
Comparative Transcriptome of Isonuclear Alloplasmic Strain Revealed the Important Role of Mitochondrial Genome in Regulating Flammulina filiformis
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
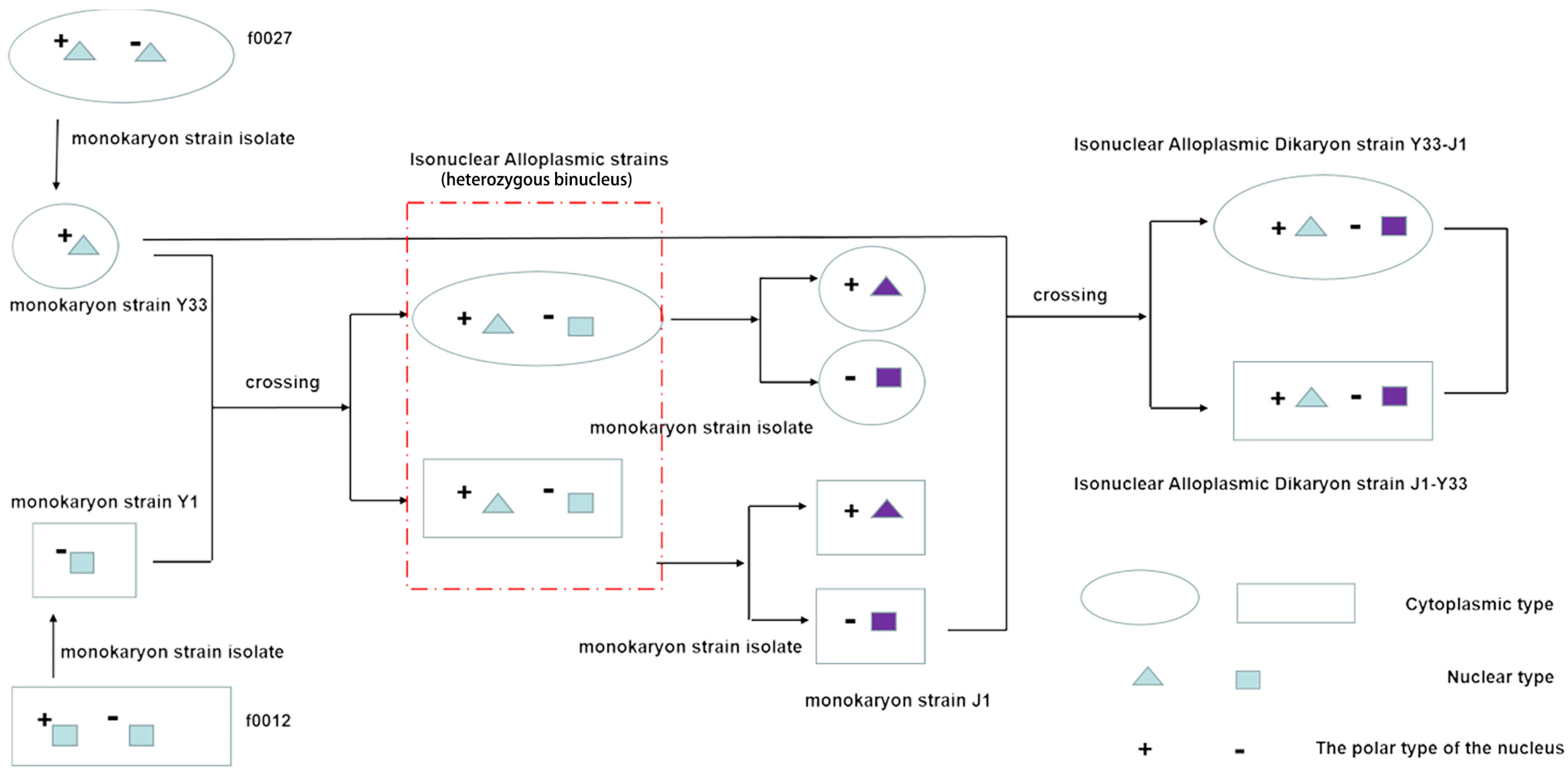
2.1. Strain Samples
2.2. Reference Genome
2.3. Culture and Collection of Samples
2.4. Total RNA Extraction and Transcriptome Sequencing
2.5. Transcriptome Analysis
2.6. Quantitative RT–PCR (qRT–PCR) Validation
3. Results
3.1. Morphological Characteristics of the Isonuclear Alloplasmic Strains in F. filiformis Mycelia and Fruit Body Morphology Observation
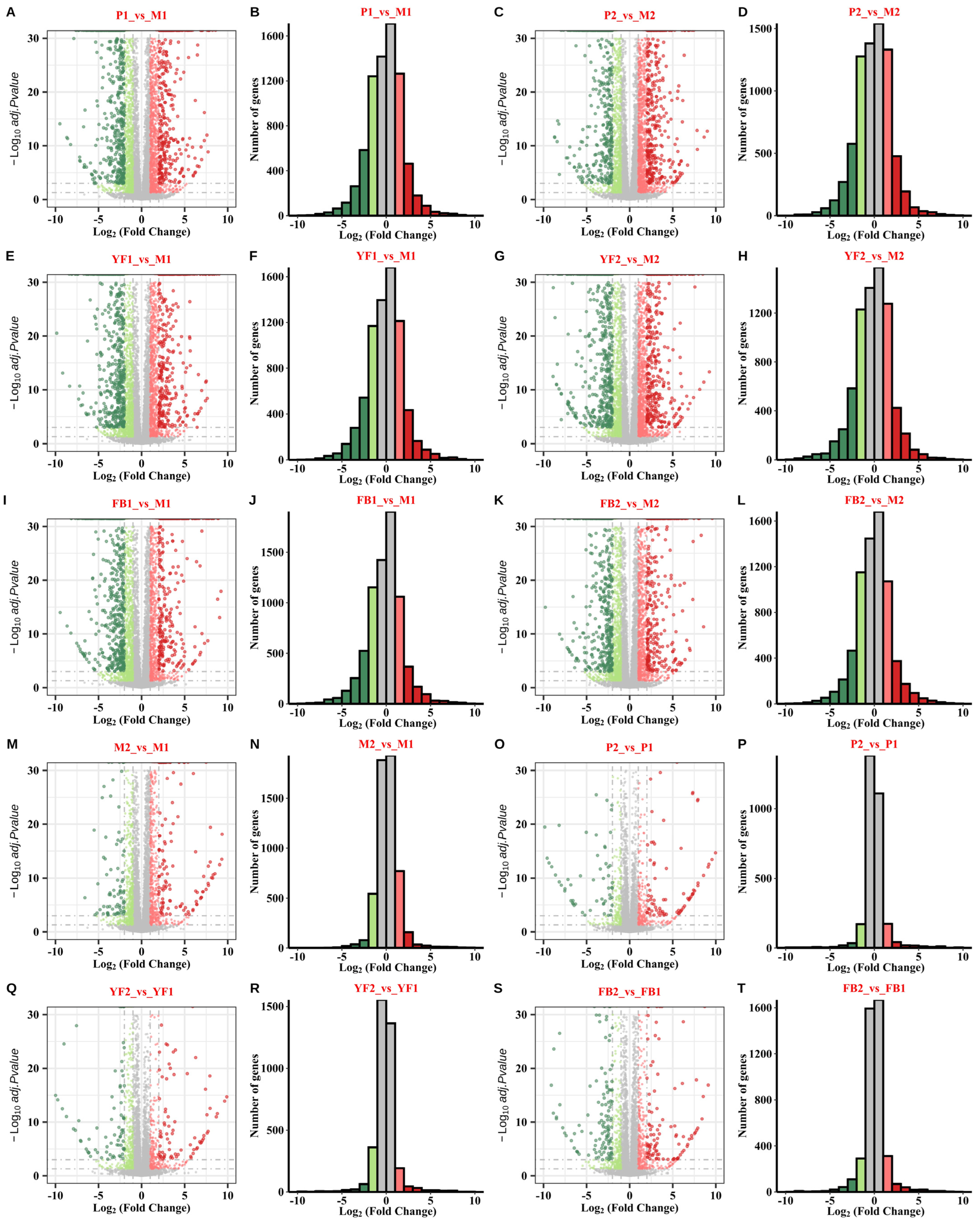
3.2. Analysis of Differentially Expressed Genes (DEGs)
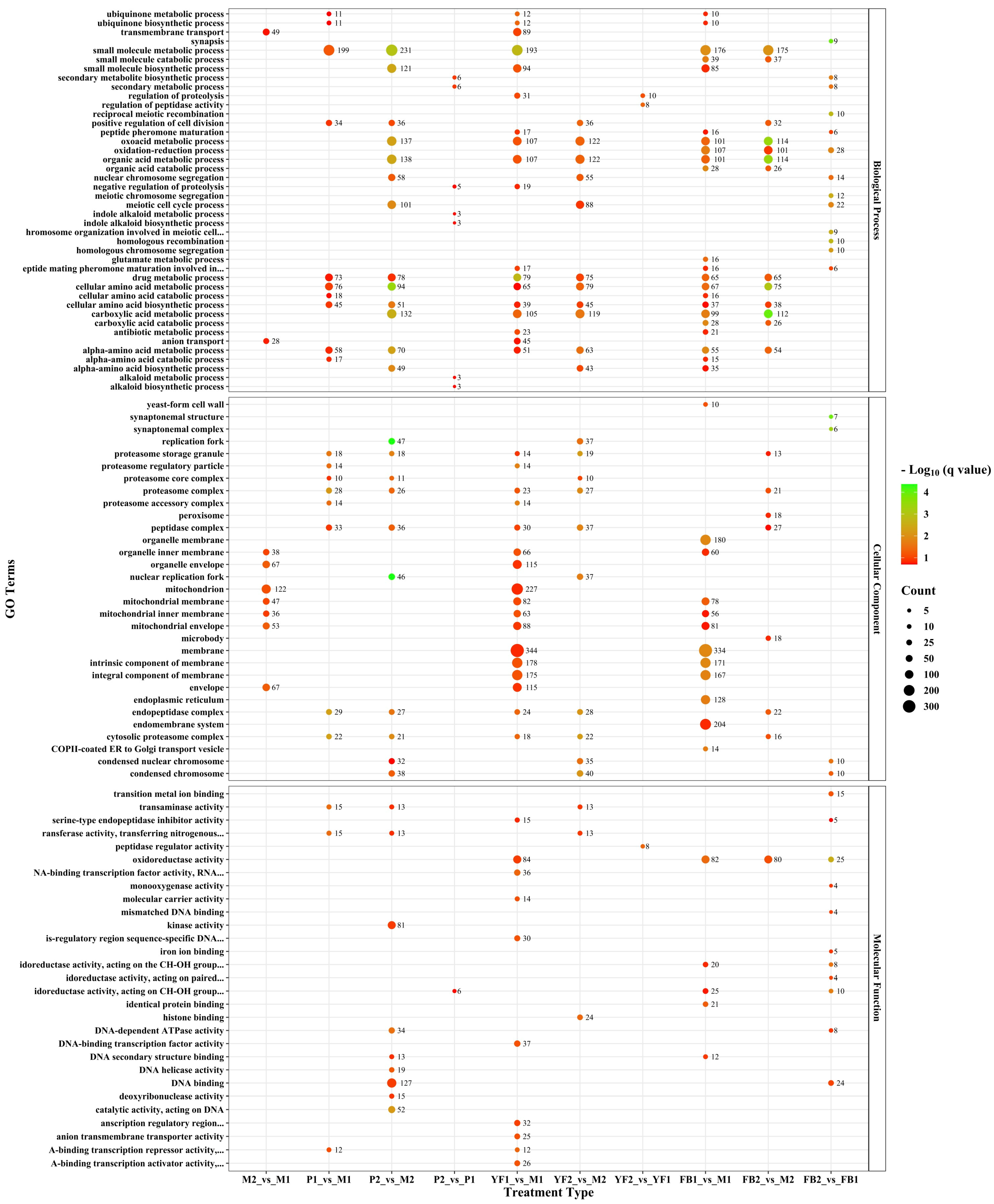
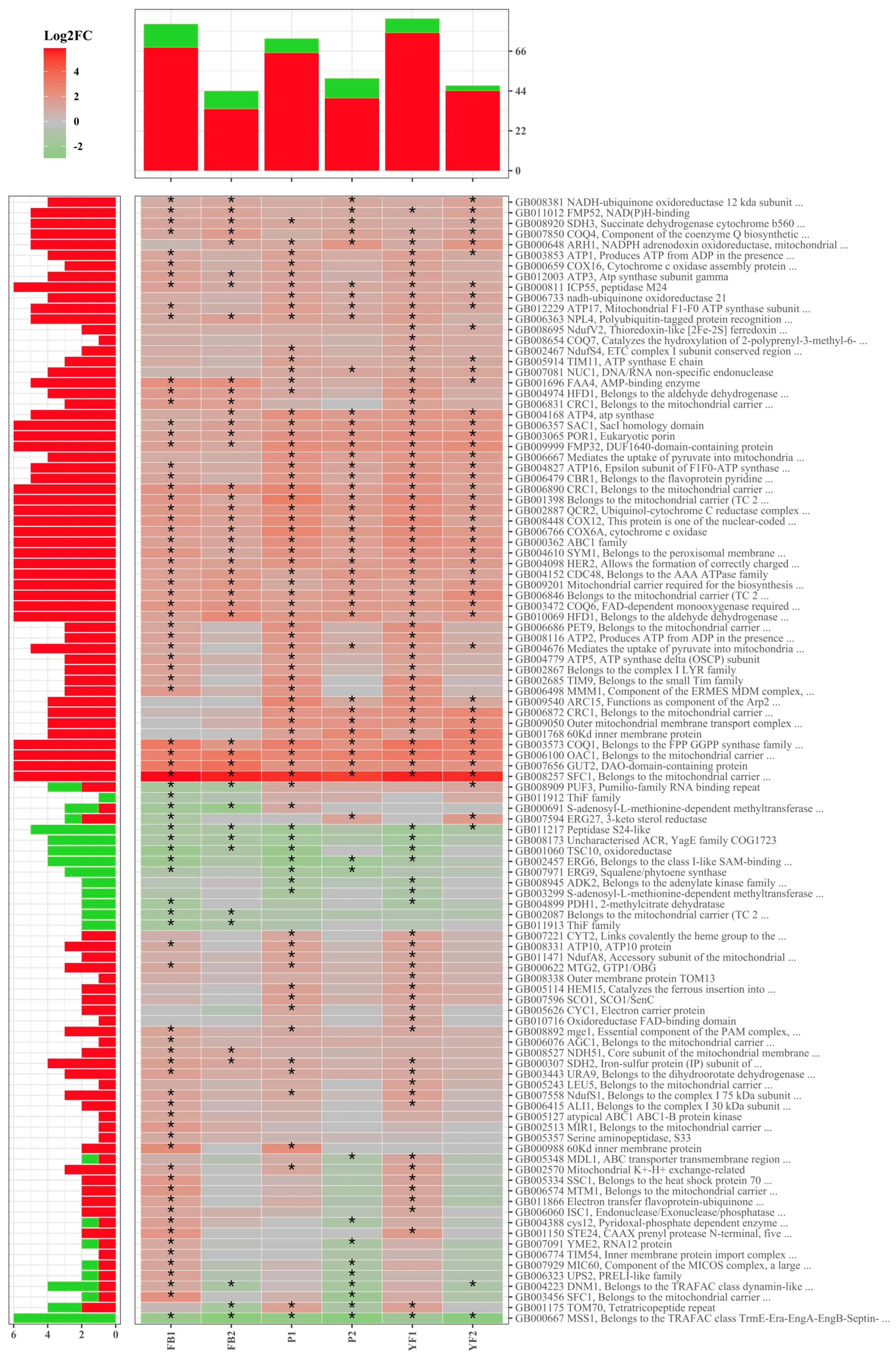
3.3. Functional Annotation of Differentially Expressed Genes
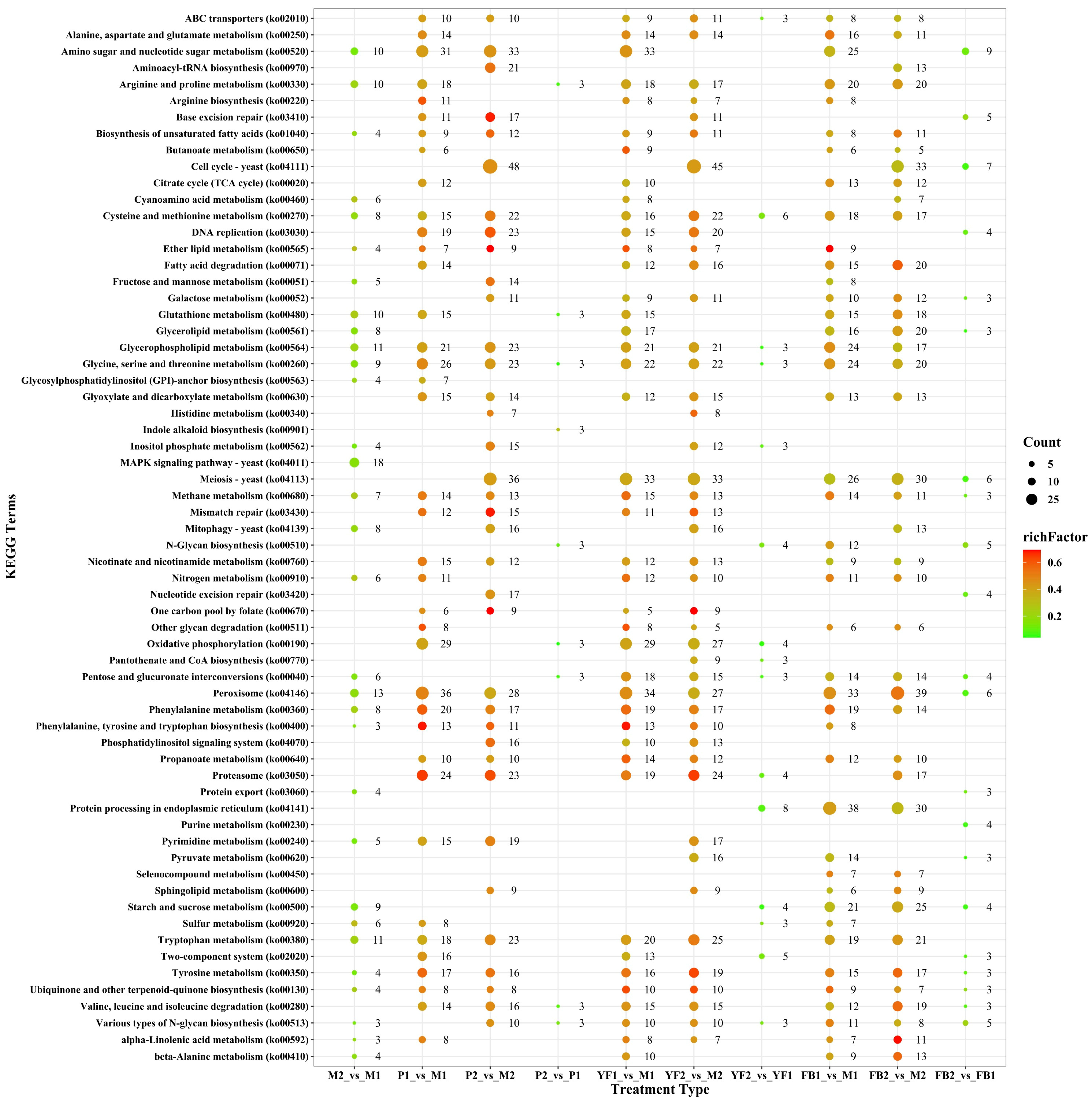
3.4. KEGG Enrichment Analysis of Differentially Expressed Genes
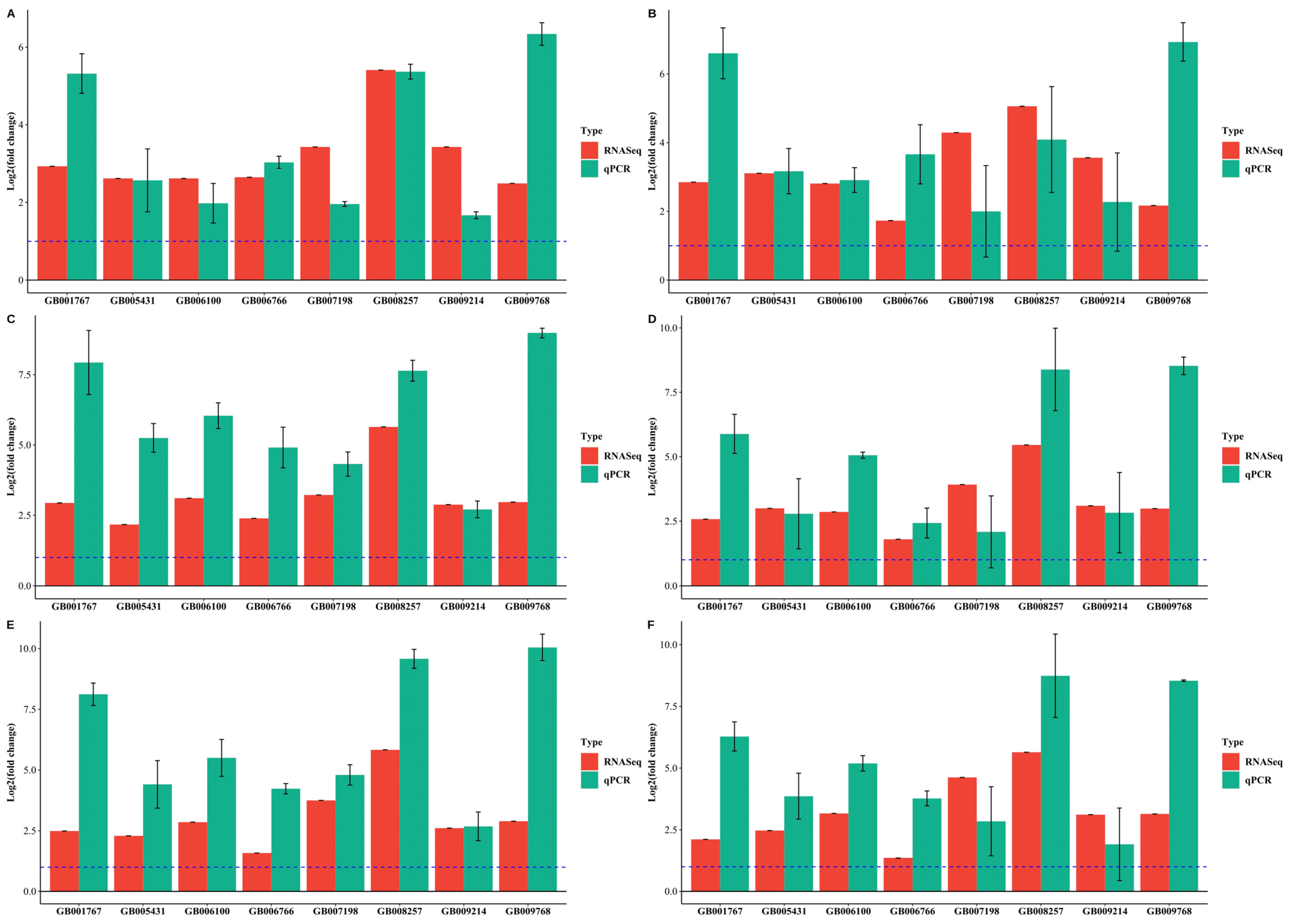
3.5. Validation of DEGs by Quantitative RT–PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, K.-A.; Gai, Y.; Fayyaz, A.; Zhang, G.; Liu, M.; Wang, Z. Genomic and morphological characteristics of the cold–adapted bacteria Mycetocola saprophilus provide insights into the pathogenesis of soft rot in Flammulina velutipes. Biotechnol. Biotechnol. Equip. 2020, 34, 885–897. [Google Scholar] [CrossRef]
- Rezaeian, S.; Pourianfar, H.R. A comparative study on bioconversion of different agro wastes by wild and cultivated strains of Flammulina velutipes. Waste Biomass Valorization 2017, 8, 2631–2642. [Google Scholar] [CrossRef]
