density genetic map construction and mapping of the homologous transformation sterility gene (hts) in wheat using gbs markers. BMC Plant Biol. 2018, 18, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Colasuonno, P.; Gadaleta, A.; Giancaspro, A.; Nigro, D.; Giove, S.; Incerti, O.; Mangini, G.; Signorile, A.; Simeone, R.; Blanco, A. Development of a high-density SNP-based linkage map and detection of yellow pigment content QTLs in durum wheat. Mol. Breed. 2014, 34, 1563–1578. [Google Scholar] [CrossRef]
- Xu, X.; Pan, S.K.; Cheng, S.F.; Zhang, B.; Mu, D.S.; Ni, P.X.; Zhang, G.Y.; Yang, S.; Li, R.Q.; Wang, J. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar]
- Felcher, K.J.; Coombs, J.J.; Massa, A.N.; Hansey, C.N.; Hamilton, J.P.; Veilleux, R.E.; Buell, C.R.; Douches, D.S. Integration of two diploid potato linkage maps with the potato genome sequence. PLoS ONE 2012, 7, e36347. [Google Scholar] [CrossRef] [Green Version]
- Hackett, C.A.; Mclean, K.; Bryan, G.J. Linkage analysis and QTL mapping using SNP dosage data in a tetraploid potato mapping population. PLoS ONE 2013, 8, e63939. [Google Scholar] [CrossRef] [Green Version]
- Endelman, J.B.; Jansky, S.H. Genetic mapping with an inbred line-derived F2 population in potato. Theor. Appl. Genet. 2016, 129, 935–943. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robus, simple genotyping by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Orsouw, N.J.; Hogers, R.C.; Janssen, A.; Yalcin, F.; Snoeijers, S.; Verstege, E.; Schneiders, H.; van der Poel, H.; Van Oeveren, J.; Verstegen, H.; et al. Complexity reduction of polymorphic sequences (CRoPS): A novel approach for large-scale polymorphism discovery in complex genomes. PLoS ONE 2007, 2, e1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Sun, X.W.; Liu, D.Y.; Zhang, X.F.; Li, W.B.; Liu, H.; Hong, W.G.; Jiang, C.B.; Guan, N.; Ma, C.X.; Zeng, H.P. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.W.; Lu, L.; Zhu, B.Y.; Xu, Q.; Qi, X.H.; Chen, X.H. QTL mapping of cucumber fruit flesh thickness by SLAF-seq. Sci. Rep. 2015, 5, 15829. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.P.; Huang, L.; Gao, A.N.; Yang, X.M.; Liu, W.H.; Li, X.Q.; Li, L.H. A high-density genetic map for P genome of Agropyron Gaertn. based on specific-locus amplified fragment sequencing (SLAF-seq). Planta 2015, 242, 1335–1347. [Google Scholar] [CrossRef]
- Zhao, X.X.; Huang, L.K.; Zhang, X.Q.; Wang, J.P.; Yan, D.F.; Li, J.; Tang, L.; Li, X.L.; Shi, T.W. Construction of high-density genetic linkage map and identification of flowering-time QTLs in orchardgrass using SSRs and SLAF-seq. Sci. Rep. 2016, 6, 29345. [Google Scholar] [CrossRef] [Green Version]
- Ken, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Depristo, M.A.; Banks, E.; Poplin, R.E.; Garimella, K.V.; Maguire, J.R.; Hartl, C. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.Y.; Ma, C.X.; Hong, W.G.; Huang, L.; Liu, M.; Liu, H.; Zeng, H.P.; Deng, D.J.; Xin, H.G.; Song, J.; et al. Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS ONE 2014, 9, e98855. [Google Scholar] [CrossRef] [PubMed]
