mRNA-Enhanced Cell Therapy and Cardiovascular Regeneration



Abstract
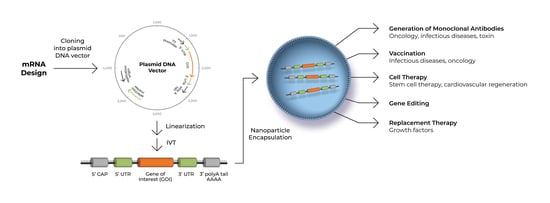
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Employing mRNA to Generate iPSCs for Stem Cell Therapy
3. Employing mRNA to Directly Generate or Enhance Therapeutic Cells
4. Inflammatory Signaling in Nuclear Reprogramming and Transdifferentiation
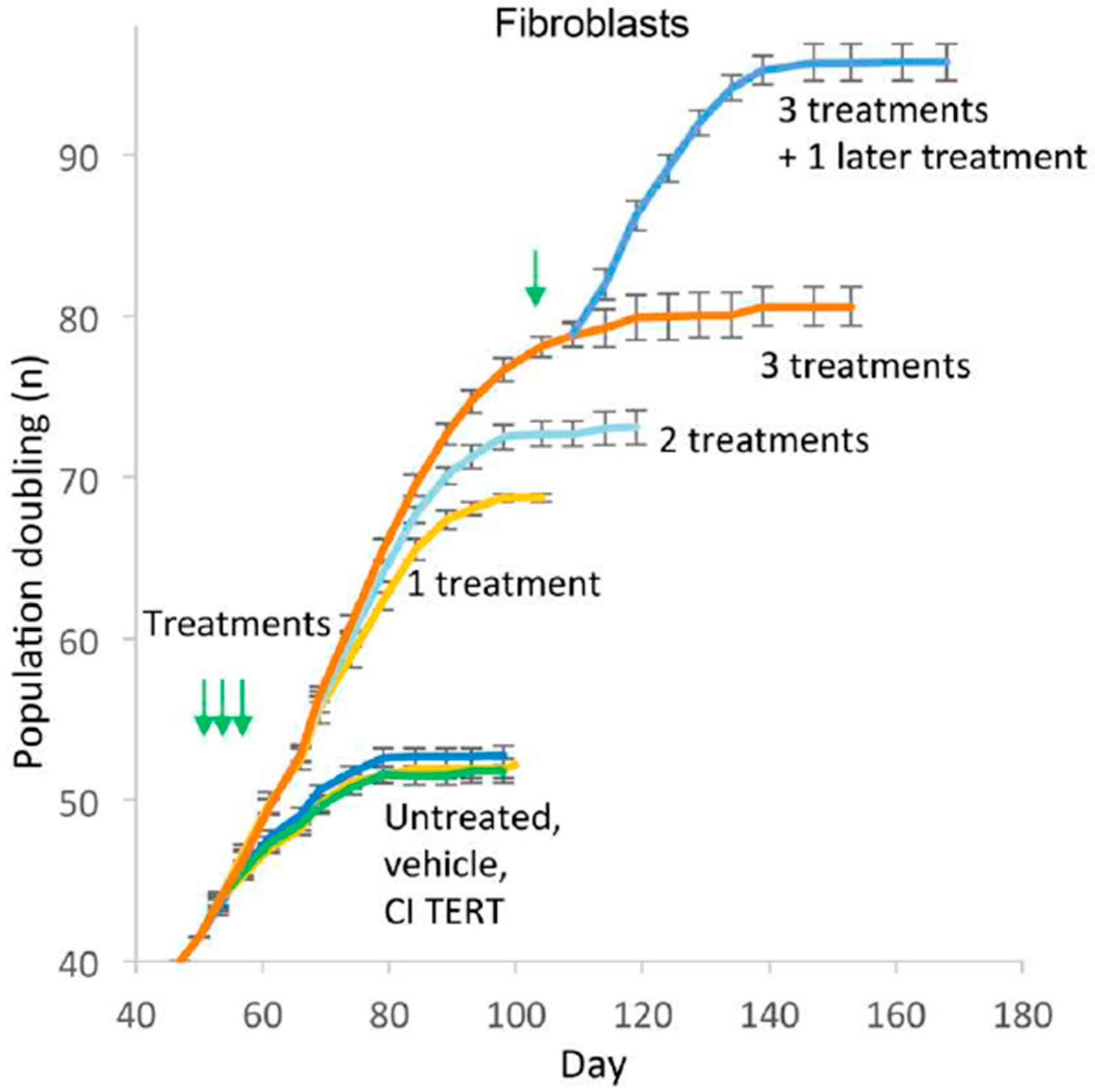
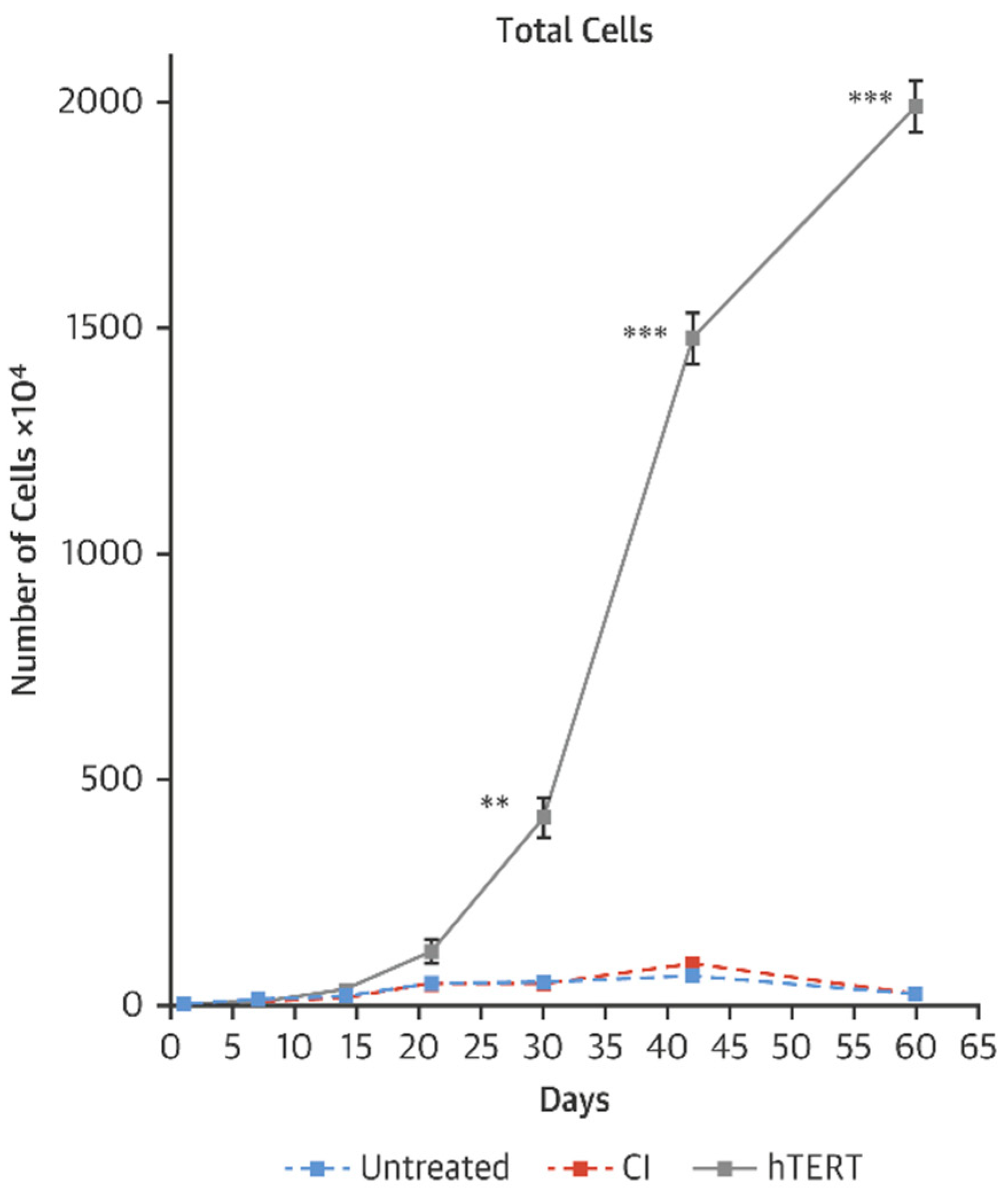
5. Employing mRNA to Reverse Cardiovascular Aging
Novel Therapeutic for Age-Related Diseases
6. Employing mRNA for Cardiovascular Regeneration
7. Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- COVID-19 Treatment and Vaccine Tracker. Available online: https://covid-19tracker.milkeninstitute.org/#vaccine_RNA-based-vaccine (accessed on 30 November 2020).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.; Wang, W.; Song, Z.; Hu, Y.; Tao, Z.; Tian, J.; Pei, Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. mRNA Vaccine against SARS-CoV-2–Preliminary report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 2020, 586, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Ezkurdia, I.; Juan, D.; Rodriguez, J.M.; Frankish, A.; Diekhans, M.; Harrow, J.; Vazquez, J.; Valencia, A.; Tress, M.L. Multiple evidence strands suggest that there may be as few as 19 000 human protein-coding genes. Hum. Mol. Genet. 2014, 23, 5866–5878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanghvi, Y.S. A status update of modified oligonucleotides for chemotherapeutics applications. Curr. Protoc. Nucleic Acid. Chem. 2011, 46, 4.1.1–4.1.22. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Sumaria, C.S.; Pradeepkumar, P.I. Exploring chemical modifications for siRNA therapeutics: A structural and functional outlook. Chem. Med. Chem. 2010, 5, 328–349. [Google Scholar] [CrossRef]
- Zou, S.; Scarfo, K.; Nantz, M.H.; Hecker, J.G. Lipid-mediated delivery of RNA is more efficient than delivery of DNA in non-dividing cells. Int. J. Pharm. 2010, 389, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Jirikowski, G.F.; Sanna, P.P.; Maciejewski-Lenoir, D.; Bloom, F.E. Reversal of diabetes insipidus in Brattleboro rats: Intrahypothalamic injection of vasopressin mRNA. Science 1992, 255, 996–998. [Google Scholar] [CrossRef]
- Shin, H.; Salzano, G.; Torchilin, V.P. Recent advances in RNA therapeutics and RNA delivery systems based on nanoparticles. Advan. Ther. 2018, 1, 1800065. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar] [PubMed]