- Attaran Dowom, S.; Rezaeian, S.; Pourianfar, H.R. Agronomic and environmental factors affecting cultivation of the winter mushroom or Enokitake: Achievements and prospects. Appl. Microbiol. Biotechnol. 2019, 103, 2469–2481. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.-M.; Tang, Y.-J.; Ma, K.; Li, B.; Zeng, X.; Liu, X.-B.; Li, Y.; Yang, Z.L.; Xu, W.-N. Genome-wide analysis and prediction of genes involved in the biosynthesis of polysaccharides and bioactive secondary metabolites in high-temperature-tolerant wild Flammulina filiformis. BMC Genom. 2020, 21, 719. [Google Scholar]
- Wang, P.M.; Liu, X.B.; Dai, Y.C.; Horak, E.; Steffen, K.; Yang, Z.L. Phylogeny and species delimitation of Flammulina: Taxonomic status of winter mushroom in East Asia and a new European species identified using an integrated approach. Mycol. Prog. 2018, 17, 1013–1030. [Google Scholar] [CrossRef]
- Xu, H.; Liu, L.; Cao, C.; Lu, W.; Zhu, Z.; Guo, Z.; Li, M.; Wang, X.; Huang, D.; Wang, S. Wound healing activity of a skin substitute from residues of culinary–medicinal winter mushroom Flammulina velutipes (agaricomycetes) cultivation. Int. J. Med. Mushrooms 2019, 21, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Bellettini, M.B.; Fiorda, F.A.; Maieves, H.A.; Teixeira, G.L.; Ávila, S.; Hornung, P.S.; Júnior, A.M.; Ribani, R.H. Factors affecting mushroom Pleurotus spp. Saudi J. Biol. Sci. 