- Vision, T.J.; Brown, D.G.; Shmoys, D.B.; Durrett, R.T.; Tanksley, S.D. Selective mapping: A strategy for optimizing the construction of high-density linkage maps. Genetics 2000, 155, 407–420. [Google Scholar]
- Foisset, N.; Delourme, R.; Barret, P.; Hubert, N.; Landry, B.S.; Renard, M. Molecular-mapping analysis in Brassica napus using isozyme, RAPD and RFLF markers on a doubled-haploid progeny. Theor. Appl. Genet. 1996, 93, 1017–1025. [Google Scholar] [CrossRef]
- Grattapaglia, D.; Sederoff, R. Genetic linkage maps of eucalyptus grandis and eucalyptus urophylla using a pseudo-testcross: Mapping strategy and RAPD markers. Genetics 1994, 137, 1121–1137. [Google Scholar]
- Venkat, S.K.; Bommisetty, P.; Patil, M.S.; Reddy, L.; Chennareddy, A. The genetic linkage maps of anthurium species based on RAPD, ISSR and SRAP markers. Sci. Hortic. 2014, 178, 132–137. [Google Scholar] [CrossRef]
- Liu, T.; Guo, L.L.; Pan, Y.L.; Zhao, Q.; Wang, J.H.; Song, Z.Q. Construction of the first high-density genetic linkage map of salvia miltiorrhiza using specific length amplified fragment (SLAF) sequencing. Sci. Rep. 2016, 6, 24070. [Google Scholar] [CrossRef] [Green Version]
- Carlier, J.D.; Reis, A.; Duval, M.F.; Coppens d’Eeckenbrugge, G.; Leitao, J.M. Genetic maps of RAPD, AFLP and ISSR markers in Ananas bracteatus and A. comosus using the pseudo-testcross strategy. Plant Breed. 2010, 123, 186–192. [Google Scholar] [CrossRef]
- Ubi, B.E.; Fujimori, M.; Mano, Y.; Komatsu, T. A genetic linkage map of rhodesgrass based on an F1 pseudo-testcross population. Plant Breed. 2010, 123, 247–253. [Google Scholar] [CrossRef]
- Cloutier, S.; Ragupathy, R.; Niu, Z.; Duguid, S. SSR-based linkage map of flax (Linum usitatissimum L.) and mapping of QTLs underlying fatty acid composition traits. Mol. Breed. 2011, 28, 437–451. [Google Scholar] [CrossRef]
- Escudero, M.; Hahn, M.; Hipp, A.L. RAD-seq linkage mapping and patterns of segregation distortion in sedges: Meiosis as a driver of karyotypic evolution in organisms with holocentric chromosomes. J. Evol. Biol. 2018, 31, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Moreira, F.M.; Madini, A.; Marino, R.; Zulini, L.; Stefanini, M.; Velasco, R.; Kozma, P.; Grando, M.S. Genetic linkage maps of two interspecific grape crosses (vitisspp.) used to localize quantitative trait loci for downy mildew resistance. Tree Genet. Genomes 2011, 7, 153–167. [Google Scholar] [CrossRef]
- Shappley, Z.W.; Jenkins, J.N.; Meredith, W.R.; Mccarty, J.C. An RFLP linkage map of upland cotton, Gossypium hirsutum L. Theor. Appl. Genet. 1998, 97, 756–761. [Google Scholar] [CrossRef]
