
With or without You: Co-Chaperones Mediate Health and Disease by Modifying Chaperone Function and Protein Triage


Abstract
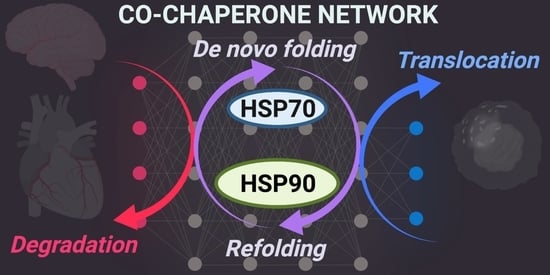
:
1. Introduction
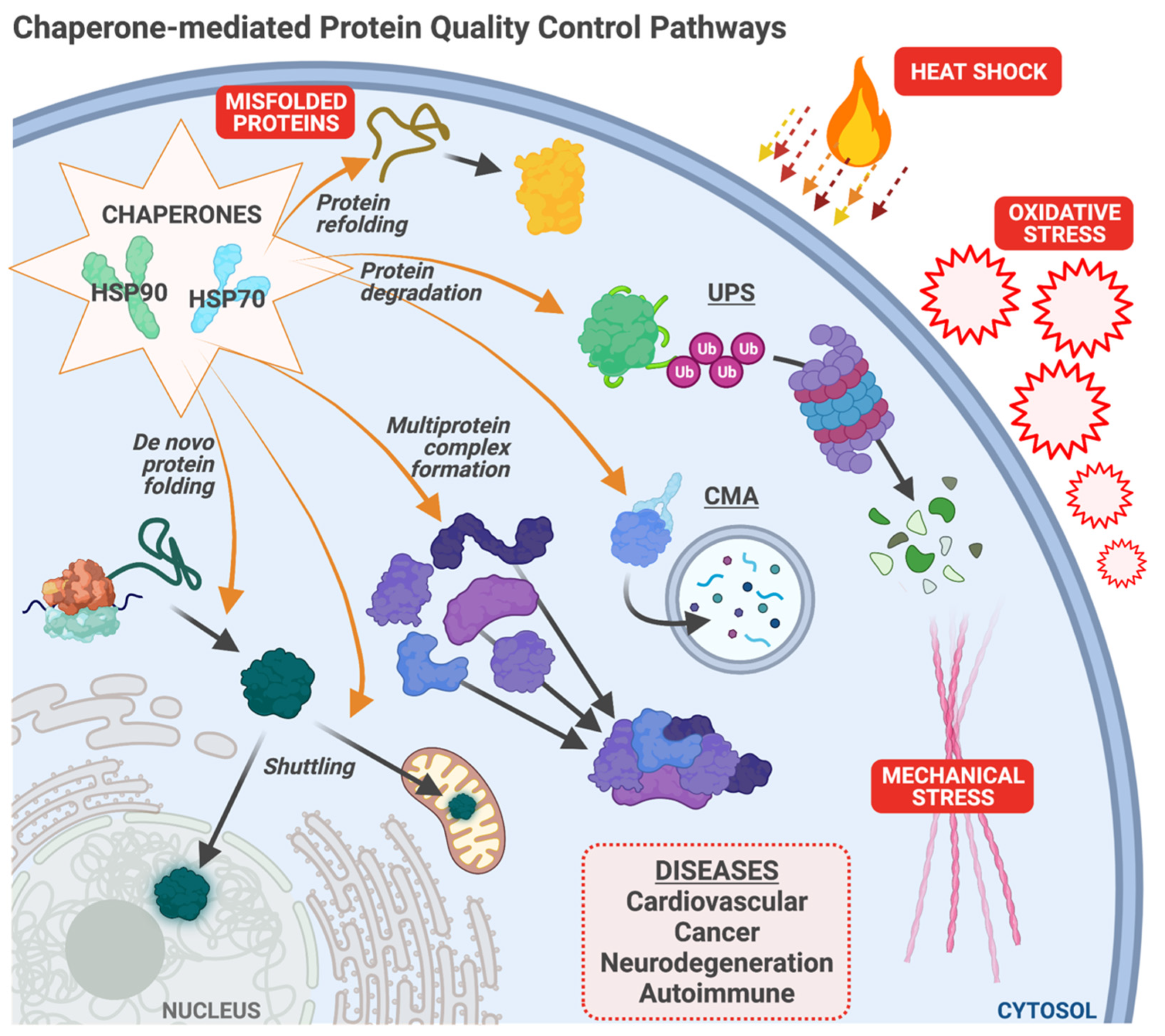
1.1. Protein Quality Control
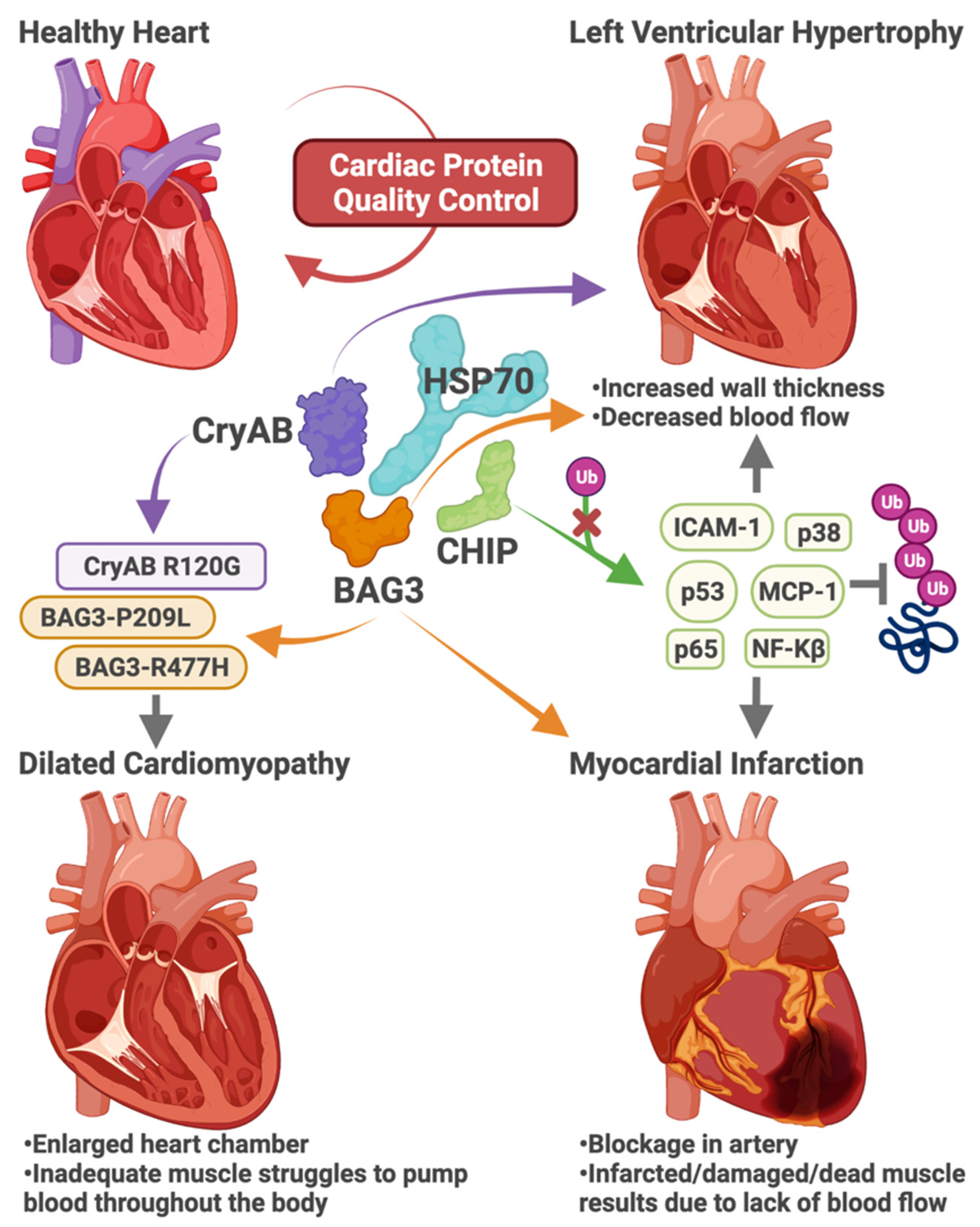
1.2. Cardiac Stress
1.3. Neurodegeneration
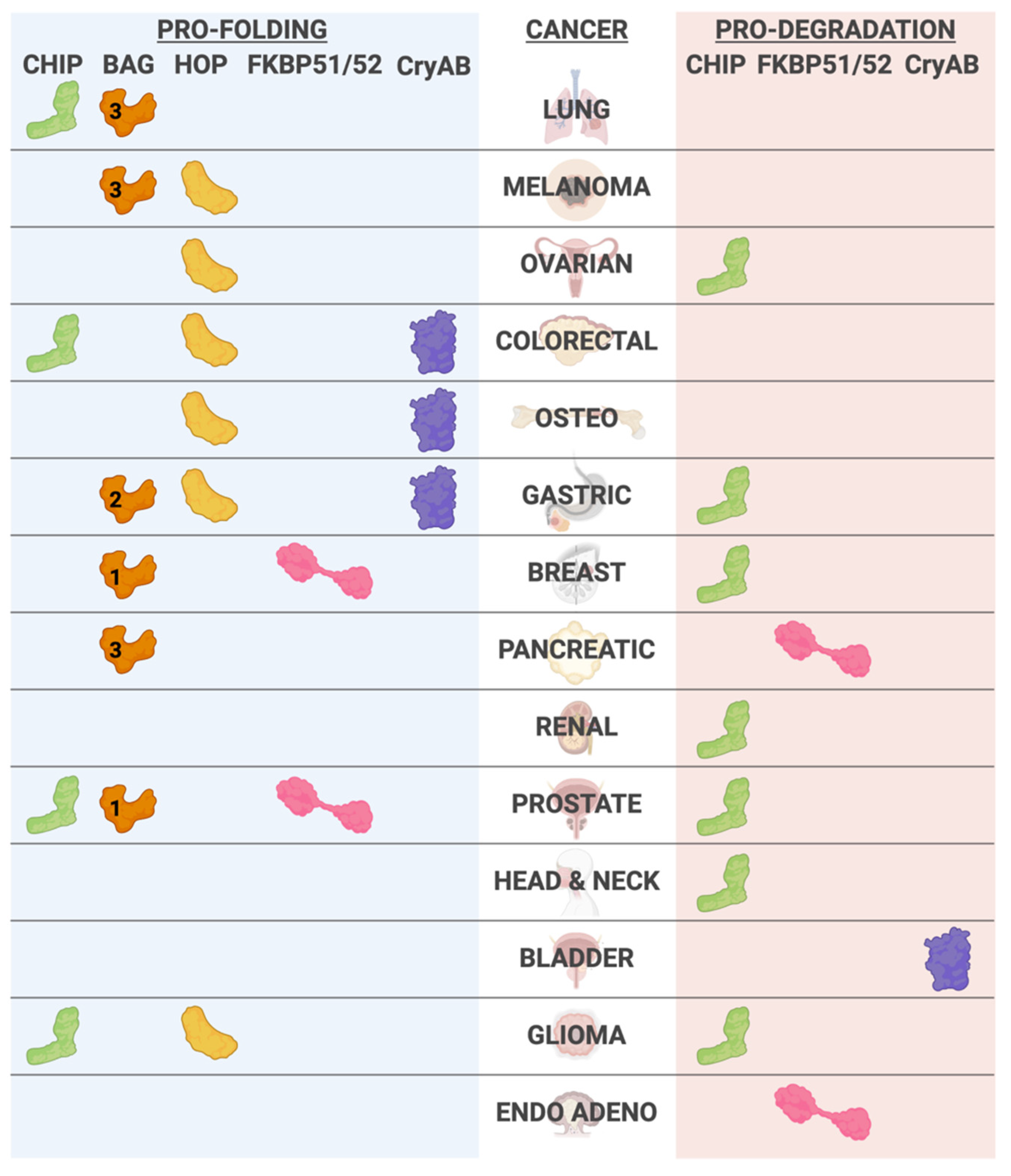
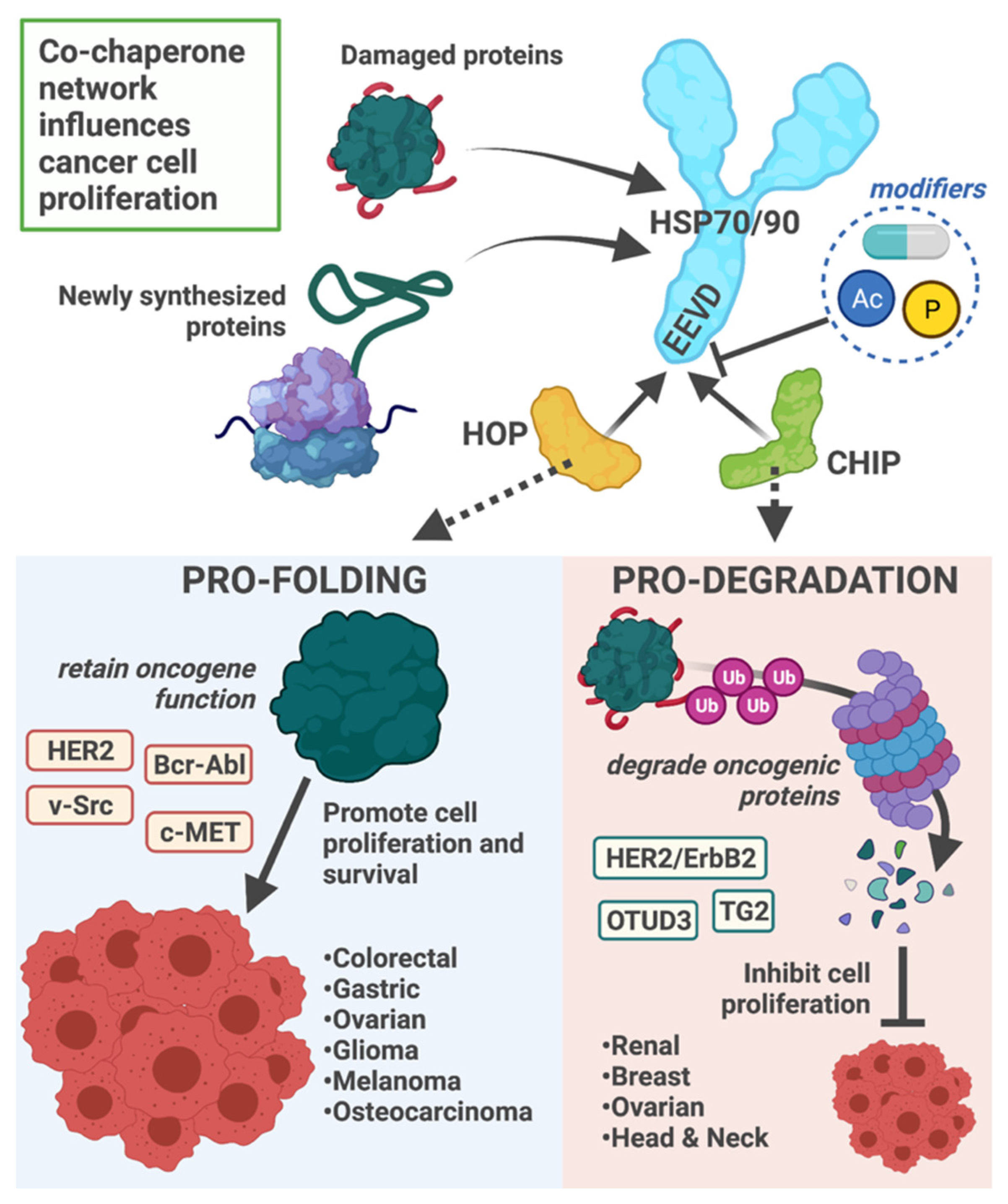
1.4. Cancer
1.5. Autoimmune Disorders
2. CHIP
2.1. Function, Expression, and Regulation
2.2. Cardioprotection
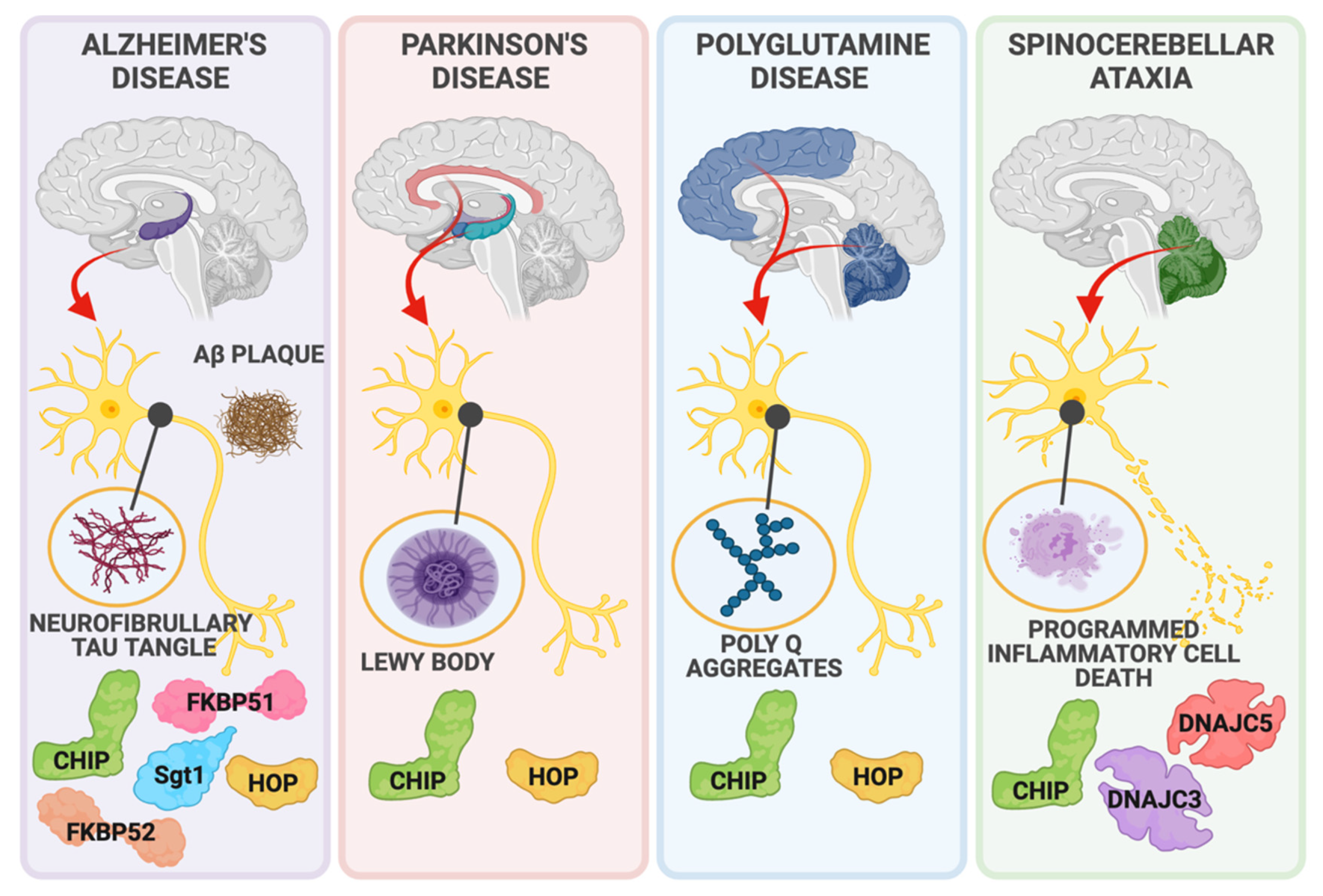
2.3. Neurodegenerative Diseases
2.3.1. Parkinson’s Disease
2.3.2. Alzheimer’s Disease
2.3.3. Spinocerebellar Ataxias
2.3.4. Polyglutamine Diseases
2.4. Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | CHIP’s Role | Target | Reference |
|---|---|---|---|
| Breast Cancer | TS | HER2/ ErbB2 | [131] |
| Ovarian cancer | TS | OTUD3 | [129,133] |
| Renal cancer | TS | TG2 | [134] |
| Head and Neck cancer | TS | unknown | [124] |
| Lung cancer | OG | p21 | [137] |
| Colorectal Cancer | OG | MAPK and AKT | [139] |
| Prostate cancer | TS | unknown | [128] |
| OG | Akt | [138] | |
| Glioblastoma/Glioma | TS | EGFR | [122,130] |
| OG | unknown | [140] |
2.5. Autoimmune Diseases
3. BAG Family Proteins
3.1. Heart Disease
3.2. Cancer
4. HOP/Stress-Inducible Phosphoprotein 1
4.1. Neurodegeneration
4.1.1. Alzheimer’s Disease
4.1.2. Parkinson’s Disease
4.1.3. Huntington’s Disease and Amyotrophic Lateral Sclerosis (ALS)
4.2. Cancer
5. FKBP51 & FKBP52
5.1. Alzheimer’s Disease
5.2. Cancer
5.3. Therapeutics
6. CryAB
6.1. Cardioprotection
6.2. Cancer
6.3. Multiple Sclerosis
7. Sgt1
Neurodegenerative Diseases
8. HSP40/DNAJ Protein Family
8.1. DNAJC3
8.2. DNAJC5
8.3. Rheumatoid Arthritis
9. Targeting Co-Chaperones with Small Molecules for Therapies
9.1. CHIP
9.2. BAG1
9.3. BAG2
9.4. BAG3
9.5. CryAB
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jackson, S.E. Hsp90: Structure and Function. In Molecular Chaperones; Jackson, S., Ed.; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2013; pp. 155–240. ISBN 978-3-642-34552-4. [Google Scholar]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 Chaperone Network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.K.; Pratt, W.B.; Lieberman, A.P.; Osawa, Y. Targeting Hsp70 Facilitated Protein Quality Control for Treatment of Polyglutamine Diseases. Cell. Mol. Life Sci. CMLS 2020, 77, 977–996. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Kim, H.J.; Lim, J.H.; Lee, S.H. Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy. Cells 2020, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Ranek, M.J.; Stachowski, M.J.; Kirk, J.A.; Willis, M.S. The Role of Heat Shock Proteins and Co-Chaperones in Heart Failure. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, M.B.; Winter, C.B.; Locke-Winter, C.R.; Banerjee, A.; Harken, A.H. Cardiac Preconditioning Does Not Require Myocardial Stunning. Ann. Thorac. Surg. 1993, 55, 395–400. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Lam, T.; Davogustto, G. Cardiac Metabolism in Perspective. Compr. Physiol. 2016, 6, 1675–1699. [Google Scholar] [CrossRef]
- Latchman, D.S. Heat Shock Proteins and Cardiac Protection. Cardiovasc. Res. 2001, 51, 637–646. [Google Scholar] [CrossRef] [Green Version]
- Pattison, J.S.; Robbins, J. Protein Misfolding and Cardiac Disease: Establishing Cause and Effect. Autophagy 2008, 4, 821–823. [Google Scholar] [CrossRef] [Green Version]
- Zhazykbayeva, S.; Pabel, S.; Mügge, A.; Sossalla, S.; Hamdani, N. The Molecular Mechanisms Associated with the Physiological Responses to Inflammation and Oxidative Stress in Cardiovascular Diseases. Biophys. Rev. 2020, 12, 947. [Google Scholar] [CrossRef]
- Delogu, G.; Signore, M.; Mechelli, A.; Famularo, G. Heat Shock Proteins and Their Role in Heart Injury. Curr. Opin. Crit. Care 2002, 8, 411–416. [Google Scholar] [CrossRef]
- Gong, R.; Li, X.-Y.; Chen, H.-J.; Xu, C.-C.; Fang, H.-Y.; Xiang, J.; Wu, Y.-Q. Role of Heat Shock Protein 22 in the Protective Effect of Geranylgeranylacetone in Response to Oxidized-LDL. Drug Des. Dev. Ther. 2019, 13, 2619–2632. [Google Scholar] [CrossRef] [Green Version]
