Epigenetic Variation at a Genomic Locus Affecting Biomass Accumulation under Low Nitrogen in Arabidopsis thaliana

 , , and
, , and 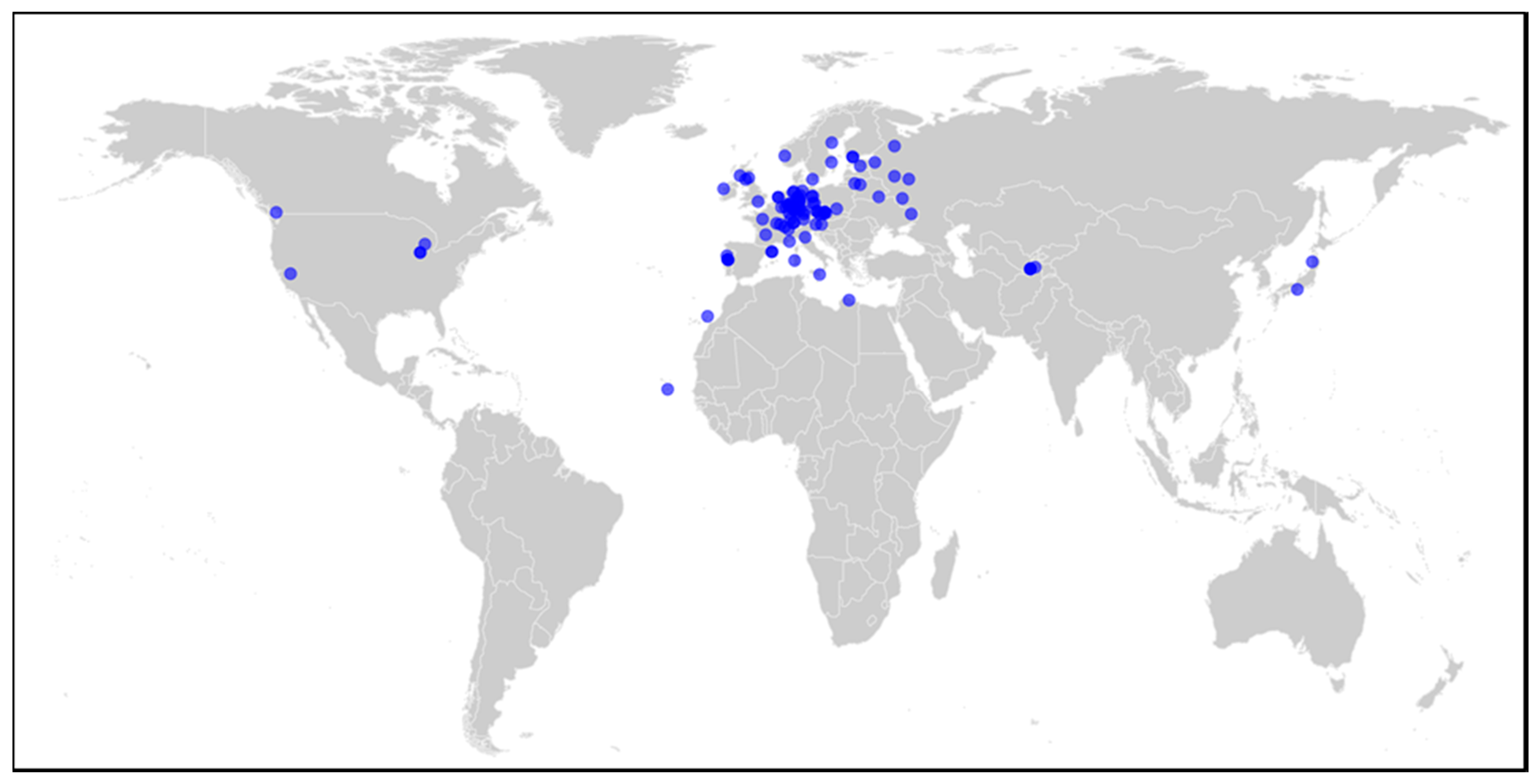{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Materials
2.2. Plant Culture of Selected Mutant Lines and Accessions
2.3. Phenotyping
2.4. Genotyping and Sequencing
2.5. RNA Extraction, cDNA Synthesis and RT-qPCR
2.6. Statistical Analyses
3. Results
3.1. Genome-Wide Association Mapping
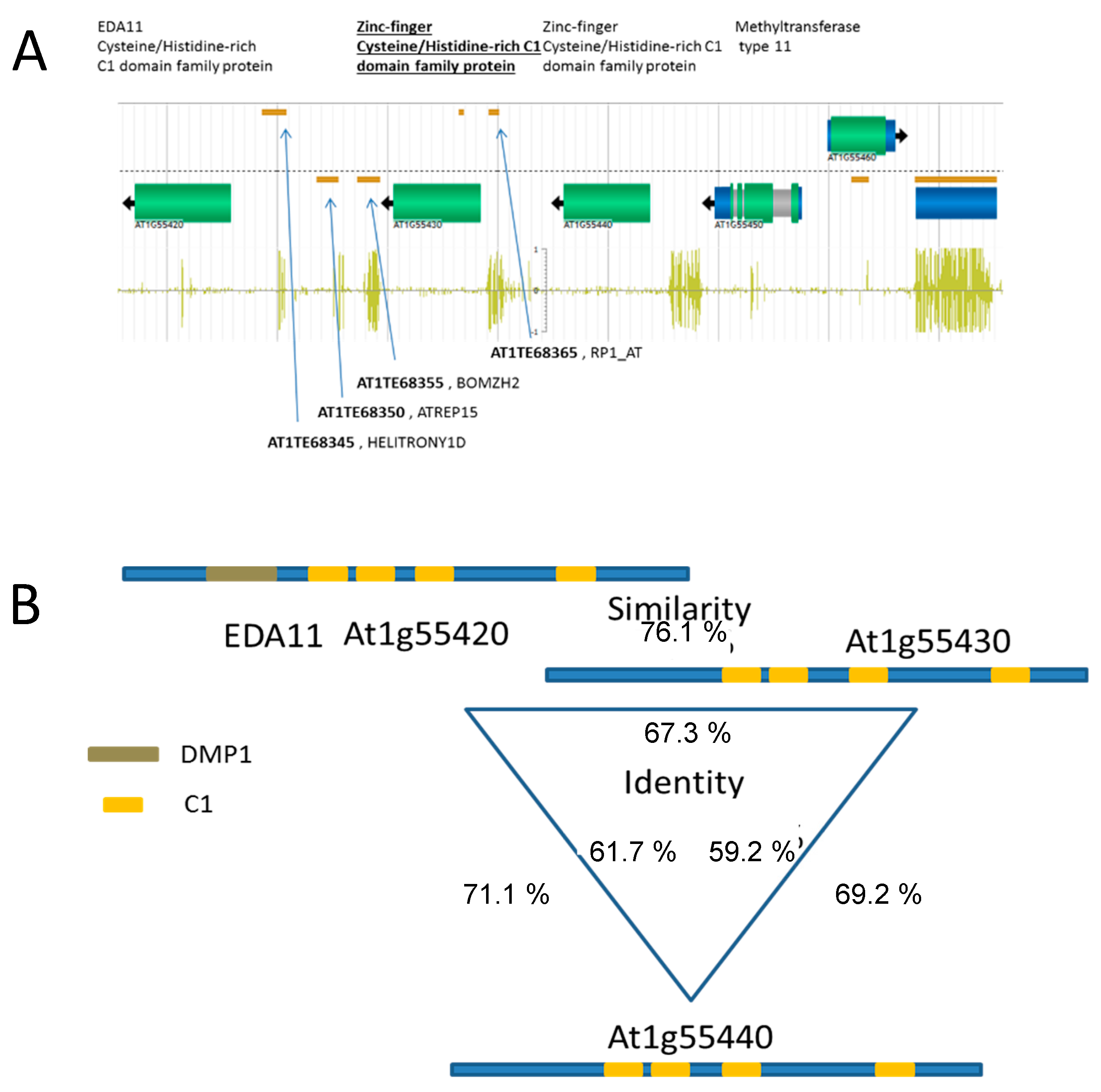
3.2. The Identified Region Contains Sequences Encoding Zinc-Finger Transcription Factors
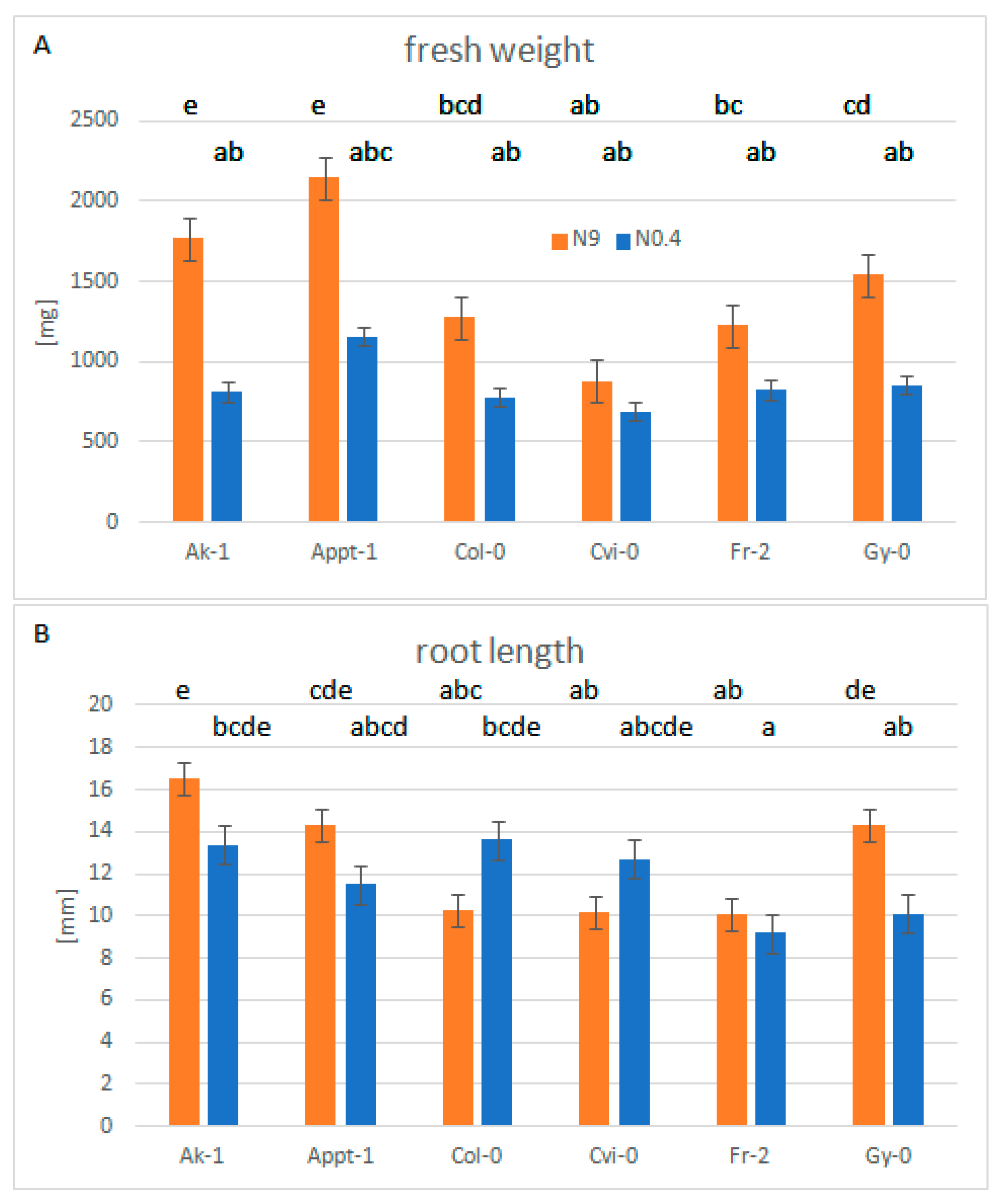
3.3. Plant Growth of Selected Accessions under Nitrogen Limiting Conditions
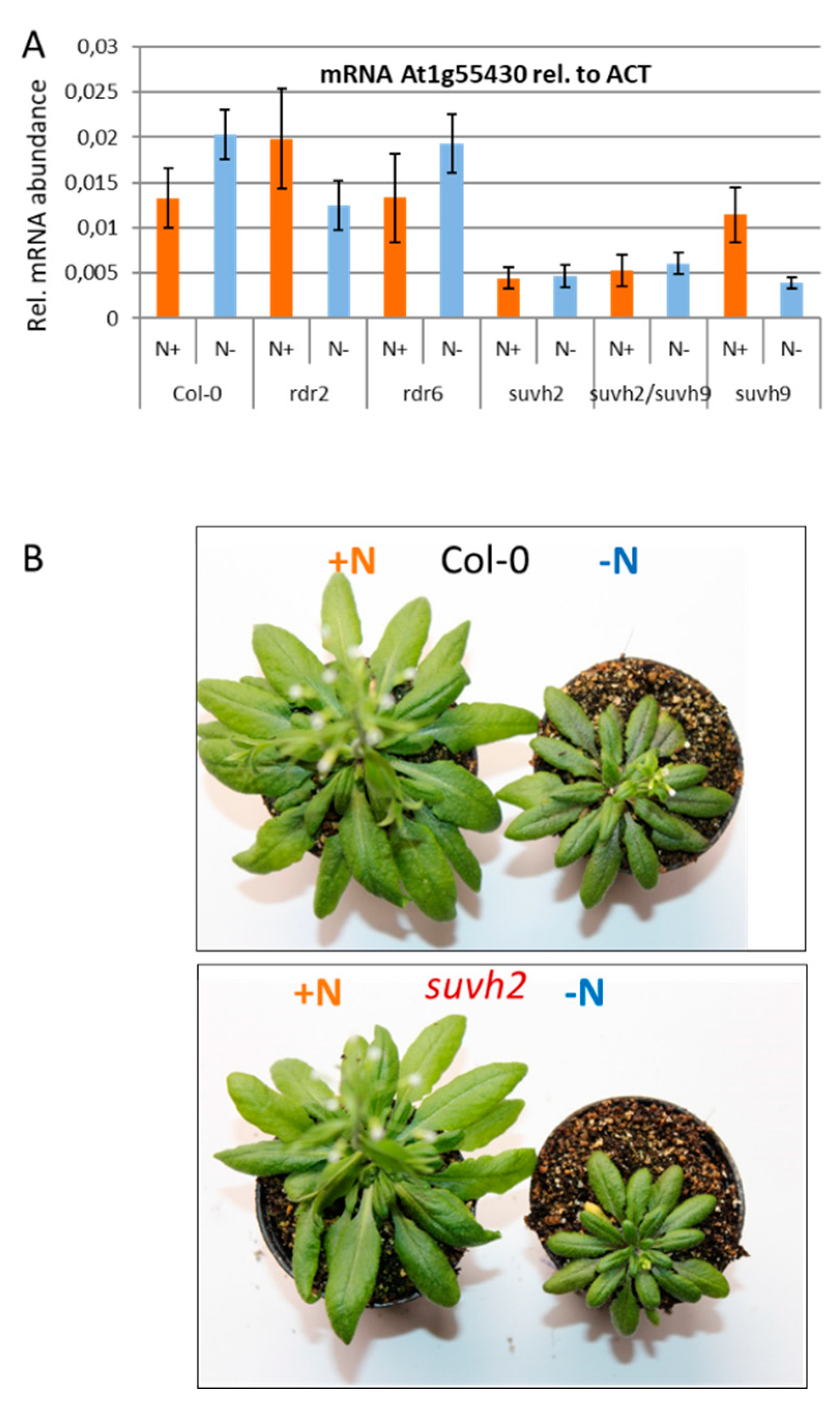
3.4. Expression Analysis of the Candidate Genes under Nitrogen Limiting Conditions
3.5. Analysis of Mutants Deregulated for Their DNA Methylation Pattern in the QTL Region
4. Discussion
4.1. Quantitative Trait Locus for Biomass Production under Nitrogen Limitation
4.2. DNA Methylation in the Promotor Region Correlates with the Transcriptional Response of the Candidate Gene(s)
4.3. Transfer of Gained Knowledge to Other Crop Plants
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McAllister, C.H.; Beatty, P.H.; Good, A.G. Engineering nitrogen use efficient crop plants: The current status. Plant. Biotechnol. J. 2012, 10, 1011–1025. [Google Scholar] [CrossRef]
- Hansen, B.; Thorling, L.; Schullehner, J.; Termansen, M.; Dalgaard, T. Groundwater nitrate response to sustainable nitrogen management. Sci. Rep. 2017, 7, 8566. [Google Scholar] [CrossRef]
- Fredes, I.; Moreno, S.; Diaz, F.P.; Gutierrez, R.A. Nitrate signaling and the control of Arabidopsis growth and development. Curr. Opin. Plant. Biol. 2019, 47, 112–118. [Google Scholar] [CrossRef]