2019, 26, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Tamai, Y.; Yajima, T. Influence of light on the morphological changes that take place during the development of the Flammulina velutipes fruit body. Mycoscience 2004, 45, 333–339. [Google Scholar] [CrossRef]
- Liu, J.Y.; Men, J.L.; Chang, M.C.; Feng, C.P.; Yuan, L.G. iTRAQ-based quantitative proteome revealed metabolic changes of Flammulina velutipes mycelia in response to cold stress. J. Proteom. 2017, 156, 75–84. [Google Scholar] [CrossRef]
- Yan, J.-J.; Tong, Z.-J.; Liu, Y.-Y.; Li, Y.-N.; Zhao, C.; Mukhtar, I.; Tao, Y.-X.; Chen, B.-Z.; Deng, Y.-J.; Xie, B.-G. Comparative transcriptomics of Flammulina filiformis suggests a high CO2 concentration inhibits early pileus expansion by decreasing cell division control pathways. Int. J. Mol. Sci. 2019, 20, 5923. [Google Scholar]
- Ye, L.; He, X.; Su, C.; Feng, H.; Meng, G.; Chen, B.; Wu, X. The Effect of Mitochondria on Ganoderma lucidum Growth and Bioactive Components Based on Transcriptomics. J. Fungi 2022, 8, 1182. [Google Scholar] [CrossRef] [PubMed]
- Konrad, A.; Thompson, O.; Waterston, R.H.; Moerman, D.G.; Keightley, P.D.; Bergthorsson, U.; Katju, V. Mitochondrial mutation rate, spectrum and heteroplasmy in Caenorhabditis elegans spontaneous mutation accumulation lines of differing population size. Mol. Biol. Evol. 2017, 34, 1319–1334. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. Mitochondria at work: New insights into regulation and dysregulation of cellular energy supply and metabolism. Biomedicines 2020, 8, 526. [Google Scholar] [CrossRef]
