microRNAs as Early Biomarkers of Alzheimer’s Disease: A Synaptic Perspective



Abstract
:1. Introduction
2. Synaptic Function and Synaptic Alteration in AD
3. miRNAs as Mediators of Synaptic Dysfunction in AD
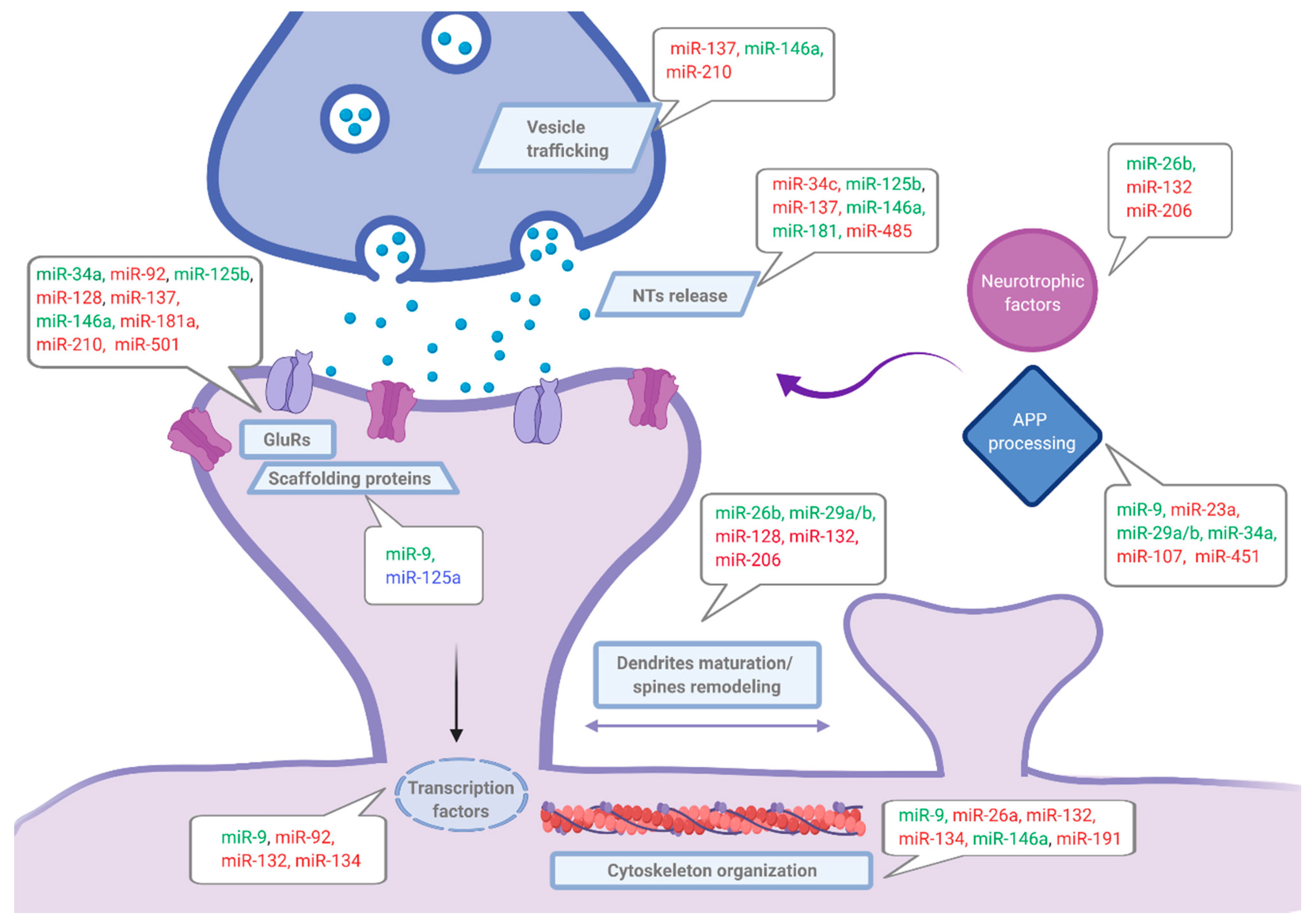
3.1. Synaptic Role of miRNAs
3.2. miRNAs and Synaptic Dysfunction in AD
3.3. Role of miRNAs in Aβ/Tau Mediated Synaptic Dysfunction and Neuroinflammation
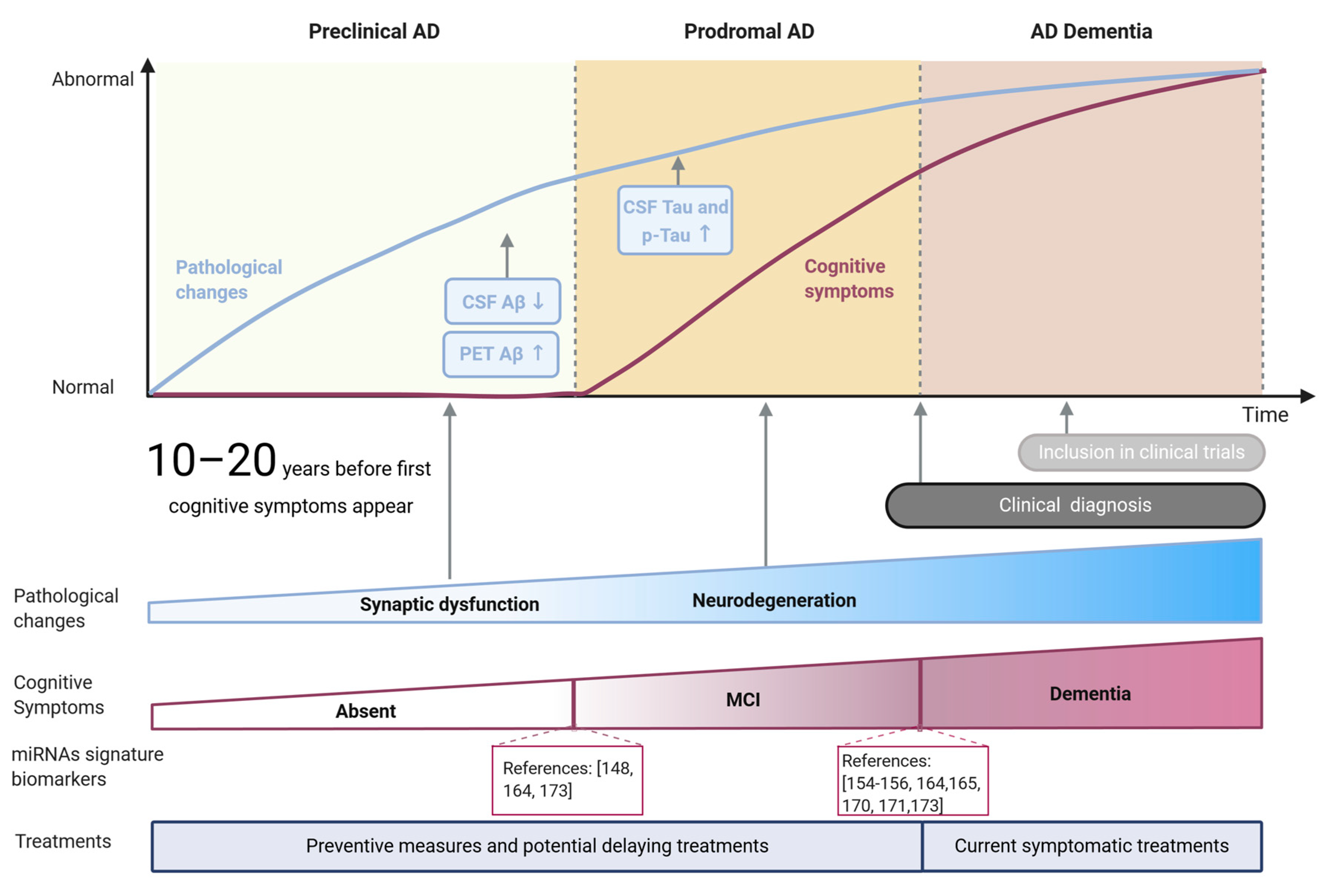
4. miRNAs as Biomarkers of AD
4.1. Current Biomarkers for AD
4.2. Synaptic-Related miRNAs as Early Biomarkers for AD
4.3. Potential and Limitations of Synaptic-Related miRNAs as Early AD Biomarkers
5. miRNAs as Therapeutic Targets
6. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prince, M.; Comas-Herrera, A.; Knapp, M.; Guerchet, M.; Karagiannidou, M. World Alzheimer Report 2016 Improving Healthcare for People Living with Dementia Coverage, Quality and Costs Now and In the Future; World Alzheimer: London, UK, 2016. [Google Scholar]
- World Health Organization. Dementia Fact Sheet. Available online: http://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 8 August 2018).