- Hashikawa, N.; Ido, M.; Morita, Y.; Hashikawa-Hobara, N. Effects from the Induction of Heat Shock Proteins in a Murine Model Due to Progression of Aortic Atherosclerosis. Sci. Rep. 2021, 11, 7025. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; van Marion, D.M.S.; Zhang, D.; Brundel, B.J.J.M. Heat Shock Protein Inducer GGA*-59 Reverses Contractile and Structural Remodeling via Restoration of the Microtubule Network in Experimental Atrial Fibrillation. J. Mol. Cell. Cardiol. 2019, 134, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Jan, R.-L.; Yang, S.-C.; Liu, Y.-C.; Yang, R.-C.; Tsai, S.-P.; Huang, S.-E.; Yeh, J.-L.; Hsu, J.-H. Extracellular Heat Shock Protein HSC70 Protects against Lipopolysaccharide-Induced Hypertrophic Responses in Rat Cardiomyocytes. Biomed. Pharmacother. 2020, 128, 110370. [Google Scholar] [CrossRef]
- Zhou, C.; Bai, J.; Jiang, C.; Ye, L.; Pan, Y.; Zhang, H. Geranylgeranylacetone Attenuates Myocardium Ischemic/Reperfusion Injury through HSP70 and Akt/GSK-3β/ENOS Pathway. Am. J. Transl. Res. 2017, 9, 386–395. [Google Scholar] [PubMed]
- Lim, J.; Yue, Z. Neuronal Aggregates: Formation, Clearance, and Spreading. Dev. Cell 2015, 32, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.F.; Gan, Z.Y.; Komander, D.; Dewson, G. Ubiquitin Signalling in Neurodegeneration: Mechanisms and Therapeutic Opportunities. Cell Death Differ. 2021, 28, 570–590. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendor, J.; Logan, T.; Edwards, R.H. The Function of α-Synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Bunting, E.L.; Hamilton, J.; Tabrizi, S.J. Polyglutamine Diseases. Curr. Opin. Neurobiol. 2021, 72, 39–47. [Google Scholar] [CrossRef]
- Fan, H.-C.; Ho, L.-I.; Chi, C.-S.; Chen, S.-J.; Peng, G.-S.; Chan, T.-M.; Lin, S.-Z.; Harn, H.-J. Polyglutamine (PolyQ) Diseases: Genetics to Treatments. Cell Transplant. 2014, 23, 441–458. [Google Scholar] [CrossRef] [Green Version]
- Reis, S.D.; Pinho, B.R.; Oliveira, J.M.A. Modulation of Molecular Chaperones in Huntington’s Disease and Other Polyglutamine Disorders. Mol. Neurobiol. 2017, 54, 5829–5854. [Google Scholar] [CrossRef] [Green Version]
- Akbar, U.; Ashizawa, T. Ataxia. Neurol. Clin. 2015, 33, 225–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulson, H.L.; Shakkottai, V.G.; Brent Clark, H.; Orr, H.T. Polyglutamine Spinocerebellar Ataxias—From Genes to Potential Treatments. Nat. Rev. Neurosci. 2017, 18, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Albakova, Z.; Armeev, G.A.; Kanevskiy, L.M.; Kovalenko, E.I.; Sapozhnikov, A.M. HSP70 Multi-Functionality in Cancer. Cells 2020, 9, 587. [Google Scholar] [CrossRef] [Green Version]
- Boysen, M.; Kityk, R.; Mayer, M.P. Hsp70- and Hsp90-Mediated Regulation of the Conformation of P53 DNA Binding Domain and P53 Cancer Variants. Mol. Cell 2019, 74, 831–843.e4. [Google Scholar] [CrossRef]
- Dahiya, V.; Agam, G.; Lawatscheck, J.; Rutz, D.A.; Lamb, D.C.; Buchner, J. Coordinated Conformational Processing of the Tumor Suppressor Protein P53 by the Hsp70 and Hsp90 Chaperone Machineries. Mol. Cell 2019, 74, 816–830.e7. [Google Scholar] [CrossRef] [PubMed]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jäättelä, M.; Penninger, J.M.; Garrido, C.; et al. Heat-Shock Protein 70 Antagonizes Apoptosis-Inducing Factor. Nat. Cell Biol. 2001, 3, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Han, C.; Huang, H.; Xin, Y.; Xu, Y.; Luo, L.; Yin, Z. Heat Shock Protein 70 Together with Its Co-Chaperone CHIP Inhibits TNF-α Induced Apoptosis by Promoting Proteasomal Degradation of Apoptosis Signal-Regulating Kinase1. Apoptosis 2010, 15, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stokes, J.; Singh, U.P.; Scissum Gunn, K.; Acharya, A.; Manne, U.; Mishra, M. Targeting Hsp70: A Possible Therapy for Cancer. Cancer Lett. 2016, 374, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Xiao, H.; Cao, L. Recent Advances in Heat Shock Proteins in Cancer Diagnosis, Prognosis, Metabolism and Treatment. Biomed. Pharmacother. 2021, 142, 112074. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Wang, T.; Guzman, M.L.; Peter, R.I.; Chiosis, G. Chaperome Networks—Redundancy and Implications for Cancer Treatment. Adv. Exp. Med. Biol. 2020, 1243, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Xu, H.; Chen, Z.; Xiong, F.; Zhang, B.; Chen, K.; Jiang, H.; Luo, C.; Zhang, H. 17-AAG Synergizes with Belinostat to Exhibit a Negative Effect on the Proliferation and Invasion of MDA-MB-231 Breast Cancer Cells. Oncol. Rep. 2020, 43, 1928–1944. [Google Scholar] [CrossRef]