- Vidal, E.A.; Gutierrez, R.A. A systems view of nitrogen nutrient and metabolite responses in Arabidopsis. Curr. Opin. Plant. Biol. 2008, 11, 521–529. [Google Scholar] [CrossRef]
- Li, H.; Hu, B.; Chu, C. Nitrogen use efficiency in crops: Lessons from Arabidopsis and rice. J. Exp. Bot. 2017, 68, 2477–2488. [Google Scholar] [CrossRef]
- Menz, J.; Range, T.; Trini, J.; Ludewig, U.; Neuhäuser, B. Molecular basis of differential nitrogen use efficiencies and nitrogen source preferences in contrasting Arabidopsis accessions. Sci. Rep. 2018, 8, 3373. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, R.A. Systems biology for enhanced plant nitrogen nutrition. Science 2012, 336, 1673–1675. [Google Scholar] [CrossRef]
- Meyer, R.C.; Gryczka, C.; Neitsch, C.; Muller, M.; Brautigam, A.; Schlereth, A.; Schon, H.; Weigelt-Fischer, K.; Altmann, T. Genetic diversity for nitrogen use efficiency in Arabidopsis thaliana. Planta 2019, 250, 41–57. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Giehl, R.F.H.; Meyer, R.C.; Altmann, T.; von Wiren, N. Natural variation of BSK3 tunes brassinosteroid signaling to regulate root foraging under low nitrogen. Nat. Commun. 2019, 10, 2378. [Google Scholar] [CrossRef] [Green Version]
- Loudet, O.; Chaillou, S.; Merigout, P.; Talbotec, J.; Daniel-Vedele, F. Quantitative trait loci analysis of nitrogen use efficiency in Arabidopsis. Plant. Physiol. 2003, 131, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.F.; Gurazada, S.G.; Zhai, J.; Li, S.; Simon, S.A.; Matzke, M.A.; Chen, X.; Meyers, B.C. RNA polymerase V-dependent small RNAs in Arabidopsis originate from small, intergenic loci including most SINE repeats. Epigenetics 2012, 7, 781–795. [Google Scholar] [CrossRef] [Green Version]