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.-J.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Van Hoesen, G.W.; Damasio, A.R.; Barnes, C.L. Alzheimer’s disease: Cell-specific pathology isolates the hippocampal formation. Science 1984, 225, 1168–1170. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Pooler, A.M.; Polydoro, M.; Wegmann, S.; Nicholls, S.B.; Spires-Jones, T.L.; Hyman, B.T. Propagation of tau pathology in Alzheimer’s disease: Identification of novel therapeutic targets. Alzheimers Res. Ther. 2013, 5, 49. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Androuin, A.; Potier, B.; Nägerl, U.V.; Cattaert, D.; Danglot, L.; Thierry, M.; Youssef, I.; Triller, A.; Duyckaerts, C.; El Hachimi, K.H.; et al. Evidence for altered dendritic spine compartmentalization in Alzheimer’s disease and functional effects in a mouse model. Acta Neuropathol. 2018, 135, 839–854. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Hoogenraad, C.C. The Postsynaptic Architecture of Excitatory Synapses: A More Quantitative View. Annu. Rev. Biochem. 2007, 76, 823–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, K.M.; Kater, S.B. Dendritic spines: Cellular specializations imparting both stability and flexibility to synaptic function. Annu. Rev. Neurosci. 1994, 17, 341–371. [Google Scholar] [CrossRef]
- O’brien, R.J.; Xu, D.; Petralia, R.S.; Steward, O.; Huganir, R.L.; Worley, P. Synaptic Clustering of AMPA Receptors by the Extracellular Immediate-Early Gene Product Narp. Neuron 1999, 23, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Kandel, E.R. The molecular biology of memory: CAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain. 2012, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [Green Version]
- Huganir, R.L.; Nicoll, R.A. AMPARs and synaptic plasticity: The last 25 years. Neuron 2013, 80, 704–717. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Sheng, M. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 2004, 5, 771–781. [Google Scholar] [CrossRef]
- Chowdhury, D.; Hell, J.W. Homeostatic synaptic scaling: Molecular regulators of synaptic AMPA-type glutamate receptors. F1000Research 2018, 7, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruska, M.; Henderson, N.T.; Xia, N.L.; Le Marchand, S.J.; Dalva, M.B. Anchoring and synaptic stability of PSD-95 is driven by ephrin-B3. Nat. Neurosci. 2015, 18, 1594–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, C.; Dresbach, T. Neuroligins and neurexins: Linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006, 29, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Mani, G.; Park, B.S.; Jacques, J.; Murdoch, G.; Whetsell, W.; Kaye, J.; Manczak, M. Differential loss of synaptic proteins in Alzheimer’s disease: Implications for synaptic dysfunction. J. Alzheimer’s Dis. 2005, 7, 103–117. [Google Scholar] [CrossRef]
- Miñano-Molina, A.J.; España, J.; Martín, E.; Barneda-Zahonero, B.; Fadó, R.; Solé, M.; Trullás, R.; Saura, C.A.; Rodríguez-Alvarez, J. Soluble oligomers of amyloid-β peptide disrupt membrane trafficking of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor contributing to early synapse dysfunction. J. Biol. Chem. 2011, 286, 27311–27321. [Google Scholar] [CrossRef] [Green Version]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J.; Rubin, E.H.; Morris, J.C. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Pickett, E.K.; Koffie, R.M.; Wegmann, S.; Henstridge, C.M.; Herrmann, A.G.; Colom-Cadena, M.; Lleo, A.; Kay, K.R.; Vaught, M.; Soberman, R.; et al. Non-Fibrillar Oligomeric Amyloid-within Synapses. J. Alzheimer’s Dis. 2016, 53, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Ittner, A.; Ittner, L.M. Dendritic Tau in Alzheimer’s Disease. Neuron 2018, 99, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid β-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol. 2006, 572, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Bilousova, T.; Miller, C.A.; Poon, W.W.; Vinters, H.V.; Corrada, M.; Kawas, C.; Hayden, E.Y.; Teplow, D.B.; Glabe, C.; Albay, R., III; et al. Synaptic amyloid-b oligomers precede p-tau and differentiate high pathology control cases. Am. J. Pathol. 2016, 186, 185–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Ovsepian, S.V.; O’Leary, V.B.; Zaborszky, L.; Ntziachristos, V.; Dolly, J.O. Synaptic vesicle cycle and amyloid β: Biting the hand that feeds. Alzheimer’s Dement. 2018, 14, 502–513. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR Removal Underlies Aβ-Induced Synaptic Depression and Dendritic Spine Loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Parameshwaran, K.; Dhanasekaran, M.; Suppiramaniam, V. Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp. Neurol. 2008, 210, 7–13. [Google Scholar] [CrossRef]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Sanz Clemente, A.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Ab Oligomer-Induced Aberrations in Synapse Composition, Shape, and Density Provide a Molecular Basis for Loss of Connectivity in Alzheimer’s Disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Tan, L.; Liu, J.; Yu, J.-T. The Essential Role of Soluble Aβ Oligomers in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 1905–1924. [Google Scholar] [CrossRef]
- Baglietto-Vargas, D.; Prieto, G.A.; Limon, A.; Forner, S.; Rodriguez-Ortiz, C.J.; Ikemura, K.; Ager, R.R.; Medeiros, R.; Trujillo-Estrada, L.; Martini, A.C.; et al. Impaired AMPA signaling and cytoskeletal alterations induce early synaptic dysfunction in a mouse model of Alzheimer’s disease. Aging Cell 2018, 17, e12791. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.-Q. Amyloid- /Fyn-Induced Synaptic, Network, and Cognitive Impairments Depend on Tau Levels in Multiple Mouse Models of Alzheimer’s Disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-β toxicity in alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.M.; Selkoe, D.J. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 2004, 44, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, E.C.; Teravskis, P.J.; Dummer, B.W.; Zhao, X.; Huganir, R.L.; Liao, D. Tau phosphorylation and tau mislocalization mediate soluble Ab oligomer-induced AMPA glutamate receptor signaling deficits. Eur. J. Neurosci. 2014, 39, 1214–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.; Kim, H.J.; Kim, Y.; Hwang, H.S.; Kasai, H.; Kim, J.H.; Park, J.W. Nanoscale imaging reveals miRNA-mediated control of functional states of dendritic spines. Proc. Natl. Acad. Sci. USA 2019, 116, 9616–9621. [Google Scholar] [CrossRef] [Green Version]
- Luarte, A.; Henzi, R.; Fernández, A.; Gaete, D.; Cisternas, P.; Pizarro, M.; Batiz, L.F.; Villalobos, I.; Masalleras, M.; Vergara, R.; et al. Astrocyte-Derived Small Extracellular Vesicles Regulate Dendritic Complexity through miR-26a-5p Activity. Cells 2020, 9, 930. [Google Scholar] [CrossRef] [Green Version]