- Mohammadian, M.; Zeynali, S.; Azarbaijani, A.F.; Khadem Ansari, M.H.; Kheradmand, F. Cytotoxic Effects of the Newly-Developed Chemotherapeutic Agents 17-AAG in Combination with Oxaliplatin and Capecitabine in Colorectal Cancer Cell Lines. Res. Pharm. Sci. 2017, 12, 517–525. [Google Scholar] [CrossRef]
- Pontes, F.S.C.; Pontes, H.A.R.; de Souza, L.L.; de Jesus, A.S.; Joaquim, A.M.C.; Miyahara, L.A.N.; Fonseca, F.P.; Pinto Junior, D.S. Effect of 17-Allylamino-17-Demethoxygeldanamycin (17-AAG) on Akt Protein Expression Is More Effective in Head and Neck Cancer Cell Lineages That Retain PTEN Protein Expression. J. Oral Pathol. Med. 2018, 47, 253–259. [Google Scholar] [CrossRef]
- Park, H.-K.; Yoon, N.G.; Lee, J.-E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.-H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the Full Potential of Hsp90 Inhibitors as Cancer Therapeutics through Simultaneous Inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, P.T.; Subramanian, C.; Zhu, Q.; Zhang, H.; Zhao, H.; Gallagher, R.; Timmermann, B.N.; Blagg, B.S.J.; Cohen, M.S. Novel HSP90 Inhibitors Effectively Target Functions of Thyroid Cancer Stem Cell Preventing Migration and Invasion. Surgery 2016, 159, 142–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Q.; Ning, J.-Y.; Dai, X.; Gao, Y.-D.; Su, L.; Zhao, B.-X.; Miao, J.-Y. Discovery of Novel HSP90 Inhibitors That Induced Apoptosis and Impaired Autophagic Flux in A549 Lung Cancer Cells. Eur. J. Med. Chem. 2018, 145, 551–558. [Google Scholar] [CrossRef]
- Kryeziu, K.; Bruun, J.; Guren, T.K.; Sveen, A.; Lothe, R.A. Combination Therapies with HSP90 Inhibitors against Colorectal Cancer. Biochim. Biophys. Acta BBA-Rev. Cancer 2019, 1871, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Edkins, A.L.; Price, J.T.; Pockley, A.G.; Blatch, G.L. Heat Shock Proteins as Modulators and Therapeutic Targets of Chronic Disease: An Integrated Perspective. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160521. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Bonam, S.R.; Schall, N.; Kuhn, L.; Hammann, P.; Chaloin, O.; Madinier, J.-B.; Briand, J.-P.; Page, N.; Muller, S. Blocking Nuclear Export of HSPA8 after Heat Shock Stress Severely Alters Cell Survival. Sci. Rep. 2018, 8, 16820. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Neckers, L. Hsp90 in Cancer: Transcriptional Roles in the Nucleus. Adv. Cancer Res. 2016, 129, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, J.E.; Shen, N.; Zeng, A.; Korm, S.; Feng, H. Failure to Guard: Mitochondrial Protein Quality Control in Cancer. Int. J. Mol. Sci. 2021, 22, 8306. [Google Scholar] [CrossRef]
- Galigniana, M.D.; Echeverría, P.C.; Erlejman, A.G.; Piwien-Pilipuk, G. Role of Molecular Chaperones and TPR-Domain Proteins in the Cytoplasmic Transport of Steroid Receptors and Their Passage through the Nuclear Pore. Nucl. Austin Tex. 2010, 1, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Gvozdenov, Z.; Kolhe, J.; Freeman, B.C. The Nuclear and DNA-Associated Molecular Chaperone Network. Cold Spring Harb. Perspect. Biol. 2019, 11, a034009. [Google Scholar] [CrossRef] [Green Version]
- Havalová, H.; Ondrovičová, G.; Keresztesová, B.; Bauer, J.A.; Pevala, V.; Kutejová, E.; Kunová, N. Mitochondrial HSP70 Chaperone System-The Influence of Post-Translational Modifications and Involvement in Human Diseases. Int. J. Mol. Sci. 2021, 22, 8077. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xiao, J.; Wang, Y.; Song, X.; Huang, L.; Ren, Z.; Kitazato, K.; Wang, Y. Posttranslational Modification and beyond: Interplay between Histone Deacetylase 6 and Heat-Shock Protein 90. Mol. Med. Camb. Mass 2021, 27, 110. [Google Scholar] [CrossRef]
- Mazaira, G.I.; Piwien Pilipuk, G.; Galigniana, M.D. Corticosteroid Receptors as a Model for the Hsp90•immunophilin-Based Transport Machinery. Trends Endocrinol. Metab. TEM 2021, 32, 827–838. [Google Scholar] [CrossRef]