- Kariko, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Presnyak, V.; Alhusaini, N.; Chen, Y.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, M.; Choi, J.; Cottrell, K.A.; Lavagnino, Z.; Thomas, E.N.; Pavlovic-Djuranovic, S.; Szczesny, P.; Piston, D.W.; Zaher, H.S.; Puglisi, J.D.; et al. A short translational ramp determines the efficiency of protein synthesis. Nat. Comm. 2019, 10, 5774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, C.L.; DeBenedette, M.A.; Tcherepanova, I.Y.; Gamble, A.; Lewis, W.E.; Cope, A.B.; Kuruc, J.D.; McGee, K.S.; Kearney, M.F.; Coffin, J.M.; et al. Immunogenicity of AGS-004 dendritic cell therapy in patients treated during acute HIV infection. AIDS Res. Hum. Retrovir. 2018, 34, 111–122. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Trans. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Zhou, L.Y.; Qin, Z.; Zhu, Y.; He, Z.; Xu, T. Current RNA-based therapeutics in clinical trials. Curr. Gene Ther. 2019, 19, 172–196. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Filareto, A.; Parker, S.; Darabi, R.; Borges, L.; Iacovino, M.; Schaaf, T.; Mayerhofer, T.; Chamberlain, J.S.; Ervasti, J.M.; McIvor, R.S.; et al. An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat. Commun. 2013, 4, 1549. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.S.; Tang, C.; Rao, M.S.; Weissman, I.L.; Wu, J.C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 2013, 19, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, C.; Moon, J.; Chung, Y.; Chang, M.; Han, B.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell. Stem. Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Jia, F.; Wilson, K.D.; Sun, N.; Gupta, D.M.; Huang, M.; Li, Z.; Panetta, N.J.; Chen, Z.Y.; Robbins, R.C.; Kay, M.A.; et al. A nonviral minicircle vector for deriving human iPS cells. Nat. Methods 2010, 7, 197–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Chau, K.F.; Vodyanik, M.A.; Jiang, J.; Jiang, Y. Efficient feeder-free episomal reprogramming with small molecules. PLoS ONE 2011, 6, e17557. [Google Scholar] [CrossRef]
- Yakubov, E.; Rechavi, G.; Rozenblatt, S.; Givol, D. Reprogramming of human fibroblasts to pluripotent stem cells using mRNA of four transcription factors. Biochem. Biophys. Res. Comm. 2010, 394, 189–193. [Google Scholar] [CrossRef]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steichen, C.; Luce, E.; Maluenda, J.; Tosca, L.; Moreno-Gimeno, I.; Desterke, C.; Dianat, N.; Goulinet-Mainot, S.; Awan-Toor, S.; Burks, D.; et al. Messenger RNA–versus retrovirus-based induced pluripotent stem cell reprogramming strategies: Analysis of genomic integrity. Stem Cells Transl. Med. 2014, 3, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Rossi, D.J. Reprogramming human fibroblasts to pluripotency using modified mRNA. Nat. Protoc. 