- Daugaard, I.; Hansen, T.B. Biogenesis and Function of Ago-Associated RNAs. Trends Genet. 2017, 33, 208–219. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Vasudevan, S.; Steitz, J.A. AU-Rich-Element-Mediated Upregulation of Translation by FXR1 and Argonaute 2. Cell 2007, 128, 1105–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dajas-Bailador, F.; Bonev, B.; Garcez, P.; Stanley, P.; Guillemot, F.; Papalopulu, N. microRNA-9 regulates axon extension and branching by targeting Map1b in mouse cortical neurons. Nat. Neurosci. 2012, 15, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Olde Loohuis, N.F.M.; Kos, A.; Martens, G.J.M.; Van Bokhoven, H.; Nadif Kasri, N.; Aschrafi, A. MicroRNA networks direct neuronal development and plasticity. Cell Mol. Life Sci. 2012, 69, 89–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Battum, E.Y.; Verhagen, M.G.; Vangoor, V.R.; Fujita, Y.; Derijck, A.A.H.A.; O’Duibhir, E.; Giuliani, G.; de Gunst, T.; Adolfs, Y.; Lelieveld, D.; et al. An Image-Based miRNA Screen Identifies miRNA-135s As Regulators of CNS Axon Growth and Regeneration by Targeting Krüppel-like Factor 4. J. Neurosci. 2018, 38, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimer’s Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef]
- Lugli, G.; Torvik, V.I.; Larson, J.; Smalheiser, N.R. Expression of microRNAs and their precursors in synaptic fractions of adult mouse forebrain. J. Neurochem. 2008, 106, 650–661. [Google Scholar] [CrossRef]
- Kye, M.-J.; Liu, T.; Levy, S.F.; Xu, N.L.; Groves, B.B.; Bonneau, R.; Lao, K.; Kosik, K.S. Somatodendritic microRNAs identified by laser capture and multiplex RT-PCR. RNA 2007, 13, 1224–1234. [Google Scholar] [CrossRef] [Green Version]
- Lugli, G.; Larson, J.; Martone, M.E.; Jones, Y.; Smalheiser, N.R. Dicer and eIF2c are enriched at postsynaptic densities in adult mouse brain and are modified by neuronal activity in a calpain-dependent manner. J. Neurochem. 2003, 94, 896–905. [Google Scholar] [CrossRef]
- Sambandan, S.; Akbalik, G.; Kochen, L.; Rinne, J.; Kahlstatt, J.; Glock, C.; Tushev, G.; Alvarez-Castelao, B.; Heckel, A.; Schuman, E.M. Activity-dependent spatially localized miRNA maturation in neuronal dendrites. Science 2017, 355, 634–637. [Google Scholar] [CrossRef]
- Parra-Damas, A.; Saura, C.A. Synapse-to-Nucleus Signaling in Neurodegenerative and Neuropsychiatric Disorders. Biol. Psychiatry 2019, 86, 87–96. [Google Scholar] [CrossRef]
- Bicker, S.; Lackinger, M.; Weiß, K.; Schratt, G. MicroRNA-132, -134, and -138: A microRNA troika rules in neuronal dendrites. Cell Mol. Life Sci. 2014, 71, 3987–4005. [Google Scholar] [CrossRef] [PubMed]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Zampa, F.; Bicker, S.; Schratt, G. Activity-Dependent Pre-miR-134 Dendritic Localization Is Required for Hippocampal Neuron Dendritogenesis. Front. Mol. Neurosci. 2018, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fiore, R.; Rajman, M.; Schwale, C.; Bicker, S.; Antoniou, A.; Bruehl, C.; Draguhn, A.; Schratt, G. MiR-134-dependent regulation of Pumilio-2 is necessary for homeostatic synaptic depression. EMBO J. 2014, 33, 2231–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippi, G.; Steinert, J.R.; Marczylo, E.L.; D’Oro, S.; Fiore, R.; Forsythe, I.D.; Schratt, G.; Zoli, M.; Nicotera, P.; Young, K.W. Targeting of the Arpc3 actin nucleation factor by miR-29a/b regulates dendritic spine morphology. J. Cell Biol. 2011, 194, 889–904. [Google Scholar] [CrossRef]
- Hu, Z.; Yu, D.; Gu, Q.H.; Yang, Y.; Tu, K.; Zhu, J.; Li, Z. MiR-191 and miR-135 are required for long-lasting spine remodelling associated with synaptic long-term depression. Nat. Commun. 2014, 5, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Giusti, S.A.; Vogl, A.M.; Brockmann, M.M.; Vercelli, C.A.; Rein, M.L.; Trümbach, D.; Wurst, W.; Cazalla, D.; Stein, V.; Deussing, J.M.; et al. MicroRNA-9 controls dendritic development by targeting REST. Elife 2014, 3, 1–22. [Google Scholar] [CrossRef]
- Sim, S.-E.; Lim, C.-S.; Kim, J.-I.; Seo, D.; Chun, H.; Yu, N.-K.; Lee, J.; Kang, S.J.; Ko, H.-G.; Choi, J.-H.; et al. The Brain-Enriched MicroRNA miR-9-3p Regulates Synaptic Plasticity and Memory. J. Neurosci. 2016, 36, 8641–8652. [Google Scholar] [CrossRef]
- Letellier, M.; Elramah, S.; Mondin, M.; Soula, A.; Penn, A.; Choquet, D.; Landry, M.; Thoumine, O.; Favereaux, A. miR-92a regulates expression of synaptic GluA1-containing AMPA receptors during homeostatic scaling. Nat. Neurosci. 2014, 17, 1040–1042. [Google Scholar] [CrossRef]
- Hu, Z.; Zhao, J.; Hu, T.; Luo, Y.; Zhu, J.; Li, Z. miR-501-3p mediates the activity-dependent regulation of the expression of AMPA receptor subunit GluA1. J. Cell Biol. 2015, 208, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Olde Loohuis, N.F.M.; Ba, W.; Stoerchel, P.H.; Kos, A.; Jager, A.; Schratt, G.; Martens, G.J.M.; van Bokhoven, H.; Nadif Kasri, N.; Aschrafi, A. MicroRNA-137 Controls AMPA-Receptor-Mediated Transmission and mGluR-Dependent LTD. Cell Rep. 2015, 11, 1876–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Q.; Ruan, H.; Gilbert, J.; Wang, G.; Ma, Q.; Yao, W.-D.; Man, H.-Y. MicroRNA miR124 is required for the expression of homeostatic synaptic plasticity. Nat. Commun. 2015, 6, 10045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocchi, A.; Moretti, D.; Lignani, G.; Colombo, E.; Scholz-Starke, J.; Baldelli, P.; Tkatch, T.; Benfenati, F. Neurite-Enriched MicroRNA-218 Stimulates Translation of the GluA2 Subunit and Increases Excitatory Synaptic Strength. Mol. Neurobiol. 2019, 56, 5701–5714. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.M.; Rodrigues, B.; Fernandes, J.; Santos, S.D.; Carreto, L.; Santos, M.A.S.; Pinheiro, P.; Carvalho, A.L. MicroRNA-186-5p controls GluA2 surface expression and synaptic scaling in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2019, 116, 5727–5736. [Google Scholar] [CrossRef] [Green Version]
- Alsharafi, W.A.; Xiao, B.; Li, J. MicroRNA-139-5p negatively regulates NR2A-containing NMDA receptor in the rat pilocarpine model and patients with temporal lobe epilepsy. Epilepsia 2016, 57, 1931–1940. [Google Scholar] [CrossRef]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.-F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of Synaptic Structure and Function by FMRP-Associated MicroRNAs miR-125b and miR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Corbel, C.; Hernandez, I.; Wu, B.; Kosik, K.S. Developmental attenuation of N-methyl-D-aspartate receptor subunit expression by microRNAs. Neural Dev. 2015, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Engler-Chiurazzi, E.B.; Cavendish, J.Z.; Povroznik, J.M.; Russell, A.E.; Quintana, D.D.; Mathers, P.H.; Simpkins, J.W. Over-expression of miR-34a induces rapid cognitive impairment and Alzheimer’s disease-like pathology. Brain Res. 2019, 1721, 146327. [Google Scholar] [CrossRef]