- Smedlund, K.B.; Sanchez, E.R.; Hinds, T.D. FKBP51 and the Molecular Chaperoning of Metabolism. Trends Endocrinol. Metab. TEM 2021, 32, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zou, M.-H. Role of the Mitochondrial Protein Import Machinery and Protein Processing in Heart Disease. Front. Cardiovasc. Med. 2021, 8, 749756. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, C.A.; Connell, P.; Wu, Y.; Hu, Z.; Thompson, L.J.; Yin, L.Y.; Patterson, C. Identification of CHIP, a Novel Tetratricopeptide Repeat-Containing Protein That Interacts with Heat Shock Proteins and Negatively Regulates Chaperone Functions. Mol. Cell. Biol. 1999, 19, 4535–4545. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The Co-Chaperone Carboxyl Terminus of Hsp70-Interacting Protein (CHIP) Mediates Alpha-Synuclein Degradation Decisions between Proteasomal and Lysosomal Pathways. J. Biol. Chem. 2005, 280, 23727–23734. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Windheim, M.; Roe, S.M.; Peggie, M.; Cohen, P.; Prodromou, C.; Pearl, L.H. Chaperoned Ubiquitylation--Crystal Structures of the CHIP U Box E3 Ubiquitin Ligase and a CHIP-Ubc13-Uev1a Complex. Mol. Cell 2005, 20, 525–538. [Google Scholar] [CrossRef]
- Jiang, J.; Ballinger, C.A.; Wu, Y.; Dai, Q.; Cyr, D.M.; Höhfeld, J.; Patterson, C. CHIP Is a U-Box-Dependent E3 Ubiquitin Ligase: Identification of Hsc70 as a Target for Ubiquitylation. J. Biol. Chem. 2001, 276, 42938–42944. [Google Scholar] [CrossRef] [Green Version]
- Murata, S.; Minami, Y.; Minami, M.; Chiba, T.; Tanaka, K. CHIP Is a Chaperone-Dependent E3 Ligase That Ubiquitylates Unfolded Protein. EMBO Rep. 2001, 2, 1133–1138. [Google Scholar] [CrossRef]
- Stankiewicz, M.; Nikolay, R.; Rybin, V.; Mayer, M.P. CHIP Participates in Protein Triage Decisions by Preferentially Ubiquitinating Hsp70-Bound Substrates. FEBS J. 2010, 277, 3353–3367. [Google Scholar] [CrossRef] [PubMed]
- Kundrat, L.; Regan, L. Balance between Folding and Degradation for Hsp90-Dependent Client Proteins: A Key Role for CHIP. Biochemistry 2010, 49, 7428–7438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.; Ruckova, E.; Halada, P.; Coates, P.J.; Hrstka, R.; Lane, D.P.; Vojtesek, B. C-Terminal Phosphorylation of Hsp70 and Hsp90 Regulates Alternate Binding to Co-Chaperones CHIP and HOP to Determine Cellular Protein Folding/Degradation Balances. Oncogene 2013, 32, 3101–3110. [Google Scholar] [CrossRef]
- Kim Chiaw, P.; Hantouche, C.; Wong, M.J.H.; Matthes, E.; Robert, R.; Hanrahan, J.W.; Shrier, A.; Young, J.C. Hsp70 and DNAJA2 Limit CFTR Levels through Degradation. PLoS ONE 2019, 14, e0220984. [Google Scholar] [CrossRef] [Green Version]
- Pastorek, M.; Muller, P.; Coates, P.J.; Vojtesek, B. Intrinsic Proteotoxic Stress Levels Vary and Act as a Predictive Marker for Sensitivity of Cancer Cells to Hsp90 Inhibition. PLoS ONE 2018, 13, e0202758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.H.; Park, J.-H.; Lee, E.J.; Vo, T.T.L.; Choi, H.; Kim, J.Y.; Jang, J.K.; Wee, H.-J.; Lee, H.S.; Jang, S.H.; et al. ARD1-Mediated Hsp70 Acetylation Balances Stress-Induced Protein Refolding and Degradation. Nat. Commun. 2016, 7, 12882. [Google Scholar] [CrossRef] [Green Version]
- Finka, A.; Mattoo, R.U.H.; Goloubinoff, P. Experimental Milestones in the Discovery of Molecular Chaperones as Polypeptide Unfolding Enzymes. Annu. Rev. Biochem. 2016, 85, 715–742. [Google Scholar] [CrossRef] [Green Version]
- Zierer, B.K.; Rübbelke, M.; Tippel, F.; Madl, T.; Schopf, F.H.; Rutz, D.A.; Richter, K.; Sattler, M.; Buchner, J. Importance of Cycle Timing for the Function of the Molecular Chaperone Hsp90. Nat. Struct. Mol. Biol. 2016, 23, 1020–1028. [Google Scholar] [CrossRef]
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Höhfeld, J.; Patterson, C. The Co-Chaperone CHIP Regulates Protein Triage Decisions Mediated by Heat-Shock Proteins. Nat. Cell Biol. 2001, 3, 93–96. [Google Scholar] [CrossRef]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 Co-Chaperone CHIP Targets Immature CFTR for Proteasomal Degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [CrossRef]