2013, 8, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P.; Tomishima, M.J.; Chambers, S.M.; Mica, Y.; Reed, E.; Menon, J.; Tabar, V.; Mo, Q.; Studer, L.; Sadelain, M. Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 12759–12764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Verma, V.; Nandihalli, M.; Ramachandra, C.J.A.; Sequiera, G.L.; Sudibyo, Y.; Chung, Y.; Sun, W.; Shim, W. A Systemic evaluation of cardiac differentiation from mRNA reprogrammed human induced pluripotent stem cells. PLoS ONE 2014, 9, e103485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Lin, R.; Hong, X.; Ng, A.H.; Lee, C.N.; Neumeyer, J.; Wang, G.; Wang, X.; Ma, M.; Pu, W.T.; et al. Robust differentiation of human pluripotent stem cells into endothelial cells via temporal modulation of ETV2 with modified mRNA. BioRxiv 2020, 6. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.T.; Cooke, J.P. Therapeutic transdifferentiation of human fibroblasts into endothelial cells using forced expression of lineage-specific transcription factors. J. Tissue Eng. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Mueller, P.; Wolfien, M.; Ekat, K.; Lang, C.I.; Koczan, D.; Wolkenhauer, O.; Hahn, O.; Peters, K.; Lang, H.; David, R.; et al. RNA-based strategies for cardiac reprogramming of human mesenchymal stromal cells. Cells 2020, 9, 504. [Google Scholar] [CrossRef] [Green Version]
- Lui, K.O.; Zangi, L.; Silva, E.A.; Bu, L.; Sahara, M.; Li, R.A.; Mooney, D.J.; Chien, K.R. Driving vascular endothelial cell fate of human multipotent Isl1+ heart progenitors with VEGF modified mRNA. Cell Res. 2013, 23, 1172–1186. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Suryaprakash, S.; Lao, Y.; Leong, K.W. Engineering mesenchymal stem cells for regenerative medicine and drug delivery. Methods 2015, 84, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Bolli, R.; Hare, J.M.; March, K.L.; Pepine, C.J.; Willerson, J.T.; Perin, E.C.; Yang, P.C.; Henry, T.D.; Traverse, J.H.; Mitrani, R.D.; et al. Rationale and design of the Concert-HF trial (combination of mesenchymal and c-kit(+) cardiac stem cells as regenerative therapy for heart failure). Circ. Res. 2018, 122, 1703–1715. [Google Scholar] [CrossRef]
- Wang, S.K.; Green, L.A.; Drucker, N.A.; Motaganahalli, R.L.; Fajardo, A.; Murphy, M.P. Rationale and design of the Clinical and Histologic Analysis of Mesenchymal Stromal Cells in AmPutations (CHAMP) trial investigating the therapeutic mechanism of mesenchymal stromal cells in the treatment of critical limb ischemia. J. Vasc. Surg. 2018, 68, 176–181.e1. [Google Scholar] [CrossRef] [PubMed]