- Deng, M.; Zhang, Q.; Wu, Z.; Ma, T.; He, A.; Zhang, T.; Ke, X.; Yu, Q.; Han, Y.; Lu, Y. Mossy cell synaptic dysfunction causes memory imprecision via miR-128 inhibition of STIM2 in Alzheimer’s disease mouse model. Aging Cell 2020, 19, e13144. [Google Scholar] [CrossRef] [Green Version]
- Pulkkinen, K.; Malm, T.; Turunen, M.; Koistinaho, J.; Ylä-Herttuala, S. Hypoxia induces microRNA miR-210 in vitro and in vivo. FEBS Lett. 2008, 582, 2397–2401. [Google Scholar] [CrossRef] [Green Version]
- Pelkey, K.A.; Barksdale, E.; Craig, M.T.; Yuan, X.; Sukumaran, M.; Vargish, G.A.; Mitchell, R.M.; Wyeth, M.S.; Petralia, R.S.; Chittajallu, R.; et al. Pentraxins coordinate excitatory synapse maturation and circuit integration of parvalbumin interneurons. Neuron 2015, 85, 1257–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Yu, J.; Wu, Z.; Si, W.; Li, X.; Liu, Y.; Zhou, J.; Deng, R.; Chen, D. MicroRNA-210-5p contributes to cognitive impairment in early vascular dementia rat model through targeting snap25. Front. Mol. Neurosci. 2018, 11, 388. [Google Scholar] [CrossRef] [PubMed]
- Siegert, S.; Seo, J.; Kwon, E.J.; Rudenko, A.; Cho, S.; Wang, W.; Flood, Z.; Martorell, A.J.; Ericsson, M.; Mungenast, A.E. The schizophrenia risk gene product miR-137 alters presynaptic plasticity. Nat. Neurosci. 2015, 18, 1008–1016. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.E.; Lee, P.R.; Chen, S.; Li, W.; Fields, R.D. MicroRNA regulation of homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 11650–11655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajgor, D.; Purkey, A.M.; Sanderson, J.L.; Welle, T.M.; Garcia, J.D.; Dell’Acqua, M.L.; Smith, K.R. Local miRNA-Dependent Translational Control of GABAAR Synthesis during Inhibitory Long-Term Potentiation. Cell Rep. 2020, 31, 107785. [Google Scholar] [CrossRef]
- Hebert, S.S.; Horre, K.; Nicolai, L.; Papadopoulou, A.S.; Mandemakers, W.; Silahtaroglu, A.N.; Kauppinen, S.; Delacourte, A.; De Strooper, B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/ -secretase expression. Proc. Natl Acad Sci. USA 2008, 105, 6415–6420. [Google Scholar] [CrossRef] [Green Version]
- Schonrock, N.; Ke, Y.D.; Humphreys, D.; Staufenbiel, M.; Ittner, L.M.; Preiss, T.; Rgen Gö Tz, J.; Götz, J. Neuronal microrna deregulation in response to Alzheimer’s disease amyloid-β. PLoS ONE 2010, 5, e11070. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The Expression of MicroRNA miR-107 Decreases Early in Alzheimer’s Disease and May Accelerate Disease Progression through Regulation of -Site Amyloid Precursor Protein-Cleaving Enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef]
- Zhang, Y.; Xing, H.; Guo, S.; Zheng, Z.; Wang, H.; Xu, D. MicroRNA-135b has a neuroprotective role via targeting of β-site APP-cleaving enzyme 1. Exp. Ther. Med. 2016, 12, 809–814. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.R.; Wang, J.; Zhang, X.B.; Geng, Y.; Hu, Z.; Rudd, J.A.; Ling, S.; Chen, W.; Han, S. The miR-124 regulates the expression of BACE1/beta-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 2012, 209, 94–105. [Google Scholar] [CrossRef]
- Schonrock, N.; Humphreys, D.T.; Preiss, T.; Götz, J. Target gene repression mediated by miRNAs miR-181c and miR-9 both of which are down-regulated by amyloid-β. J. Mol. Neurosci. 2012, 46, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.; Chertkow, H.; Schipper, H.M.; Yuan, Z.; Shetty, V.; Jenkins, S.; Jones, T.; Wang, E. Increased microRNA-34c abundance in Alzheimer’s disease circulating blood plasma. Front. Mol. Neurosci. 2014, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucci, C.; Mesquita-Ribeiro, R.; Rathbone, A.; Dajas-Bailador, F. Spatiotemporal regulation of GSK3β levels by miRNA-26a controls axon development in cortical neurons. Development 2020, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetere, G.; Barbato, C.; Pezzola, S.; Yuan, Z.; Shetty, V.; Jenkins, S.; Jones, T.; Wang, E. Selective inhibition of miR-92 in hippocampal neurons alters contextual fear memory. Hippocampus 2014, 24, 1458–1465. [Google Scholar] [CrossRef]
- Liu, H.; Chu, W.; Gong, L.; Gao, X.; Wang, W. MicroRNA-26b is upregulated in a double transgenic mouse model of Alzheimer’s disease and promotes the expression of amyloid-β by targeting insulin-like growth factor 1. Mol. Med. Rep. 2016, 13, 2809–2814. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-T.; Chu, K.; Jung, K.-H.; Kim, J.H.; Huh, J.-Y.; Yoon, H.; Park, D.-K.; Lim, J.-Y.; Kim, J.-M.; Jeon, D.; et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann. Neurol. 2012, 72, 269–277. [Google Scholar] [CrossRef]
- Chu, T.; Shu, Y.; Qu, Y.; Gao, S.; Zhang, L. miR-26b inhibits total neurite outgrowth, promotes cells apoptosis and downregulates neprilysin in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2018, 11, 3383–3390. [Google Scholar]
- Li, H.; Mao, S.; Wang, H.; Zen, K.; Zhang, C.; Li, L. MicroRNA-29a modulates axon branching by targeting doublecortin in primary neurons. Protein Cell. 2014, 5, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Rehfeld, F.; Maticzka, D.; Grosser, S.; Knauff, P.; Eravci, M.; Vida, I.; Backofen, R.; Wulczyn, F.G. The RNA-binding protein ARPP21 controls dendritic branching by functionally opposing the miRNA it hosts. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Impey, S.; Davare, M.; Lasiek, A.; Fortin, D.; Ando, H.; Varlamova, O.; Obrietan, K.; Soderling, T.R.; Goodman, R.H.; Wayman, G.A. An activity-induced microRNA controls dendritic spine formation by regulating Rac1-PAK signaling. Mol. Cell Neurosci. 2010, 43, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhang, R.; Lu, K.; Yu, W.; Xie, B.; Cui, D.; Jiang, L.; Zhang, Q.; Xu, S. Deregulation of miRNA-181c potentially contributes to the pathogenesis of AD by targeting collapsin response mediator protein 2 in mice. J. Neurol Sci. 2016, 367, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Kos, A.; Olde Loohuis, N.; Meinhardt, J.; van Bokhoven, H.; Kaplan, B.B.; Martens, G.J.; Aschrafi, A. MicroRNA-181 promotes synaptogenesis and attenuates axonal outgrowth in cortical neurons. Cell Mol. Life Sci. 2016, 73, 3555–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Wang, H.; Chen, K.; Cheng, P.; Gao, S.; Liu, J.; Li, X.; Sun, X. MicroRNA-34c Downregulation Ameliorates Amyloid-β-Induced Synaptic Failure and Memory Deficits by Targeting VAMP2. J. Alzheimer’s Dis. 2015, 48, 673–686. [Google Scholar] [CrossRef]
- Prada, I.; Gabrielli, M.; Turola, E.; Iorio, A.; D’Arrigo, G.; Parolisi, R.; De Luca, M.; Pacifici, M.; Bastoni, M.; Lombardi, M.; et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018, 135, 529–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.L.; Shen, C.K.J. Modulation of mGluR-dependent MAP1B translation and AMPA receptor endocytosis by microRNA miR-146a-5p. J. Neurosci. 2013, 33, 9013–9020. [Google Scholar] [CrossRef] [Green Version]