- McDonough, H.; Patterson, C. CHIP: A Link between the Chaperone and Proteasome Systems. Cell Stress Chaperones 2003, 8, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Kundrat, L.; Regan, L. Identification of Residues on Hsp70 and Hsp90 Ubiquitinated by the Cochaperone CHIP. J. Mol. Biol. 2010, 395, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Qian, S.-B.; McDonough, H.; Boellmann, F.; Cyr, D.M.; Patterson, C. CHIP-Mediated Stress Recovery by Sequential Ubiquitination of Substrates and Hsp70. Nature 2006, 440, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Ranek, M.J.; Oeing, C.; Sanchez-Hodge, R.; Kokkonen-Simon, K.M.; Dillard, D.; Aslam, M.I.; Rainer, P.P.; Mishra, S.; Dunkerly-Eyring, B.; Holewinski, R.J.; et al. CHIP Phosphorylation by Protein Kinase G Enhances Protein Quality Control and Attenuates Cardiac Ischemic Injury. Nat. Commun. 2020, 11, 5237. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Han, S.Y.; Seong, D.; Han, H.-J.; Song, J. Multifaceted C-Terminus of HSP70-Interacting Protein Regulates Tumorigenesis via Protein Quality Control. Arch. Pharm. Res. 2019, 42, 63–75. [Google Scholar] [CrossRef]
- Zhan, S.; Wang, T.; Ge, W. Multiple Functions of the E3 Ubiquitin Ligase CHIP in Immunity. Int. Rev. Immunol. 2017, 36, 300–312. [Google Scholar] [CrossRef]
- Zhang, S.; Hu, Z.-W.; Mao, C.-Y.; Shi, C.-H.; Xu, Y.-M. CHIP as a Therapeutic Target for Neurological Diseases. Cell Death Dis. 2020, 11, 727. [Google Scholar] [CrossRef] [PubMed]
- Scaglione, K.M.; Zavodszky, E.; Todi, S.V.; Patury, S.; Xu, P.; Rodríguez-Lebrón, E.; Fischer, S.; Konen, J.; Djarmati, A.; Peng, J.; et al. Ube2w and Ataxin-3 Coordinately Regulate the Ubiquitin Ligase CHIP. Mol. Cell 2011, 43, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Liu, S.; Cai, W.; Wen, J.; Fu, Y.; Peng, J.; Zheng, Z. The Carboxyl Terminus of Heat Shock Protein 70-Interacting Protein (CHIP) Participates in High Glucose-Induced Cardiac Injury. Free Radic. Biol. Med. 2017, 106, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, Z.; He, X.-R.; Michael, L.H.; Patterson, C. CHIP, a Cochaperone/Ubiquitin Ligase That Regulates Protein Quality Control, Is Required for Maximal Cardioprotection after Myocardial Infarction in Mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2836–H2842. [Google Scholar] [CrossRef] [Green Version]
- Schisler, J.C.; Rubel, C.E.; Zhang, C.; Lockyer, P.; Cyr, D.M.; Patterson, C. CHIP Protects against Cardiac Pressure Overload through Regulation of AMPK. J. Clin. Investig. 2013, 123, 3588–3599. [Google Scholar] [CrossRef] [Green Version]
- Rosser, M.F.N.; Washburn, E.; Muchowski, P.J.; Patterson, C.; Cyr, D.M. Chaperone Functions of the E3 Ubiquitin Ligase CHIP. J. Biol. Chem. 2007, 282, 22267–22277. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Shin, S.; Seo, J.; Lee, E.-W.; Jeong, M.; Lee, M.-S.; Han, H.-J.; Song, J. C-Terminus of HSC70-Interacting Protein (CHIP) Inhibits Adipocyte Differentiation via Ubiquitin- and Proteasome-Mediated Degradation of PPARγ. Sci. Rep. 2017, 7, 40023. [Google Scholar] [CrossRef]
- Zheng, Z.-G.; Cheng, H.-M.; Zhou, Y.-P.; Zhu, S.-T.; Thu, P.M.; Li, H.-J.; Li, P.; Xu, X. Dual Targeting of SREBP2 and ERRα by Carnosic Acid Suppresses RANKL-Mediated Osteoclastogenesis and Prevents Ovariectomy-Induced Bone Loss. Cell Death Differ. 2020, 27, 2048–2065. [Google Scholar] [CrossRef]
- Sumanasekera, W.K.; Tien, E.S.; Davis, J.W.; Turpey, R.; Perdew, G.H.; Vanden Heuvel, J.P. Heat Shock Protein-90 (Hsp90) Acts as a Repressor of Peroxisome Proliferator-Activated Receptor-Alpha (PPARalpha) and PPARbeta Activity. Biochemistry 2003, 42, 10726–10735. [Google Scholar] [CrossRef]
- Ravi, S.; Parry, T.L.; Willis, M.S.; Lockyer, P.; Patterson, C.; Bain, J.R.; Stevens, R.D.; Ilkayeva, O.R.; Newgard, C.B.; Schisler, J.C. Adverse Effects of Fenofibrate in Mice Deficient in the Protein Quality Control Regulator, CHIP. J. Cardiovasc. Dev. Dis. 2018, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.-W.; Zhang, T.-P.; Wang, H.-X.; Yang, H.; Li, H.-H. CHIP Enhances Angiogenesis and Restores Cardiac Function after Infarction in Transgenic Mice. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2013, 31, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhang, T.-P.; Tian, C.; Jia, L.-X.; Du, J.; Li, H.-H. Carboxyl Terminus of Heat Shock Protein 70-Interacting Protein Inhibits Angiotensin II-Induced Cardiac Remodeling. Am. J. Hypertens. 2012, 25, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Kim, S.Y.; Tong, D.; Ferdous, A.; Piristine, H.; Dasgupta, S.; Wang, X.; French, K.M.; Villalobos, E.; et al. Xbp1s-FoxO1 Axis Governs Lipid Accumulation and Contractile Performance in Heart Failure with Preserved Ejection Fraction. Nat. Commun. 2021, 12, 1684. [Google Scholar] [CrossRef] [PubMed]