- Medina, R.; Franco, M.E.E.; Bartel, L.C.; Martínez Alcántara, V.; Saparrat, M.C.N.; Balatti, P.A. Fungal mitogenomes: Relevant features to planning plant disease management. Front. Microbiol. 2020, 11, 978. [Google Scholar] [CrossRef]
- Giordano, L.; Sillo, F.; Garbelotto, M.; Gonthier, P. Mitonuclear interactions may contribute to fitness of fungal hybrids. Sci. Rep. 2018, 8, 1706. [Google Scholar] [CrossRef]
- Kouvelis, V.N.; Hausner, G. Mitochondrial genomes and mitochondrion related gene insights to fungal evolution. Front. Microbiol. 2022, 13, 897981. [Google Scholar] [CrossRef]
- Bernt, M.; Braband, A.; Schierwater, B.; Stadler, P.F. Genetic aspects of mitochondrial genome evolution. Mol. Phylogenetics Evol. 2013, 69, 328–338. [Google Scholar] [CrossRef]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta (BBA)—Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Soledad, R.B.; Charles, S.; Samarjit, D. The secret messages between mitochondria and nucleus in muscle cell biology. Arch. Biochem. Biophys. 2019, 666, 52–62. [Google Scholar] [CrossRef]
- Hartl, F.-U.; Neupert, W. Protein sorting to mitochondria: Evolutionary conservations of folding and assembly. Science 1990, 247, 930–938. [Google Scholar] [CrossRef]
- Vedel, F.; Lalanne, É.; Sabar, M.; Chétrit, P.; De Paepe, R. The mitochondrial respiratory chain and ATP synthase complexes: Composition, structure and mutational studies. Plant Physiol. Biochem. 1999, 37, 629–643. [Google Scholar] [CrossRef]