- Muddashetty, R.S.; Nalavadi, V.C.; Gross, C.; Yao, X.; Xing, L.; Laur, O.; Warren, S.T.; Bassell, G.J. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation and mGluR signaling. Mol. Cell. 2011, 42, 673–688. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Chen, P.; Wang, X.; Yao, J.; Zhuang, S. miR-34a deficiency in APP/PS1 mice promotes cognitive function by increasing synaptic plasticity via AMPA and NMDA receptors. Neurosci. Lett. 2018, 670, 94–104. [Google Scholar] [CrossRef]
- Rodriguez-Ortiz, C.J.; Prieto, G.A.; Martini, A.C.; Forner, S.; Trujillo-Estrada, L.; LaFerla, F.M.; Baglietto-Vargas, D.; Cotman, C.W.; Kitazawa, M. miR-181a negatively modulates synaptic plasticity in hippocampal cultures and its inhibition rescues memory deficits in a mouse model of Alzheimer’s disease. Aging Cell 2020, 19, e13118. [Google Scholar] [CrossRef] [Green Version]
- Lau, P.; Bossers, K.; Janky, R.; Salta, E.; Frigerio, C.S.; Barbash, S.; Rothman, R.; Sierksma, A.S.R.; Thathiah, A.; Greenberg, D.; et al. Alteration of the microRNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 1613–1634. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.Y.; Zhou, Y.; Zhu, L.S.; Wang, X.; Pang, P.; Wang, D.Q.; Liuyang, Z.Y.; Man, H.; Lu, Y.; Zhu, L.Q.; et al. Correcting abnormalities in miR-124/PTPN1 signaling rescues tau pathology in Alzheimer’s disease. J. Neurochem. 2020, 154, 441–457. [Google Scholar] [CrossRef]
- Baby, N.; Alagappan, N.; Dheen, S.T.; Sajikumar, S. MicroRNA-134-5p inhibition rescues long-term plasticity and synaptic tagging/capture in an Aβ(1–42)-induced model of Alzheimer’s disease. Aging Cell 2020, 19, 13046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.J.; Wang, B.; Li, Q.X.; Dong, X.L.; Han, X.L.; Zhang, S.B. Effects of microRNA-206 and its target gene IGF-1 on sevoflurane-induced activation of hippocampal astrocytes in aged rats through the PI3K/AKT/CREB signaling pathway. J. Cell Physiol. 2018, 233, 4294–4306. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Hu, M.; Zhang, J.; Teng, Z.Q.; Chen, C. A novel mechanism of synaptic and cognitive impairments mediated via microRNA-30b in Alzheimer’s disease. EBioMedicine 2019, 39, 409–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Liu, D.; Huang, H.Z.; Wang, Z.-H.; Hou, T.-Y.; Yang, X.; Pang, P.; Wei, N.; Zhou, Y.-F.; Dupras, M.-J.; et al. A Novel MicroRNA-124/PTPN1 Signal Pathway Mediates Synaptic and Memory Deficits in Alzheimer’s Disease. Biol. Psychiatry 2017, 83, 395–405. [Google Scholar] [CrossRef]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; De Strooper, B. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso, A.D.C.; Chohan, M.O.; El-Akkad, E.; Gong, C.; Khatoon, S.; Liu, F.; Grundke-Iqbal, I. Molecular Basis of Tau Protein Pathology: Role of Abnormal Hyperphosphorylation; Oxford University Press: New York, NY, USA, 2007. [Google Scholar]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tan, L.; Lu, Y.; Peng, J.; Zhu, Y.; Zhang, Y.; Sun, Z. MicroRNA-138 promotes tau phosphorylation by targeting retinoic acid receptor alpha. FEBS Lett. 2015, 589, 726–729. [Google Scholar] [CrossRef] [Green Version]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef] [Green Version]
- Hébert, S.S.; Wang, W.-X.; Zhu, Q.; Nelson, P.T. A Study of Small RNAs from Cerebral Neocortex of Pathology-Verified Alzheimer’s Disease, Dementia with Lewy Bodies, Hippocampal Sclerosis, Frontotemporal Lobar Dementia, and Non-Demented Human Controls. J. Alzheimer’s Dis. 2013, 35, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; Dorval, V.; Parsi, S.; Morin, F.; Planel, E.; et al. MiR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 2015, 24, 6721–6735. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Song, Y.; Zhou, X.; Deng, Y.; Liu, T.; Weng, G.; Yu, D.; Pan, S. MicroRNA-29c targets β-site amyloid precursor protein-cleaving enzyme 1 and has a neuroprotective role in vitro and in vivo. Mol. Med. Rep. 2015, 12, 3081–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Hu, M.; Teng, Z.; Tang, Y.-P.; Chen, C. Synaptic and Cognitive Improvements by Inhibition of 2-AG Metabolism Are through Upregulation of MicroRNA-188-3p in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2014, 34, 14919–14933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Kim, J.-H.; Kwon, O.-B.; An, K.; Ryu, J.; Cho, K.; Suh, Y.-H.; Kim, H.-S. An Activity-Regulated microRNA, miR-188, Controls Dendritic Plasticity and Synaptic Transmission by Downregulating Neuropilin-2. J. Neurosci. 2012, 32, 5678–5687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilardo, E.; Barbato, C.; Ciotti, M.; Cogoni, C.; Ruberti, F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J. Biol. Chem. 2010, 285, 18344–18351. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Liu, C.; Zhu, J.; Shu, P.; Yin, B.; Gong, Y.; Qiang, B.; Yuan, J.; Peng, X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 2012, 33, 522–534. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, C.-F.; Wang, A.-H.; Lin, Q.-F. MiR-16 regulates cell death in Alzheimer’s disease by targeting amyloid precursor protein. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4020–4027. [Google Scholar]
- Delay, C.; Calon, F.; Mathews, P.; Hébert, S.S. Alzheimer-specific variants in the 3’UTR of Amyloid precursor protein affect microRNA function. Mol. Neurodegener. 2011, 6, 70. [Google Scholar] [CrossRef] [Green Version]
- Hébert, S.S.; Papadopoulou, A.S.; Smith, P.; Galas, M.C.; Planel, E.; Silahtaroglu, A.N.; Sergeant, N.; Buée, L.; de Strooper, B. Genetic ablation of dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 2010, 19, 3959–3969. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Yao, Y.; Liu, Y.; Zhou, R.; Zhang, W.; Hu, Q.; Liu, H.; Al Hamda, M.H.; Zhang, A. Regulation of ADAM10 by MicroRNA-23a Contributes to Epileptogenesis in Pilocarpine-Induced Status Epilepticus Mice. Front. Cell Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Li, W.; Zhang, Z.; Yoshimura, S.; Hao, Q.; Zhang, C.; Wang, Z. MicroRNA-144 is regulated by activator protein-1 (AP-1) and decreases expression of alzheimer disease-related a disintegrin and metalloprotease 10 (ADAM10). J. Biol. Chem. 2013, 288, 13748–13761. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, P.H.; Colombo, A.V.; Schusser, B.; Dreymueller, D.; Wetzel, S.; Schepers, U.; Herber, J.; Ludwig, A.; Kremmer, E.; Montag, D.; et al. Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. Elife 2016, 5, e12748. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics. 2013, 14, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International: London, UK, 2019. [Google Scholar]
- Landau, S.M.; Harvey, D.; Madison, C.M.; Reiman, E.M.; Foster, N.L.; Aisen, P.S.; Petersen, R.C.; Shaw, L.M.; Trojanowski, J.Q.; Jack, C.R.; et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 2010, 75, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- El Kadmiri, N.; Said, N.; Slassi, I.; El Moutawakil, B.; Nadifi, S. Biomarkers for Alzheimer Disease: Classical and Novel Candidates’ Review. Neuroscience 2018, 370, 181–190. [Google Scholar] [CrossRef]