- Kitakata, H.; Endo, J.; Hashimoto, S.; Mizuno, E.; Moriyama, H.; Shirakawa, K.; Goto, S.; Katsumata, Y.; Fukuda, K.; Sano, M. Imeglimin Prevents Heart Failure with Preserved Ejection Fraction by Recovering the Impaired Unfolded Protein Response in Mice Subjected to Cardiometabolic Stress. Biochem. Biophys. Res. Commun. 2021, 572, 185–190. [Google Scholar] [CrossRef]
- Ding, X.; Goldberg, M.S. Regulation of LRRK2 Stability by the E3 Ubiquitin Ligase CHIP. PLoS ONE 2009, 4, e5949. [Google Scholar] [CrossRef]
- Ko, H.S.; Bailey, R.; Smith, W.W.; Liu, Z.; Shin, J.-H.; Lee, Y.-I.; Zhang, Y.-J.; Jiang, H.; Ross, C.A.; Moore, D.J.; et al. CHIP Regulates Leucine-Rich Repeat Kinase-2 Ubiquitination, Degradation, and Toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 2897–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, Y.; Soda, M.; Hatakeyama, S.; Akagi, T.; Hashikawa, T.; Nakayama, K.I.; Takahashi, R. CHIP Is Associated with Parkin, a Gene Responsible for Familial Parkinson’s Disease, and Enhances Its Ubiquitin Ligase Activity. Mol. Cell 2002, 10, 55–67. [Google Scholar] [CrossRef]
- Yoo, L.; Chung, K.C. The Ubiquitin E3 Ligase CHIP Promotes Proteasomal Degradation of the Serine/Threonine Protein Kinase PINK1 during Staurosporine-Induced Cell Death. J. Biol. Chem. 2018, 293, 1286–1297. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Dickey, C.A.; Yue, M.; Lin, W.-L.; Dickson, D.W.; Dunmore, J.H.; Lee, W.C.; Zehr, C.; West, G.; Cao, S.; Clark, A.M.K.; et al. Deletion of the Ubiquitin Ligase CHIP Leads to the Accumulation, But Not the Aggregation, of Both Endogenous Phospho- and Caspase-3-Cleaved Tau Species. J. Neurosci. 2006, 26, 6985–6996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The High-Affinity HSP90-CHIP Complex Recognizes and Selectively Degrades Phosphorylated Tau Client Proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Saidi, L.-J.; Polydoro, M.; Kay, K.R.; Sanchez, L.; Mandelkow, E.-M.; Hyman, B.T.; Spires-Jones, T.L. Carboxy Terminus Heat Shock Protein 70 Interacting Protein Reduces Tau-Associated Degenerative Changes. J. Alzheimers Dis. 2015, 44, 937–947. [Google Scholar] [CrossRef] [Green Version]
- Shimura, H.; Schwartz, D.; Gygi, S.P.; Kosik, K.S. CHIP-Hsc70 Complex Ubiquitinates Phosphorylated Tau and Enhances Cell Survival. J. Biol. Chem. 2004, 279, 4869–4876. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau Degradation: The Ubiquitin-Proteasome System versus the Autophagy-Lysosome System. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-Driven 26S Proteasome Impairment and Cognitive Dysfunction Can Be Prevented Early in Disease by Activating CAMP-PKA Signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar] [CrossRef]
- Tai, H.-C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated with Dysfunction of the Ubiquitin-Proteasome System. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Dolan, P.J.; Johnson, G.V.W. A Caspase Cleaved Form of Tau Is Preferentially Degraded through the Autophagy Pathway. J. Biol. Chem. 2010, 285, 21978–21987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grune, T.; Botzen, D.; Engels, M.; Voss, P.; Kaiser, B.; Jung, T.; Grimm, S.; Ermak, G.; Davies, K.J.A. Tau Protein Degradation Is Catalyzed by the ATP/Ubiquitin-Independent 20S Proteasome under Normal Cell Conditions. Arch. Biochem. Biophys. 2010, 500, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, C.; Gendron, T.F.; Scheffel, K.; Carlomagno, Y.; Dunmore, J.; DeTure, M.; Petrucelli, L. Loss of HDAC6, a Novel CHIP Substrate, Alleviates Abnormal Tau Accumulation. Hum. Mol. Genet. 2012, 21, 2936–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickey, C.A.; Koren, J.; Zhang, Y.-J.; Xu, Y.-F.; Jinwal, U.K.; Birnbaum, M.J.; Monks, B.; Sun, M.; Cheng, J.Q.; Patterson, C.; et al. Akt and CHIP Coregulate Tau Degradation through Coordinated Interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 3622–3627. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.-H.; Schisler, J.C.; Rubel, C.E.; Tan, S.; Song, B.; McDonough, H.; Xu, L.; Portbury, A.L.; Mao, C.-Y.; True, C.; et al. Ataxia and Hypogonadism Caused by the Loss of Ubiquitin Ligase Activity of the U Box Protein CHIP. Hum. Mol. Genet. 2014, 23, 1013–1024. [Google Scholar] [CrossRef] [Green Version]