- Levy, O.; Zhao, W.; Mortensen, L.J.; Leblanc, S.; Tsang, K.; Fu, M.; Phillips, J.A.; Sagar, V.; Anandakumaran, P.; Ngai, J.; et al. mRNA-engineered mesenchymal stem cells for targeted delivery of interleukin-10 to sites of inflammation. Blood 2013, 122, e23–e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, W.; Pham, V.; Liu, L.; Riazifar, M.; Pone, E.J.; Zhang, S.X.; Ma, F.; Lu, M.; Walsh, C.M.; Zhao, W. Mesenchymal stem cells engineered to express selectin ligands and IL-10 exert enhanced therapeutic efficacy in murine experimental autoimmune encephalomyelitis. Biomaterials 2016, 77, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowakowski, A.; Andrzejewska, A.; Boltze, J.; Nitzsche, F.; Cui, L.; Jolkkonen, J.; Walczak, P.; Lukomska, B.; Janowski, M. Translation, but not transfection limits clinically relevant, exogenous mRNA based induction of alpha-4 integrin expression on human mesenchymal stem cells. Sci. Rep. 2017, 7, 1103. [Google Scholar] [CrossRef]
- Ryser, M.F.; Ugarte, F.; Thieme, S.; Bornhäuser, M.; Roesen-Wolff, A.; Brenner, S. mRNA transfection of CXCR4-GFP fusion—Simply generated by PCR-results in efficient migration of primary human mesenchymal stem cells. Tissue Eng. Part C Methods 2008, 14, 179–184. [Google Scholar] [CrossRef]
- Wiehe, J.M.; Kaya, Z.; Homann, J.M.; Wöhrle, J.; Vogt, K.; Nguyen, T.; Rottbauer, W.; Torzewski, J.; Fekete, N.; Rojewski, M.; et al. GMP-adapted overexpression of CXCR4 in human mesenchymal stem cells for cardiac repair. Int. J. Cardiol. 2013, 167, 2073–2081. [Google Scholar] [CrossRef]
- Lee, J.; Sayed, N.; Hunter, A.; Au, K.F.; Wong, W.H.; Mocarski, E.S.; Pera, R.R.; Yakubov, E.; Cooke, J.P. Activation of innate immunity is required for efficient nuclear reprogramming. Cell 2012, 151, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Zhou, G.; Gu, Q.; Chanda, P.K.; Ospino, F.; Cooke, J.P. Transdifferentiation requires iNOS activation: Role of RING1A S-nitrosylation. Circ. Res. 2016, 119, e129–e138. [Google Scholar] [CrossRef] [Green Version]
- Chanda, P.K.; Meng, S.; Lee, J.; Leung, H.E.; Chen, K.; Cooke, J.P. Nuclear S-nitrosylation defines an optimal zone for inducing pluripotency. Circulation 2019, 140, 1081–1099. [Google Scholar] [CrossRef]
- Zhou, G.; Meng, S.; Li, Y.; Ghebre, Y.T.; Cooke, J.P. Optimal ROS signaling is critical for nuclear reprogramming. Cell. Rep. 2016, 15, 919–925. [Google Scholar] [CrossRef] [Green Version]