- Chételat, G.; Arbizu, J.; Barthel, H.; Garibotto, V.; Law, I.; Morbelli, S.; van de Giessen, E.; Agosta, F.; Barkhof, F.; Brooks, D.J.; et al. Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. Lancet Neurol. 2020, 19, 951–962. [Google Scholar] [CrossRef]
- Ritchie, C.; Smailagic, N.; Noel-Storr, A.H.; Takwoingi, Y.; Flicker, L.; Mason, S.E.; Mcshane, R. Plasma and cerebrospinal fluid amyloid beta for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2014, 2014. [Google Scholar] [CrossRef]
- Preische, O.; Schultz, S.A.; Apel, A.; Kuhle, J.; Kaeser, S.A.; Barro, C.; Gräber, S.; Kuder-Buletta, E.; LaFougere, C.; Laske, C.; et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med. 2019, 25, 277–283. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, M.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Swarbrick, S.; Wragg, N.; Ghosh, S.; Stolzing, A. Systematic Review of miRNA as Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6156–6167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Jaber, V.; Alexandrov, P.N.; Vergallo, A.; Lista, S.; Hampel, H.; Lukiw, W.J. microRNA-Based Biomarkers in Alzheimer’s Disease (AD). Front. Neurosci. 2020, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Vázquez-Higuera, J.L.; Pozueta, A.; Lage, C.; Kazimierczak, M.; Bravo, M.; Calero, M.; Gonalezález, A.; Rodríguez, E.; Lleó, A.; et al. MicroRNA Profile in Patients with Alzheimer’s Disease: Analysis of miR-9-5p and miR-598 in Raw and Exosome Enriched Cerebrospinal Fluid Samples. J. Alzheimer’s Dis. 2017, 57, 483–491. [Google Scholar] [CrossRef]
- Geekiyanage, H.; Jicha, G.A.; Nelson, P.T.; Chan, C. Blood serum miRNA: Non-invasive biomarkers for Alzheimer’s disease. Exp. Neurol. 2012, 235, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbagallo, C.; Mostile, G.; Baglieri, G.; Giunta, F.; Luca, A.; Raciti, L.; Zappia, M.; Purrello, M.; Ragusa, M.; Nicoletti, A. Specific Signatures of Serum miRNAs as Potential Biomarkers to Discriminate Clinically Similar Neurodegenerative and Vascular-Related Diseases. Cell Mol. Neurobiol. 2020, 40, 531–546. [Google Scholar] [CrossRef]
- Guo, R.; Fan, G.; Zhang, J.; Wu, C.; Du, Y.; Ye, H.; Li, Z.; Wang, L.; Zhang, Z.; Zhang, L.; et al. A 9-microRNA Signature in Serum Serves as a Noninvasive Biomarker in Early Diagnosis of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 1365–1377. [Google Scholar] [CrossRef]
- Denk, J.; Oberhauser, F.; Kornhuber, J.; Wiltfang, J.; Fassbender, K.; Schroeter, M.L.; Volk, A.E.; Diehl-Schmid, J.; Prudlo, J.; Danek, A.; et al. Specific serum and CSF microRNA profiles distinguish sporadic behavioural variant of frontotemporal dementia compared with Alzheimer patients and cognitively healthy controls. PLoS ONE. 2018, 13, e0197329. [Google Scholar] [CrossRef]
- Galimberti, D.; Villa, C.; Fenoglio, C.; Serpente, M.; Ghezzi, L.; Cioffi, S.M.G.; Arighi, A.; Fumagalli, G.; Scarpini, E. Circulating miRNAs as Potential Biomarkers in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 42, 1261–1267. [Google Scholar] [CrossRef]
- Satoh, J.I.; Kino, Y.; Niida, S. MicroRNA-Seq data analysis pipeline to identify blood biomarkers for alzheimer’s disease from public data. Biomark Insights 2015, 2015, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in plasma and cerebrospinal fluid as potential markers for Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 253–259. [Google Scholar] [CrossRef]
- Müller, M.; Jäkel, L.; Bruinsma, I.B.; Claassen, J.A.; Kuiperij, H.B.; Verbeek, M.M. MicroRNA-29a Is a Candidate Biomarker for Alzheimer’s Disease in Cell-Free Cerebrospinal Fluid. Mol. Neurobiol. 2016, 53, 2894–2899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosín-Tomás, M.; Antonell, A.; Lladó, A.; Alcolea, D.; Fortea, G.; Ezquerra, M.; Lleó, A.; Martí, M.J.; Pallàs, M.; Sanchez-Valle, R.; et al. Plasma miR-34a-5p and miR-545-3p as Early Biomarkers of Alzheimer’s Disease: Potential and Limitations. Mol. Neurobiol. 2017, 54, 5550–5562. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, C.; Zhang, Y. An investigation of microRNA-103 and microRNA-107 as potential blood-based biomarkers for disease risk and progression of Alzheimer’s disease. J. Clin. Lab. Anal. 2020, 34, 23006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.L.; Yu, J.-T.T.; Liu, Q.-Y.Y.; Tan, M.-S.S.; Zhang, W.; Hu, N.; Wang, Y.-L.L.; Sun, L.; Jiang, T.; Tan, L.L. Circulating miR-125b as a biomarker of Alzheimer’s disease. J. Neurol. Sci. 2014, 336, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, K.S.; Tsivinsky, V.G.; Abdullah, L.; Crawford, F.; Umansky, S.R. Plasma microRNA biomarkers for detection of mild cognitive impairment: Biomarker validation study. Aging 2013, 5, 925–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, D.J.; Mengel, D.; Mustapic, M.; Liu, W.; Selkoe, D.J.; Kapogiannis, D.; Galasko, D.; Rissman, R.A.; Bennett, D.A.; Walsh, D.M. miR-212 and miR-132 Are Downregulated in Neurally Derived Plasma Exosomes of Alzheimer’s Patients. Front. Neurosci. 2019, 13, 1208. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Zhou, H.; Zhang, R.; Song, M.; Yu, L.; Wang, L.; Liu, Z.; Zhang, Q.; Cui, D.; Wang, X.; et al. Serum miR-206 and miR-132 as Potential Circulating Biomarkers for Mild Cognitive Impairment. J. Alzheimer’s Dis. 2015, 45, 721–731. [Google Scholar] [CrossRef]
- Ansari, A.; Maffioletti, E.; Milanesi, E.; Marizzoni, M.; Frisoni, G.B.; Blin, O.; Richardson, J.C.; Bordet, R.; Forloni, G.; Gennarelli, M.; et al. miR-146a and miR-181a are involved in the progression of mild cognitive impairment to Alzheimer’s disease. Neurobiol. Aging 2019, 82, 102–109. [Google Scholar] [CrossRef]
- Siedlecki-Wullich, D.; Català-Solsona, J.; Fábregas, C.; Hernández, I.; Clarimon, J.; Lleó, A.; Boada, M.; Saura, C.A.; Rodríguez-Álvarez, J.; Miñano-Molina, A.J. Altered microRNAs related to synaptic function as potential plasma biomarkers for Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 46. [Google Scholar] [CrossRef]
- Kumar, P.; Dezso, Z.; MacKenzie, C.; Oestreicher, J.; Agoulnik, S.; Byrne, M.; Bernier, F.; Yanagimachi, M.; Aoshima, K.; Oda, Y. Circulating miRNA biomarkers for Alzheimer’s disease. PLoS ONE 2013, 8, e69807. [Google Scholar] [CrossRef]
- Kenny, A.; McArdle, H.; Calero, M.; Rabano, A.; Madden, S.F.; Adamson, K.; Forster, R.; Spain, E.; Prehn, J.H.M.; Henshall, D.C.; et al. Elevated plasma microRNA-206 levels predict cognitive decline and progression to dementia from mild cognitive impairment. Biomolecules 2019, 9, 734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gámez-Valero, A.; Campdelacreu, J.; Vilas, D.; Ispierto, L.; Reñé, R.; Álvarez, R.; Armengol, M.P.; Borràs, F.E.; Beyer, K. Exploratory study on microRNA profiles from plasma-derived extracellular vesicles in Alzheimer’s disease and dementia with Lewy bodies. Transl. Neurodegener. 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Li, H.; Liu, W.; Zhang, L.; Tian, Q.; Li, H.; Li, M. MiR-485-3p serves as a biomarker and therapeutic target of Alzheimer’s disease via regulating neuronal cell viability and neuroinflammation by targeting AKT3. Mol. Genet. Genomic Med. 2020. [Google Scholar] [CrossRef]