- Ronnebaum, S.M.; Patterson, C.; Schisler, J.C. Emerging Evidence of Coding Mutations in the Ubiquitin-Proteasome System Associated with Cerebellar Ataxias. Hum. Genome Var. 2014, 1, 14018. [Google Scholar] [CrossRef] [Green Version]
- De Michele, G.; Galatolo, D.; Barghigiani, M.; Dello Iacovo, D.; Trovato, R.; Tessa, A.; Salvatore, E.; Filla, A.; De Michele, G.; Santorelli, F.M. Spinocerebellar Ataxia Type 48: Last but Not Least. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2020, 41, 2423–2432. [Google Scholar] [CrossRef]
- Madrigal, S.C.; McNeil, Z.; Sanchez-Hodge, R.; Shi, C.; Patterson, C.; Scaglione, K.M.; Schisler, J.C. Changes in Protein Function Underlie the Disease Spectrum in Patients with CHIP Mutations. J. Biol. Chem. 2019, 294, 19236–19245. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.-H.; Rubel, C.; Soss, S.E.; Sanchez-Hodge, R.; Zhang, S.; Madrigal, S.C.; Ravi, S.; McDonough, H.; Page, R.C.; Chazin, W.J.; et al. Disrupted Structure and Aberrant Function of CHIP Mediates the Loss of Motor and Cognitive Function in Preclinical Models of SCAR16. PLoS Genet. 2018, 14, e1007664. [Google Scholar] [CrossRef] [PubMed]
- De Michele, G.; Lieto, M.; Galatolo, D.; Salvatore, E.; Cocozza, S.; Barghigiani, M.; Tessa, A.; Baldacci, J.; Pappatà, S.; Filla, A.; et al. Spinocerebellar Ataxia 48 Presenting with Ataxia Associated with Cognitive, Psychiatric, and Extrapyramidal Features: A Report of Two Italian Families. Parkinsonism Relat. Disord. 2019, 65, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Mengel, D.; Traschütz, A.; Reich, S.; Leyva-Gutiérrez, A.; Bender, F.; Hauser, S.; Haack, T.B.; Synofzik, M. A de Novo STUB1 Variant Associated with an Early Adult-Onset Multisystemic Ataxia Phenotype. J. Neurol. 2021, 268, 3845–3851. [Google Scholar] [CrossRef] [PubMed]
- Ravel, J.-M.; Benkirane, M.; Calmels, N.; Marelli, C.; Ory-Magne, F.; Ewenczyk, C.; Halleb, Y.; Tison, F.; Lecocq, C.; Pische, G.; et al. Expanding the Clinical Spectrum of STIP1 Homology and U-Box Containing Protein 1-Associated Ataxia. J. Neurol. 2021, 268, 1927–1937. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-J.; Hu, B.; Han, Z.-Q.; Zhu, J.-H.; Fan, X.; Chen, X.-X.; Li, Z.-P.; Zhou, H. BAG2-Mediated Inhibition of CHIP Expression and Overexpression of MDM2 Contribute to the Initiation of Endometriosis by Modulating Estrogen Receptor Status. Front. Cell Dev. Biol. 2021, 8, 1530. [Google Scholar] [CrossRef]
- Miller, V.M.; Nelson, R.F.; Gouvion, C.M.; Williams, A.; Rodriguez-Lebron, E.; Harper, S.Q.; Davidson, B.L.; Rebagliati, M.R.; Paulson, H.L. CHIP Suppresses Polyglutamine Aggregation and Toxicity in Vitro and in Vivo. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 9152–9161. [Google Scholar] [CrossRef] [Green Version]
- Jana, N.R.; Dikshit, P.; Goswami, A.; Kotliarova, S.; Murata, S.; Tanaka, K.; Nukina, N. Co-Chaperone CHIP Associates with Expanded Polyglutamine Protein and Promotes Their Degradation by Proteasomes. J. Biol. Chem. 2005, 280, 11635–11640. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.J.; Knutson, T.M.; Colomer Gould, V.F.; Paulson, H.L. In Vivo Suppression of Polyglutamine Neurotoxicity by C-Terminus of Hsp70-Interacting Protein (CHIP) Supports an Aggregation Model of Pathogenesis. Neurobiol. Dis. 2009, 33, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.V.; Oppermann, F.S.; Rauen, M.J.; Fog, K.; Schmidt, T.; Schmidt, J.; Harmuth, T.; Hartmann-Petersen, R.; Thirstrup, K. Mass Spectrometry Analyses of Normal and Polyglutamine Expanded Ataxin-3 Reveal Novel Interaction Partners Involved in Mitochondrial Function. Neurochem. Int. 2018, 112, 5–17. [Google Scholar] [CrossRef]
- Pinho, B.R.; Almeida, L.M.; Duchen, M.R.; Oliveira, J.M.A. Allosteric Activation of Hsp70 Reduces Mutant Huntingtin Levels, the Clustering of N-Terminal Fragments, and Their Nuclear Accumulation. Life Sci. 2021, 285, 120009. [Google Scholar] [CrossRef]