- Lacape, J.M.; Nguyen, T.B.; Thibivilliers, S.; Bojinov, B.; Courtois, B.; Cantrell, R.G.; Burr, B.; Hau, B. A combined RFLP-SSR-AFLP map of tetraploid cotton based on a Gossypium hirsutum x Gossypium barbadense backcross population. Genome 2003, 46, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, S.; Cappadocia, M.; Landry, B.S. Analysis of RFLP mapping inaccuracy in Brassica napus L. Theor. Appl. Genet. 1997, 95, 83–91. [Google Scholar] [CrossRef]

| Sample | Total Reads | Total Base | Q30 Percentage (%) | GC Percentage (%) |
|---|---|---|---|---|
| MIN-21 | 48,849,737 | 11,944,501,020 | 95.18 | 40.28 |
| YSP-4 | 41,510,213 | 10,126,475,890 | 95.66 | 40.18 |
| offspring | 20,562,465 | 5,043,758,955 | 95.04 | 39.49 |
| Control (Rice) | 9,873,113 | 2,422,915,844 | 94.69 | 39.49 |
| Total | 2,269,981,207 | 556,709,426,104 | 95.05 | 39.51 |
| Type of Segregation Patterns | Number of SLAFs | Percentage (%) |
|---|---|---|
| Hk × hk | 51 | 0.67 |
| Lm × ll | 3898 | 54.03 |
| Nn × np | 3638 | 47.63 |
| ef × eg | 51 | 0.67 |
| Total | 7638 | 100 |
| Linkage Group (ID) | Total Marker | Total DISTANCE (cM) | Average Distance (cM) | Max Gap (cM) | Gap ≥5 cM (%) |
|---|---|---|---|---|---|
| chr1 | 282 | 107.42 | 0.38 | 12.84 | 99.29 |
| chr2 | 225 | 205.09 | 0.92 | 24.43 | 95.98 |
| chr3 | 253 | 63.48 | 0.25 | 14.38 | 98.02 |
| chr4 | 238 | 169.78 | 0.72 | 23.76 | 94.51 |
| chr5 | 243 | 76.4 | 0.32 | 10.46 | 98.35 |
| chr6 | 311 | 125.8 | 0.41 | 22.86 | 97.1 |
| chr7 | 201 | 203.86 | 1.02 | 25.19 | 95 |
| chr8 | 222 | 109.69 | 0.5 | 14.07 | 97.74 |
| chr9 | 162 | 115.41 | 0.72 | 15.34 | 96.89 |
| chr10 | 440 | 33.47 | 0.08 | 5.23 | 99.77 |
| chr11 | 194 | 156.97 | 0.81 | 21.35 | 94.82 |
| chr12 | 230 | 48.51 | 0.21 | 7.54 | 99.13 |
| Total | 3001 | 1415.88 | 0.47 | 25.19 | 97.22 |
| Linkage Group (ID) | The Number of Distorted SNP Markers |
|---|---|
| chr3 | 10 |
| chr5 | 24 |
| chr7 | 9 |
| chr8 | 3 |
| chr11 | 18 |
| chr12 | 16 |
| Total | 80 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Zhang, M.; Yu, Z.; Yang, D.; Li, J.; Wu, G.; Li, J. An SNP-Based High-Density Genetic Linkage Map for Tetraploid Potato Using Specific Length Amplified Fragment Sequencing (SLAF-Seq) Technology. Agronomy 2020, 10, 114. https://doi.org/10.3390/agronomy10010114
Yu X, Zhang M, Yu Z, Yang D, Li J, Wu G, Li J. An SNP-Based High-Density Genetic Linkage Map for Tetraploid Potato Using Specific Length Amplified Fragment Sequencing (SLAF-Seq) Technology. Agronomy. 2020; 10(1):114. https://doi.org/10.3390/agronomy10010114
Chicago/Turabian StyleYu, Xiaoxia, Mingfei Zhang, Zhuo Yu, Dongsheng Yang, Jingwei Li, Guofang Wu, and Jiaqi Li. 2020. "An SNP-Based High-Density Genetic Linkage Map for Tetraploid Potato Using Specific Length Amplified Fragment Sequencing (SLAF-Seq) Technology" Agronomy 10, no. 1: 114. https://doi.org/10.3390/agronomy10010114
APA StyleYu, X., Zhang, M., Yu, Z., Yang, D., Li, J., Wu, G., & Li, J. (2020). An SNP-Based High-Density Genetic Linkage Map for Tetraploid Potato Using Specific Length Amplified Fragment Sequencing (SLAF-Seq) Technology. Agronomy, 10(1), 114. https://doi.org/10.3390/agronomy10010114