- Hara, N.; Kikuchi, M.; Miyashita, A.; Hatsuta, H.; Saito, Y.; Kasuga, K.; Murayama, S.; Ikeuchi, T.; Kuwano, R. Serum microRNA miR-501-3p as a potential biomarker related to the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Hajian-Tilaki, K. Receiver Operating Characteristic (ROC) Curve Analysis for Medical Diagnostic Test Evaluation. Casp. J. Intern. Med. 2013, 4, 627. [Google Scholar]
- Sheinerman, K.S.; Toledo, J.B.; Tsivinsky, V.G.; Irwin, D.; Grossman, M.; Weintraub, D.; Hurtig, H.I.; Chen-Plotkin, A.; Wolk, D.A.; McCluskey, L.F.; et al. Circulating brain-enriched microRNAs as novel biomarkers for detection and differentiation of neurodegenerative diseases. Alzheimers Res. Ther. 2017, 9, 89. [Google Scholar] [CrossRef]
- Leidinger, P.; Backes, C.; Deutscher, S.; Schmitt, K.; Mueller, S.C.; Frese, K.; Haas, J.; Ruprecht, K.; Paul, F.; Stähler, C.; et al. A blood based 12-miRNA signature of Alzheimer disease patients. Genome Biol. 2013, 14, R78. [Google Scholar] [CrossRef] [Green Version]
- Jain, G.; Stuendl, A.; Rao, P.; Berulava, T.; Pena Centeno, T.; Kaurani, L.; Burkhardt, S.; Delalle, I.; Kornhuber, J.; Hüll, M.; et al. A combined miRNA–piRNA signature to detect Alzheimer’s disease. Transl. Psychiatry 2019, 9, 250. [Google Scholar] [CrossRef] [Green Version]
- Souza, V.C.; Morais, G.S.; Henriques, A.D.; Machado-Silva, W.; Perez, D.I.V.; Brito, C.J.; Camargos, E.F.; Moraes, C.F.; Nóbrega, O.T. Whole-Blood Levels of MicroRNA-9 Are Decreased in Patients With Late-Onset Alzheimer Disease. Am. J. Alzheimer’s Dis. Other Dementiasr. 2020, 35, 153331752091157. [Google Scholar] [CrossRef]
- Denk, J.; Boelmans, K.; Siegismund, C.; Lassner, D.; Arlt, S.; Jahn, H. MicroRNA Profiling of CSF Reveals Potential Biomarkers to Detect Alzheimer`s Disease. Hoheisel JD, ed. PLoS ONE 2015, 10, e0126423. [Google Scholar] [CrossRef] [Green Version]
- Lugli, G.; Cohen, A.M.; Bennett, D.A.; Shah, R.C.; Fields, C.J.; Hernandez, A.G.; Smalheiser, N.R. Plasma Exosomal miRNAs in Persons with and without Alzheimer Disease: Altered Expression and Prospects for Biomarkers. PLoS ONE 2015, 10, e0139233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endzelinš, E.; Berger, A.; Melne, V.; Bajo-Santos, C.; Sobolevska, K.; Abols, A.; Rodriguez, M.; Šantare, D.; Rudnickiha, A.; Lietuvietis, V.; et al. Detection of circulating miRNAs: Comparative analysis of extracellular vesicle-incorporated miRNAs and cell-free miRNAs in whole plasma of prostate cancer patients. BMC Cancer 2017, 17, 730. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Cechova, K.; Valis, M.; Kuca, K.; Zhang, B.; Hort, J. MicroRNAs in Alzheimer’s Disease: Diagnostic Markers or Therapeutic Agents? Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Gabr, M.T.; Brogi, S. MicroRNA-Based Multitarget Approach for Alzheimer’s Disease: Discovery of the First-In-Class Dual Inhibitor of Acetylcholinesterase and MicroRNA-15b Biogenesis. J. Med Chem. 2020, 63, 9695–9704. [Google Scholar] [CrossRef]
- Jahangard, Y.; Monfared, H.; Moradi, A.; Zare, M.; Mirnajafi-Zadeh, J.; Mowla, S.J. Therapeutic Effects of Transplanted Exosomes Containing miR-29b to a Rat Model of Alzheimer’s Disease. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Key Targets | Relevant Function | Related-miRNAs | Ref. Related miRNAs |
|---|---|---|---|
| BACE1 | TNF-α, ephrin-A2, and APP cleavage | miR-9, miR-107, miR-29a/b | [88,89,90,91,92] |
| SIRT1 | Acetylation of substrates related to learning and memory | miR-9, miR-34a/c, miR-181c, miR-132 | [93,94] |
| ADAM10 | Non-amyloidogenic APP processing | miR-23a, miR-34a, miR-107, miR-451 | [80,93] |
| GSK3B | Phosphorylation of key related targets including tau | miR-26a | [95] |
| MEF2D | Transcription factor involved in structural plasticity | miR-92 | [96] |
| CREB1 | Transcription factor involved in synaptic plasticity | miR-132, miR-134 | [63] |
| MECP2 | Transcription factor involved in synaptic plasticity | miR-132 | [63] |
| REST | Gene silencing transcription factor involved in synaptogenesis, synaptic plasticity, and structural remodeling | miR-9 | [69] |
| IGF1 | Growth factor involved in synapse maturation | miR-26b, miR-206 | [97] |
| BDNF | Neurotrophic factor involved in synaptic plasticity | miR-132, miR-206 | [98] |
| MME (NEP) | Neurite outgrowth | miR-26b | [99] |
| EFNA3 | (Ephrin-A3) Axon guidance | miR-210 | [82] |
| DCX | Axon assembly and branching | miR-29a | [100] |
| ARPP21 | Dendritic branching | miR-128 | [101] |
| p250GAP | Actin reorganization in dendritic spines. | miR-132 | [102] |
| LIMK1 | Actin cytoskeleton organization | miR-134 | [64,65] |
| PUM2 | Actin cytoskeleton organization | miR-134 | [66] |
| TMOD2 | Actin filaments organization | miR-191 | [68] |
| DPYSL2 | (CRMP-2) Axon guidance | miR-181c | [103] |
| SNAP25 | Vesicle trafficking | miR-210 | [84] |
| SV2A | Neurotransmitter release | miR-485 | [86] |
| SYN2 | Neurotransmitter release | miR-125b, miR-181 | [104] |
| VAMP2 | Neurotransmitter release | miR-34c | [105] |
| SYT1 | Vesicle trafficking/Neurotransmitter release | miR-137, miR-146a | [106] |
| ARPC3 | Negatively regulates synaptic scaling | miR-29a/b | [67] |
| MAP2 | Microtubules assembly | miR-26a | [50] |
| MAP1B | Microtubules stabilization | miR-9, miR-146a | [54,107] |
| DLG4 | (PSD-95) Scaffold protein | miR-125a | [108] |
| NLGN1 | AMPAR clustering, synaptic transmission | miR-146a | [106] |
| CAMK2A | Kinase involved in synaptic plasticity, neurotransmitter release and long-term potentiation | miR-181a | [61] |
| SAP97 | Scaffold protein | miR-9 | [70] |
| STIM2 | Negative regulator of NMDA-evoked intracellular Ca2+ | miR-128 | [81] |
| NPTX1 | AMPAR recruitment/clustering, synaptic transmission | miR-210 | [82] |
| GRIA1 | Synaptic transmission | miR-92, miR-137, miR-501, miR-34a | [71,72,73,109] |
| GRIA2 | Synaptic transmission | miR-181a | [110] |
| GRIN2A | Synaptic transmission | miR-125b | [77,78] |
| GRIN2B | Synaptic transmission | miR-34a | [80,109] |
| Source | Cohort | ↑↓ | AUC | Sen/Spec (%) | NDD Tested Specific for AD? | Other Biomarkers | Cognitive Test | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| # miR9 | CSF exosomes | HCC = 10 + 18 | CSF Aβ | [147] | |||||
| AD = 10 + 18 | ↑ | N/A | N/A | CSF tau | |||||
| Serum | HCC = 7 | MMSE | [148] | ||||||
| MCI = 7 | |||||||||
| AD = 7 | ↓ | N/A | N/A | ||||||
| # miR-23a | Serum | HCC = 15 + 30 | PD and VD/No | MMSE | [149] | ||||