- Di Donato, S. Disorders related to mitochondrial membranes: Pathology of the respiratory chain and neurodegeneration. J. Inherit. Metab. Dis. 2000, 23, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.G.; Herrmann, J.M. Transport of proteins into mitochondria. Protein J. 2019, 38, 330–342. [Google Scholar] [CrossRef]
- Ng, S.; De Clercq, I.; Van Aken, O.; Law, S.R.; Ivanova, A.; Willems, P.; Giraud, E.; Van Breusegem, F.; Whelan, J. Anterograde and retrograde regulation of nuclear genes encoding mitochondrial proteins during growth, development, and stress. Mol. Plant 2014, 7, 1075–1093. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim. Biophys. Acta (BBA)—Gene Struct. Expr. 2002, 1576, 1–14. [Google Scholar] [CrossRef]
- Vijg, J.; Suh, Y. Genetics of longevity and aging. Annu. Rev. Med. 2005, 56, 193. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.F.; Gray, M.W.; Burger, G. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 1999, 33, 351–397. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Yu, Y.; Fu, Y.; Liu, T.; Wang, Y.; Peng, W.; Wang, B.; Chen, J. Comparative analyses of Flammulina filiformis mitochondrial genomes reveal high length polymorphism in intergenic regions and multiple intron gain/loss in cox1. Int. J. Biol. Macromol. 2022, 221, 1593–1605. [Google Scholar] [CrossRef]