- Lai, L.; Reineke, E.; Hamilton, D.J.; D, M.; Cooke, J.P. Glycolytic switch is required for transdifferentiation to endothelial lineage. Circulation 2019, 139, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.J.; Jamieson, C.H.; Weissman, I.L. Stems cells and the pathways to aging and cancer. Cell 2008, 132, 681–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sfeir, A.; de Lange, T. Removal of shelterin reveals the telomere end-protection problem. Science 2012, 336, 593–597. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.; Harley, C.B. Telomere length and replicative aging in human vascular tissues. Proc. Natl. Acad. Sci. USA 1995, 92, 11190–11194. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, H.; Chang, E.; Glassford, A.J.; Cooke, J.P.; Chiu, C.P.; Tsao, P.S. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ. Res. 2001, 89, 793–798. [Google Scholar] [CrossRef]
- Ramunas, J.; Yakubov, E.; Brady, J.J.; Corbel, S.Y.; Holbrook, C.; Brandt, M.; Stein, J.; Santiago, J.G.; Cooke, J.P.; Bla, H.M. Transient delivery of modified mRNA encoding TERT rapidly extends telomeres in human cells. FASEB J. 2015, 29, 1930–1939. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhou, G.; Bruno, I.G.; Cooke, J.P. Telomerase mRNA reverses senescence in progeria cells. J. Am. Coll. Cardiol. 2017, 70, 804–805. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, G.; Bruno, I.G.; Zhang, N.; Sho, S.; Tedone, E.; Lai, T.; Cooke, J.P.; Shay, J.W. Transient introduction of human telomerase mRNA improves hallmarks of progeria cells. Aging Cell. 2019, 18, e12979. [Google Scholar] [CrossRef]
- Artandi, S.E.; DePinho, R.A. Mice without telomerase: What can they teach us about human cancer? Nat. Med. 2000, 6, 852–855. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Senescence and immortalization: Role of telomeres and telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2115. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K. RNA therapy: Current status and future potential. Chonnam. Med. J. 2020, 56, 87–93. [Google Scholar] [CrossRef]
- Connolly, B.; Isaacs, C.; Cheng, L.; Asrani, K.H.; Subramanian, R.R. SERPINA1 mRNA as a treatment for alpha-1 antitrypsin deficiency. J. Nucleic Acids 2018, 2018, 8247935. [Google Scholar] [CrossRef]
- DeRosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved efficacy in a fabry disease model. Using a systemic mRNA liver depot system as compared to enzyme replacement therapy. Mol. Ther. 2019, 27, 878–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.; Cooke, J.P. The limitless future of RNA therapeutics. Under review. 2020. [Google Scholar]
- BIONTECH. Available online: https://biontech.de/science/pipeline (accessed on 17 November 2020).