- Harmoko, R.; Fanata, W.I.; Yoo, J.Y.; Ko, K.S.; Rim, Y.G.; Uddin, M.N.; Siswoyo, T.A.; Lee, S.S.; Kim, D.Y.; Lee, S.Y.; et al. RNA-dependent RNA polymerase 6 is required for efficient hpRNA-induced gene silencing in plants. Mol. Cells 2013, 35, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Kuhlmann, M.; Mette, M.F. Developmentally non-redundant SET domain proteins SUVH2 and SUVH9 are required for transcriptional gene silencing in Arabidopsis thaliana. Plant. Mol. Biol. 2012, 79, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Estelle, M.A.; Somerville, C. Auxin-resistant mutants of Arabidopsis thaliana with an altered morphology. Mol. Gen. Genet. 1987, 206, 200–206. [Google Scholar] [CrossRef]
- Platt, A.; Horton, M.; Huang, Y.S.; Li, Y.; Anastasio, A.E.; Mulyati, N.W.; Ågren, J.; Bossdorf, O.; Byers, D.; Donohue, K. The scale of population structure in Arabidopsis thaliana. PLoS Genet. 2010, 6, e1000843. [Google Scholar] [CrossRef] [Green Version]
- Atwell, S.; Huang, Y.S.; Vilhjálmsson, B.J.; Willems, G.; Horton, M.; Li, Y.; Meng, D.; Platt, A.; Tarone, A.M.; Hu, T.T.; et al. Genome-wide association study of 107 phenotypes in a common set of Arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef]
- Horton, M.W.; Hancock, A.M.; Huang, Y.S.; Toomajian, C.; Atwell, S.; Auton, A.; Muliyati, N.W.; Platt, A.; Sperone, F.G.; Vilhjálmsson, B.J. Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat. Genet. 2012, 44, 212–216. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Arvidsson, S.; Kwasniewski, M.; Riano-Pachon, D.M.; Mueller-Roeber, B. QuantPrime—A flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinform. 2008, 9, 465. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Liu, X.; Bradbury, P.; Yu, J.; Zhang, Y.-M.; Todhunter, R.J.; Buckler, E.S.; Zhang, Z. Enrichment of statistical power for genome-wide association studies. BMC Biol. 2014, 12, 73. [Google Scholar] [CrossRef] [Green Version]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Liu, X.; Wang, J.; Li, M.; Wang, Q.; Tian, F.; Su, Z.; Pan, Y.; Liu, D.; Lipka, A.E.; et al. GAPIT Version 2: An Enhanced Integrated Tool for Genomic Association and Prediction. Plant. Genome 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, K.E.; Barboza-Barquero, L.; Tiburcio, A.F.; Alcázar, R. Genome Wide Association Mapping for the Tolerance to the Polyamine Oxidase Inhibitor Guazatine in Arabidopsis thaliana. Front. Plant. Sci. 2016, 7, 401. [Google Scholar] [CrossRef] [Green Version]
- Francisco, M.; Joseph, B.; Caligagan, H.; Li, B.; Corwin, J.A.; Lin, C.; Kerwin, R.E.; Burow, M.; Kliebenstein, D.J. Genome Wide Association Mapping in Arabidopsis thaliana Identifies Novel Genes Involved in Linking Allyl Glucosinolate to Altered Biomass and Defense. Front. Plant. Sci. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.K.; Rowe, H.C.; Corwin, J.A.; Joseph, B.; Kliebenstein, D.J. Combining genome-wide association mapping and transcriptional networks to identify novel genes controlling glucosinolates in Arabidopsis thaliana. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Plagnol, V.; Hu, T.T.; Toomajian, C.; Clark, R.M.; Ossowski, S.; Ecker, J.R.; Weigel, D.; Nordborg, M. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 2007, 39, 1151–1155. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Pagnussat, G.C.; Yu, H.J.; Ngo, Q.A.; Rajani, S.; Mayalagu, S.; Johnson, C.S.; Capron, A.; Xie, L.F.; Ye, D.; Sundaresan, V. Genetic and molecular identification of genes required for female gametophyte development and function in Arabidopsis. Development 2005, 132, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Blevins, T.; Podicheti, R.; Mishra, V.; Marasco, M.; Wang, J.; Rusch, D.; Tang, H.; Pikaard, C.S. Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in Arabidopsis. Elife 2015, 4, e09591. [Google Scholar] [CrossRef]
- Lyons, E.; Freeling, M. How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant. J. 2008, 53, 661–673. [Google Scholar] [CrossRef]
- Zhao, J.H.; Fang, Y.Y.; Duan, C.G.; Fang, R.X.; Ding, S.W.; Guo, H.S. Genome-wide identification of endogenous RNA-directed DNA methylation loci associated with abundant 21-nucleotide siRNAs in Arabidopsis. Sci. Rep. 2016, 6, 36247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, R.J.; Schultz, M.D.; Urich, M.A.; Nery, J.R.; Pelizzola, M.; Libiger, O.; Alix, A.; McCosh, R.B.; Chen, H.; Schork, N.J.; et al. Patterns of population epigenomic diversity. Nature 2013, 495, 193–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendte, J.M.; Pikaard, C.S. The RNAs of RNA-directed DNA methylation. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 140–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, D.; Vinegar, B.; Nahal, H.; Ammar, R.; Wilson, G.V.; Provart, N.J. An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2007, 2, e718. [Google Scholar] [CrossRef]
- Brady, S.M.; Orlando, D.A.; Lee, J.-Y.; Wang, J.Y.; Koch, J.; Dinneny, J.R.; Mace, D.; Ohler, U.; Benfey, P.N. A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 2007, 318, 801–806. [Google Scholar] [CrossRef]