- O’Connor, E.; McGowan, J.; McCarthy, C.G.; Amini, A.; Grogan, H.; Fitzpatrick, D.A. Whole genome sequence of the commercially relevant mushroom strain Agaricus bisporus var. bisporus ARP23. G3 Genes Genomes Genet. 2019, 9, 3057–3066. [Google Scholar]
- Liu, X.; Wu, X.; Tan, H.; Xie, B.; Deng, Y. Large inverted repeats identified by intra–specific comparison of mitochondrial genomes provide insights into the evolution of Agrocybe aegerita. Comput. Struct. Biotechnol. J. 2020, 18, 2424–2437. [Google Scholar] [CrossRef] [PubMed]
- Bashir, K.M.I.; Rheu, K.M.; Kim, M.-S.; Cho, M.-G. The complete mitochondrial genome of an edible mushroom, Sparassis crispa. Mitochondrial DNA Part B 2020, 5, 862–863. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wan, J.; Shang, J.-J.; Feng, Z.; Jin, Y.; Li, H.; Guo, T.; Wu, Y.-Y.; Bao, D.-P.; Zhang, M. The complete mitochondrial genome of the edible mushroom Grifola frondosa. Mitochondrial DNA Part B 2022, 7, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high–throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA–seq data with DESeq2. Genome Biol. 2014, 15, 131. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid. Redox Signal. 2013, 19, 1469–1480. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta (BBA)—Bioenerg. 2014, 1837, 427–443. [Google Scholar] [CrossRef]
- Lenaz, G.; Tioli, G.; Falasca, A.I.; Genova, M.L. Coenzyme Q and respiratory supercomplexes: Physiological and pathological implications. Rend. Lincei Sci. Fis. Nat. 2018, 29, 383–395. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, H.; Zhou, X. Weanling offspring of dams maintained on serine-deficient diet are vulnerable to oxidative stress. Oxidative Med. Cell. Longev. 2018, 2018, 8026496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Gillies, M.C.; Madigan, M.C.; Shen, W.; Du, J.; Grünert, U.; Zhou, F.; Yam, M.; Zhu, L. Disruption of de novo serine synthesis in Müller cells induced mitochondrial dysfunction and aggravated oxidative damage. Mol. Neurobiol. 2018, 55, 7025–7037. [Google Scholar] [CrossRef] [PubMed]
- Pourvali, K.; Abbasi, M.; Mottaghi, A. Role of superoxide dismutase 2 gene Ala16Val polymorphism and total antioxidant capacity in diabetes and its complications. Avicenna J. Med. Biotechnol. 2016, 8, 48. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Developmental Stage | Label |
|---|---|---|
| J1–Y33 | The mycelium growth stage of F. filiformis | M1 |
| Y33–J1 | The mycelium growth stage of F. filiformis | M2 |
| J1–Y33 | The primordia formation stage of F. filiformis | P1 |
| Y33–J1 | The primordia formation stage of F. filiformis | P2 |
| J1–Y33 | The young mushroom stage of F. filiformis | YF1 |
| Y33–J1 | The young mushroom stage of F. filiformis | YF2 |
| J1–Y33 | The fruiting body stage of F. filiformis | FB1 |
| Y33–J1 | The fruiting body stage of F. filiformis | FB2 |
| Label | Upregulated Genes | Downregulated Genes | DEGs |
|---|---|---|---|
| P1_vs._M1 | 2072 | 2316 | 4388 |
| P2_vs._M2 | 2152 | 2350 | 4502 |
| YF1_vs._M1 | 1999 | 2240 | 4239 |
| YF2_vs._M2 | 2090 | 2352 | 4442 |
| FB1_vs._M1 | 1777 | 2182 | 3959 |