- BIONTECH. Available online: https://www.curevac.com/en/pipeline (accessed on 17 November 2020).
- Moderna. Available online: https://www.modernatx.com/pipeline (accessed on 17 November 2020).
- Zangi, L.; Lui, K.O.; von Gise, A.; Ma, Q.; Ebina, W.; Ptaszek, L.M.; Später, D.; Xu, H.; Tabebordbar, M.; Gorbatov, R.; et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 2013, 31, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, L.; Clarke, J.C.; Yen, C.; Gregoire, F.; Albery, T.; Billger, M.; Egnell, A.; Gan, L.; Jennbacken, K.; Johansson, E.; et al. Biocompatible, purified VEGF-A mRNA improves cardiac function after intracardiac injection 1 week post-myocardial infarction in swine. Mol. Ther. Methods Clin. Dev. 2018, 9, 330–346. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.fiercebiotech.com/biotech/moderna-raises-500m-to-move-mrna-drugs-deeper-into-human-tests (accessed on 17 November 2020).
- Anttila, V.; Saraste, A.; Knuuti, J.; Jaakkola, P.; Hedman, M.; Svedlund, S.; Lagerström-Fermér, M.; Kjaer, M.; Jeppsson, A.; Gan, L. Synthetic mRNA encoding VEGF-A in patients undergoing coronary artery bypass grafting: Design of a phase 2a clinical trial. Mol. Ther. Methods Clin. Dev. 2020, 18, 464–472. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT03370887 (accessed on 17 November 2020).
- Gan, L.M.; Lagerström-Fermér, M.; Carlsson, L.G.; Arfvidsson, C.; Egnell, A.; Rudvik, A.; Kjaer, M.; Collén, A.; Thompson, J.D.; Joyal, J.; et al. Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 2019, 10, 871. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Ning, B.; Hansson, K.M.; Bruce, A.C.; Seaman, S.A.; Zhang, C.; Rikard, M.; DeRosa, C.A.; Fraser, C.L.; Wågberg, M.; et al. Modified VEGF-A mRNA induces sustained multifaceted microvascular response and accelerates diabetic wound healing. Sci. Rep. 2018, 8, 17509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pehrsson, S.; Hölttä, M.; Linhardt, G.; Danielson, R.F.; Carlsson, L. Rapid production of human VEGF-A following intradermal injection of modified VEGF-A mRNA demonstrated by cutaneous microdialysis in the rabbit and pig in vivo. Biomed. Res. Int. 2019, 2019, 3915851. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ma, Q.; King, J.S.; Sun, Y.; Xu, B.; Zhang, X.; Zohrabian, S.; Guo, H.; Cai, W.; Li, G.; et al. aYAP modRNA reduces cardiac inflammation and hypertrophy in a murine ischemia-reperfusion model. Life Sci. Alliance 2020, 3, e201900424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.L.; Leblond, A.; Turner, E.C.; Kumar, A.H.; Martin, K.; Whelan, D.; O’Sullivan, D.M.; Caplice, N.M. Synthetic chemically modified mrna-based delivery of cytoprotective factor promotes early cardiomyocyte survival post-acute myocardial infarction. Mol. Pharm. 2015, 12, 991–996. [Google Scholar] [CrossRef]
- Zangi, L.; Oliveira, M.S.; Ye, L.Y.; Ma, Q.; Sultana, N.; Hadas, Y.; Chepurko, E.; Später, D.; Zhou, B.; Chew, W.L.; et al. Insulin-like growth factor 1 receptor-dependent pathway drives epicardial adipose tissue formation after myocardial injury. Circulation 2017, 135, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Magadum, A.; Singh, N.; Kurian, A.A.; Sharkar, M.T.K.; Chepurko, E.; Zangi, L. Ablation of a single N-glycosylation site in human FSTL 1 induces cardiomyocyte proliferation and cardiac regeneration. Mol. Ther. Nucleic Acids 2018, 13, 133–143. [Google Scholar] [CrossRef]