- Gifford, M.L.; Dean, A.; Gutierrez, R.A.; Coruzzi, G.M.; Birnbaum, K.D. Cell-specific nitrogen responses mediate developmental plasticity. Proc. Natl. Acad Sci. USA 2008, 105, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Gruber, B.D.; Giehl, R.F.H.; Friedel, S.; von Wiren, N. Plasticity of the Arabidopsis Root System under Nutrient Deficiencies. Plant. Physiol. 2013, 163, 161–179. [Google Scholar] [CrossRef] [Green Version]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Xie, M.; Sun, J.; Gong, D.; Kong, Y. The Roles of Arabidopsis C1-2i Subclass of C2H2-type Zinc-Finger Transcription Factors. Genes 2019, 10, 653. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, R.V.; Mohanty, B.; Verma, V.; Wijaya, E.; Kumar, P.P. A hormone-responsive C1-domain-containing protein At5g17960 mediates stress response in Arabidopsis thaliana. PLoS ONE 2015, 10, e0115418. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Lv, H.; Xia, G.; Wang, M. Does diacylglycerol serve as a signaling molecule in plants? Plant. Signal. Behav. 2012, 7, 472–475. [Google Scholar] [CrossRef]
- Angkawijaya, A.E.; Nguyen, V.C.; Nakamura, Y. Lysophosphatidic acid acyltransferases 4 and 5 are involved in glycerolipid metabolism and nitrogen starvation response in Arabidopsis. New Phytol. 2019, 224, 336–351. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, X.; Song, L.; An, C. ABI4 activates DGAT1 expression in Arabidopsis seedlings during nitrogen deficiency. Plant. Physiol. 2011, 156, 873–883. [Google Scholar] [CrossRef] [Green Version]
- Yoshitake, Y.; Sato, R.; Madoka, Y.; Ikeda, K.; Murakawa, M.; Suruga, K.; Sugiura, D.; Noguchi, K.; Ohta, H.; Shimojima, M. Arabidopsis Phosphatidic Acid Phosphohydrolases Are Essential for Growth under Nitrogen-Depleted Conditions. Front. Plant. Sci. 2017, 8, 1847. [Google Scholar] [CrossRef] [Green Version]