| AD = 15 + 30 | ↑ | 0.71 | 57/83 | ||||||
| # miR-26a | Serum | HCC = 9 + 86 | MMSE | [150] | |||||
| AD = 19 + 121 | ↓ | 0.75 | 85/57 | ||||||
| # miR-26b | Serum | HCC = 44 | FTD/No | CSF Aβ | MMSE | [151] | |||
| AD = 48 | ↑ | 0.97 | 89/89 | CSF tau | |||||
| CSF | HCC = 18 | FTD/Yes | CSF Aβ | MMSE | [152] | ||||
| AD = 22 | 0.82 | N/A | CSF tau | ||||||
| Blood | HCC = 22 | MMSE | [153] | ||||||
| AD = 48 | ↑ | 0.81 | N/A | ||||||
| # miR-29a | Serum | HCC = 15 + 30 | MMSE | [149] | |||||
| AD = 15 + 30 | ↑ | 0.71 | 43/97 | ||||||
| Serum | HCC = 7 | MMSE | [148] | ||||||
| MCI = 7 | |||||||||
| AD = 7 | ↓ | N/A | N/A | ||||||
| CSF | HCC = 10 | CSF Aβ | MMSE | [154] | |||||
| AD = 10 | ↑ | N/A | N/A | CSF tau | |||||
| CSF | HCC = 20 | MMSE | [155] | ||||||
| AD = 18 | ↑ | N/A | N/A | ||||||
| miR-29b | Serum | HCC = 44 | FTD/Yes | CSF Aβ | MMSE | [151] | |||
| AD = 48 | ↑ | 0.83 | 93/- | CSF tau | |||||
| Serum | HCC = 7 | MMSE | [148] | ||||||
| MCI = 7 | |||||||||
| AD = 7 | ↓ | N/A | N/A | ||||||
| CSF | HCC = 10 | CSF Aβ | MMSE | [154] | |||||
| AD = 10 | ↑ | N/A | N/A | CSF tau | |||||
| # miR-34a | Plasma | HCC = 21 + 15 | PD/Yes | CSF Aβ | MMSEGDS | [156] | |||
| MCI = 21 + 15 | CSF tau | ||||||||
| AD = 21 + 15 | ↓ | 0.79 | 80/71 | ApoE 4 | |||||
| Plasma | HCC = 27 | MMSE | [94] | ||||||
| AD = 25 | ↑ | 0.81 | 84/74 | ||||||
| CSF and plasma | HCC = 10 | CSF Aβ | MMSE | [154] | |||||
| AD = 10 | ↓ | N/A | N/A | CSF tau | |||||
| miR-34c | Plasma | HCC = 27 | MMSE | [94] | |||||
| AD = 25 | ↑ | 0.99 | 92/96 | ||||||
| miR-92 | Serum | HCC = 44 | FTD/No | CSF Aβ | MMSE | [151] | |||
| AD = 48 | ↓ | 0.8 | N/A | CSF tau | |||||
| miR-107 | Plasma | HCC = 120 | PD/No | MMSE | [157] | ||||
| AD = 120 | ↓ | 0.74 | 77/59 | ||||||
| miR-125a | CSF | HCC = 44 | FTD/Yes | CSF Aβ | MMSE | [151] | |||
| AD = 48 | ↑ | 0–84 | 74/82 | CSF tau | |||||
| CSF | HCC = 20 | MMSE | [155] | ||||||
| AD = 18 | ↑ | N/A | N/A | ||||||
| # miR-125b | Serum | HCC = 155 | MMSE | [158] | |||||
| AD = 105 | ↑ | 0.85 | 80/68 | ||||||
| # Serum | HCC = 15 + 30 | MMSE | [149] | ||||||
| AD = 15 + 30 | ↑ | 0.71 | 63/76 | ||||||
| CSF | HCC = 18 | FTD/Yes | CSF Aβ | MMSE | [152] | ||||
| AD = 22 | 0.82 | N/A | CSF tau | ||||||
| CSF | HCC = 10 | CSF Aβ | MMSE | [154] | |||||
| AD = 10 | ↓ | N/A | N/A | CSF tau | |||||
| miR-128 | Plasma | HCC = 50 | MMSE | [159] | |||||
| MCI = 50 | ↑ | 0.97 | 84/96 | ||||||
| # miR-132 | Plasma | HCC = 50 | MMSE | [159] | |||||
| MCI = 50 | ↑ | 0.97 | 88/98 | ||||||
| Plasma | HCC = 31 | CSF Aβ CSF tau | MMSE | [160] | |||||
| AD-MCI = 16 | |||||||||
| AD = 16 | ↓ | 0.77 | N/A | ||||||
| Serum | HCC = 44 | CSF Aβ | MMSE | [151] | |||||
| AD = 48 | ↓ | 0.79 | N/A | CSF tau | |||||
| Serum | HCC = 76 | [161] | |||||||
| MCI = 66 | ↓ | 0.91 | 70/100 | ||||||
| miR-134 | Plasma | HCC = 50 | MMSE | [159] | |||||
| MCI = 50 | ↑ | 0.92 | 86/82 | ||||||
| miR-137 | Serum | HCC = 7 MCI = 7 AD = 7 | ↓ | N/A | N/A | MMSE | [148] | ||
| miR146a | Serum | HCC = 44 | FTD/No | CSF Aβ | MMSE | [151] | |||
| AD = 48 | ↓ | 0.87 | N/A | CSF tau | |||||
| Blood | sMCI = 25 | CSF Aβ CSF tau ApoE MRI | MMSE GDS | [162] | |||||
| pMCI = 19 | ↑ | N/A | N/A | ||||||
| CSF and plasma | HCC = 10 | CSF Aβ | MMSE | [154] | |||||
| AD = 10 | ↓ | N/A | N/A | CSF tau | |||||
| miR-181a | Blood | sMCI = 24 | CSF Aβ CSF tau ApoE MRI | MMSE GDS | [162] | ||||
| pMCI = 17 | ↑ | N/A | N/A | ||||||
| # miR-181c | Serum | HCC = 9 + 86 | MMSE | [150] | |||||
| AD = 19 + 121 | ↓ | 0.78 | 72/73 | ||||||
| Serum | HCC = 155 | MMSE | [158] | ||||||
| AD = 105 | ↑ | 0.74 | 75/64 | ||||||
| Plasma | HCC = 14 + 24 | ↑ | FTD/Yes | MMSE GDS | [163] | ||||
| MCI = 26 | 0.84 | 85/86 | |||||||
| AD = 56 | 0.77 | 70/86 | |||||||
| Serum | HCC = 7 | MMSE | [148] | ||||||
| MCI = 7 | |||||||||
| AD = 7 | ↓ | N/A | N/A | ||||||
| # miR-191 | Plasma | HCC = 20 + 17 | [164] | ||||||
| MCI = 9 | |||||||||
| AD = 11 + 20 | 0.95 | 95/76 | |||||||
| # miR-206 | Serum | HCC = 76 | ↓ | [161] | |||||
| MCI = 66 | 0.91 | 70/100 | |||||||
| Plasma | HCC = 31 MCI = 30 AD = 25 | ↑ | N/A | N/A | MMSE | [165] | |||
| # miR-210 | Plasma | HCC = 14 + 24 | ↑ | FTD/Yes | MMSE GDS | [163] | |||
| MCI = 26 | 0.74 | 77/71 | |||||||
| AD = 56 | 0.80 | 81/71 | |||||||
| miR-451 | Plasma Evs | HCC = 15 | DLB/Yes | ApoE | MMSE | [166] | |||
| AD = 10 | ↓ | 0.95 | |||||||
| miR-485-3p | Serum | HCC = 62 | MMSE | [167] | |||||
| AD = 89 | ↑ | 0.93 | 84/97 | ||||||
| miR-501 | Serum | HCC = 22 | ApoE | MMSE | [168] | ||||
| AD = 36 | ↓ | 0.82 | 53/100 |
| Source | Cohort | AUC | Sen/Spec (%) | NDD Tested/Specific for AD? | Other Biomarkers | Cognitive Test | Ref. | |
|---|---|---|---|---|---|---|---|---|
| miR-9/miR-874 miR-329/miR-181a miR-99/let-7e | Plasma | HC = 50 AD = 50 | 0.96 | 88/96 | PD-FTD/No | CSF Aβ CSF tau | MMSE | [170] |
| miR-23a, miR-29a, miR-125b, miR-22 | Serum | HCC = 30 AD = 30 | 0.84 | 80/72 | MMSE | [149] | ||
| miR-26a-5p, miR-107, miR-26b-5p, miR-112, miR-161, let-7d-3p, miR-5010-3p, miR-1285-5p, miR-151a-3p, miR-103a-3p, miR-532-5p, let-7f-5p. | Blood | HCC = 22 + 21 MCI = 18 AD = 94 | 0.84 0.93 | 81/88 95/95 | PD-MS/Yes | MMSE | [171] | |
| miR-206, miR-132 | Serum | HCC = 76 MCI = 66 | 0.98 | 85/98 | [161] | |||
| miR-30a-5p, miR-34c, miR-27a-3p, piR_019949, piR_020364 | CSF exosomes | HCC = 38 + 44 MCI = 17 AD = 23 + 19 | 0.83 | CSF Aβ CSF tau | MMSE | [172] | ||
| miR26b, miR125b | CSF | HCC = 18 AD = 22 | 0.80 | CSF Aβ CSF tau | MMSE | [152] | ||
| miR-191, miR-15b | Plasma | HCC = 20 + 17 MCI = 9 AD = 11 + 20 | 0.96 | 95/82 | MMSE | [164] | ||
| miR-92a-3p, miR-181c-5p, miR-210-3p | Plasma | HCC = 14 + 24 MCI = 26 AD = 56 | 0.90 0.85 | 85/86 93/71 | FTD/Yes | MMSE GDS | [163] | |
| miR-26a-5p, miR-181c-3p, miR-126-5p, miR-22-3p, miR-148b-5p, miR-106b3p, miR-6119-5p, miR-1246 and miR-660-5p | Serum | HCC = 9 + 86 AD = 19 + 121 | 0.99 | 93/99 | MMSE CDR | [150] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siedlecki-Wullich, D.; Miñano-Molina, A.J.; Rodríguez-Álvarez, J. microRNAs as Early Biomarkers of Alzheimer’s Disease: A Synaptic Perspective. Cells 2021, 10, 113. https://doi.org/10.3390/cells10010113
Siedlecki-Wullich D, Miñano-Molina AJ, Rodríguez-Álvarez J. microRNAs as Early Biomarkers of Alzheimer’s Disease: A Synaptic Perspective. Cells. 2021; 10(1):113. https://doi.org/10.3390/cells10010113
Chicago/Turabian StyleSiedlecki-Wullich, Dolores, Alfredo J. Miñano-Molina, and José Rodríguez-Álvarez. 2021. "microRNAs as Early Biomarkers of Alzheimer’s Disease: A Synaptic Perspective" Cells 10, no. 1: 113. https://doi.org/10.3390/cells10010113
APA StyleSiedlecki-Wullich, D., Miñano-Molina, A. J., & Rodríguez-Álvarez, J. (2021). microRNAs as Early Biomarkers of Alzheimer’s Disease: A Synaptic Perspective. Cells, 10(1), 113. https://doi.org/10.3390/cells10010113