- Ji, R.; Akashi, H.; Drosatos, K.; Liao, X.; Jiang, H.; Kennel, P.J.; Brunjes, D.L.; Castillero, E.; Zhang, X.; Deng, L.Y.; et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight 2017, 2, e82922. [Google Scholar] [CrossRef] [Green Version]
- Reforgiato, M.R.; Milano, G.; Fabriàs, G.; Casas, J.; Gasco, P.; Paroni, R.; Samaja, M.; Ghidoni, R.; Caretti, A.; Signorelli, P. Inhibition of ceramide de novo synthesis as a postischemic strategy to reduce myocardial reperfusion injury. Basic Res. Cardiol. 2016, 111, 12. [Google Scholar] [CrossRef]
- Klevstig, M.; Ståhlman, M.; Lundqvist, A.; Täng, M.S.; Fogelstrand, P.; Adiels, M.; Andersson, L.; Kolesnick, R.; Jeppsson, A.; Borén, J.; et al. Targeting acid sphingomyelinase reduces cardiac ceramide accumulation in the post-ischemic heart. J. Mol. Cell. Cardiol. 2016, 93, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Hadas, Y.; Vincek, A.S.; Youssef, E.; Żak, M.M.; Chepurko, E.; Sultana, N.; Sharkar, M.T.K.; Guo, N.; Komargodski, R.; Kurian, A.A.; et al. Altering sphingolipid metabolism attenuates cell death and inflammatory response after myocardial infarction. Circulation 2020, 141, 916–930. [Google Scholar] [CrossRef]
- Magadum, A.; Singh, N.; Kurian, A.A.; Munir, I.; Mehmood, T.; Brown, K.; Sharkar, M.T.K.; Chepurko, E.; Sassi, Y.; Oh, J.G.; et al. Pkm2 regulates cardiomyocyte cell cycle and promotes cardiac regeneration. Circulation 2020, 141, 1249–1265. [Google Scholar] [CrossRef] [PubMed]
- Hamma, T.; Ferre-D’Amare, A.R. Structure of protein L7Ae bound to a K-turn derived from an archaeal box H/ACA sRNA at 1.8 A resolution. Structure 2004, 12, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Wroblewska, L.; Kitada, T.; Endo, K.; Siciliano, V.; Stillo, B.; Saito, H.; Weiss, R. Mammalian synthetic circuits with RNA binding proteins for RNA-only delivery. Nat. Biotechnol. 2015, 33, 839–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar] [CrossRef]
- Williams, A.H.; Liu, N.; van Rooij, E.; Olson, E.N. MicroRNA control of muscle development and disease. Curr. Opin. Cell. Biol. 2009, 24, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Magadum, A.; Kaur, K.; Zangi, L. mRNA-Based Protein Replacement Therapy for the Heart. Mol. Ther. 2019, 27, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.; Zangi, L. Modified mRNA as a therapeutic tool for the heart. Cardiovasc. Drugs Ther. 2020, 34, 871–880. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chanda, P.K.; Sukhovershin, R.; Cooke, J.P. mRNA-Enhanced Cell Therapy and Cardiovascular Regeneration. Cells 2021, 10, 187. https://doi.org/10.3390/cells10010187
Chanda PK, Sukhovershin R, Cooke JP. mRNA-Enhanced Cell Therapy and Cardiovascular Regeneration. Cells. 2021; 10(1):187. https://doi.org/10.3390/cells10010187
Chicago/Turabian StyleChanda, Palas K., Roman Sukhovershin, and John P. Cooke. 2021. "mRNA-Enhanced Cell Therapy and Cardiovascular Regeneration" Cells 10, no. 1: 187. https://doi.org/10.3390/cells10010187
APA StyleChanda, P. K., Sukhovershin, R., & Cooke, J. P. (2021). mRNA-Enhanced Cell Therapy and Cardiovascular Regeneration. Cells, 10(1), 187. https://doi.org/10.3390/cells10010187