- Caldo, K.M.P.; Acedo, J.Z.; Panigrahi, R.; Vederas, J.C.; Weselake, R.J.; Lemieux, M.J. Diacylglycerol Acyltransferase 1 Is Regulated by Its N-Terminal Domain in Response to Allosteric Effectors. Plant. Physiol. 2017, 175, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.J.; Scheibe, M.; Wongpalee, S.P.; Liu, W.L.; Cornett, E.M.; Vaughan, R.M.; Li, X.Q.; Chen, W.; Xue, Y.; Zhong, Z.H.; et al. A DNA methylation reader complex that enhances gene transcription. Science 2018, 362, 1182–1186. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuhlmann, M.; Meyer, R.C.; Jia, Z.; Klose, D.; Krieg, L.-M.; von Wirén, N.; Altmann, T. Epigenetic Variation at a Genomic Locus Affecting Biomass Accumulation under Low Nitrogen in Arabidopsis thaliana. Agronomy 2020, 10, 636. https://doi.org/10.3390/agronomy10050636
Kuhlmann M, Meyer RC, Jia Z, Klose D, Krieg L-M, von Wirén N, Altmann T. Epigenetic Variation at a Genomic Locus Affecting Biomass Accumulation under Low Nitrogen in Arabidopsis thaliana. Agronomy. 2020; 10(5):636. https://doi.org/10.3390/agronomy10050636
Chicago/Turabian StyleKuhlmann, Markus, Rhonda C. Meyer, Zhongtao Jia, Doreen Klose, Lisa-Marie Krieg, Nicolaus von Wirén, and Thomas Altmann. 2020. "Epigenetic Variation at a Genomic Locus Affecting Biomass Accumulation under Low Nitrogen in Arabidopsis thaliana" Agronomy 10, no. 5: 636. https://doi.org/10.3390/agronomy10050636
APA StyleKuhlmann, M., Meyer, R. C., Jia, Z., Klose, D., Krieg, L. -M., von Wirén, N., & Altmann, T. (2020). Epigenetic Variation at a Genomic Locus Affecting Biomass Accumulation under Low Nitrogen in Arabidopsis thaliana. Agronomy, 10(5), 636. https://doi.org/10.3390/agronomy10050636