| FB2_vs._M2 | 1811 | 2032 | 3843 |
| M2_vs._M1 | 1032 | 686 | 1718 |
| P2_vs._P1 | 290 | 256 | 546 |
| YF2_vs._YF1 | 314 | 482 | 796 |
| FB2_vs._FB1 | 479 | 486 | 965 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Li, T.; Song, L.; Kong, J.; Niu, Q.; Wang, Y.; Wu, C.; Deng, B.; Wang, H.; Gai, Y. Comparative Transcriptome of Isonuclear Alloplasmic Strain Revealed the Important Role of Mitochondrial Genome in Regulating Flammulina filiformis. Agronomy 2023, 13, 998. https://doi.org/10.3390/agronomy13040998
Liu J, Li T, Song L, Kong J, Niu Q, Wang Y, Wu C, Deng B, Wang H, Gai Y. Comparative Transcriptome of Isonuclear Alloplasmic Strain Revealed the Important Role of Mitochondrial Genome in Regulating Flammulina filiformis. Agronomy. 2023; 13(4):998. https://doi.org/10.3390/agronomy13040998
Chicago/Turabian StyleLiu, Jingyu, Tianle Li, Linhao Song, Jinchao Kong, Qichen Niu, Yiting Wang, Chenjian Wu, Bing Deng, Hongkai Wang, and Yunpeng Gai. 2023. "Comparative Transcriptome of Isonuclear Alloplasmic Strain Revealed the Important Role of Mitochondrial Genome in Regulating Flammulina filiformis" Agronomy 13, no. 4: 998. https://doi.org/10.3390/agronomy13040998
APA StyleLiu, J., Li, T., Song, L., Kong, J., Niu, Q., Wang, Y., Wu, C., Deng, B., Wang, H., & Gai, Y. (2023). Comparative Transcriptome of Isonuclear Alloplasmic Strain Revealed the Important Role of Mitochondrial Genome in Regulating Flammulina filiformis. Agronomy, 13(4), 998. https://doi.org/10.3390/agronomy13040998