- Wang, T.; Yang, J.; Xu, J.; Li, J.; Cao, Z.; Zhou, L.; You, L.; Shu, H.; Lu, Z.; Li, H.; et al. CHIP Is a Novel Tumor Suppressor in Pancreatic Cancer through Targeting EGFR. Oncotarget 2014, 5, 1969–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajiro, M.; Hirota, R.; Nakajima, Y.; Kawanowa, K.; So-ma, K.; Ito, I.; Yamaguchi, Y.; Ohie, S.; Kobayashi, Y.; Seino, Y.; et al. The Ubiquitin Ligase CHIP Acts as an Upstream Regulator of Oncogenic Pathways. Nat. Cell Biol. 2009, 11, 312–319. [Google Scholar] [CrossRef]
- Xiao, M.; Yan, M.; Zhang, J.; Xu, Q.; Chen, W. Carboxy-Terminus Hsc70 Interacting Protein Exerts a Tumor Inhibition Function in Head and Neck Cancer. Oncol. Rep. 2017, 38, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Kim, J.I.; Kim, H.S.; Park, S.D.; Jang, K.W. The Antitumor Effect of C-Terminus of Hsp70-Interacting Protein via Degradation of c-Met in Small Cell Lung Cancer. Korean J. Thorac. Cardiovasc. Surg. 2017, 50, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Jang, K.W.; Lee, J.E.; Kim, S.Y.; Kang, M.-W.; Na, M.H.; Lee, C.S.; Song, K.S.; Lim, S.P. The C-Terminus of Hsp70-Interacting Protein Promotes Met Receptor Degradation. J. Thorac. Oncol. 2011, 6, 679–687. [Google Scholar] [CrossRef] [Green Version]
- Nyhan, M.J.; O’Sullivan, G.C.; McKenna, S.L. Role of the VHL (von Hippel–Lindau) Gene in Renal Cancer: A Multifunctional Tumour Suppressor. Biochem. Soc. Trans. 2008, 36, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Brautigan, D.L.; Larner, J.M. Aurora Kinase A Promotes AR Degradation via the E3 Ligase CHIP. Mol. Cancer Res. 2017, 15, 1063–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Li, C.; Li, H.; Yuan, L.; Dai, H.; Peng, Z.; Deng, Z.; Chang, Z.; Cui, C.-P.; Zhang, L. Ubiquitin Ligase CHIP Regulates OTUD3 Stability and Suppresses Tumour Metastasis in Lung Cancer. Cell Death Differ. 2020, 27, 3177–3195. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Deng, Q.; Zhou, J.; Zou, J.; Zhang, Y.; Tan, P.; Zhang, W.; Cui, H. CSN6 Controls the Proliferation and Metastasis of Glioblastoma by CHIP-Mediated Degradation of EGFR. Oncogene 2017, 36, 1134–1144. [Google Scholar] [CrossRef]
- Xu, W.; Marcu, M.; Yuan, X.; Mimnaugh, E.; Patterson, C.; Neckers, L. Chaperone-Dependent E3 Ubiquitin Ligase CHIP Mediates a Degradative Pathway for c-ErbB2/Neu. Proc. Natl. Acad. Sci. USA 2002, 99, 12847–12852. [Google Scholar] [CrossRef] [Green Version]
- Giordano, S.H.; Temin, S.; Kirshner, J.J.; Chandarlapaty, S.; Crews, J.R.; Davidson, N.E.; Esteva, F.J.; Gonzalez-Angulo, A.M.; Krop, I.; Levinson, J.; et al. Systemic Therapy for Patients With Advanced Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2014, 32, 2078–2099. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Lv, Y.; Li, H.; Gao, H.; Song, S.; Zhang, Y.; Xing, G.; Kong, X.; Wang, L.; Li, Y.; et al. Deubiquitylase OTUD3 Regulates PTEN Stability and Suppresses Tumorigenesis. Nat. Cell Biol. 2015, 17, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Min, B.; Park, H.; Lee, S.; Li, Y.; Choi, J.-M.; Lee, J.Y.; Kim, J.; Choi, Y.D.; Kwon, Y.-G.; Lee, H.-W.; et al. CHIP-Mediated Degradation of Transglutaminase 2 Negatively Regulates Tumor Growth and Angiogenesis in Renal Cancer. Oncogene 2016, 35, 3718–3728. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Deb, S.; Paul, I.; Chatterjee, A.; Mandal, T.; Chatterjee, U.; Ghosh, M.K. The Chaperone-Assisted E3 Ligase C Terminus of Hsc70-Interacting Protein (CHIP) Targets PTEN for Proteasomal Degradation. J. Biol. Chem. 2012, 287, 15996–16006. [Google Scholar] [CrossRef] [Green Version]
- Esser, C.; Scheffner, M.; Höhfeld, J. The Chaperone-Associated Ubiquitin Ligase CHIP Is Able to Target P53 for Proteasomal Degradation. J. Biol. Chem. 2005, 280, 27443–27448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, K.; Sarkar, S.; Du, K.; Brautigan, D.L.; Abbas, T.; Larner, J.M. The E3 Ligase CHIP Mediates P21 Degradation to Maintain Radioresistance. Mol. Cancer Res. 2017, 15, 651–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Zang, J.; Dai, H.-J.; Li, F.; Guo, F. Ubiquitin Ligase CHIP Functions as an Oncogene and Activates the AKT Signaling Pathway in Prostate Cancer. Int. J. Oncol. 2018, 53, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhou, J.; Dai, H.; Liu, F.; Li, W.; Wang, W.; Guo, F. CHIP Functions as an Oncogene by Promoting Colorectal Cancer Metastasis via Activation of MAPK and AKT Signaling and Suppression of E-Cadherin. J. Transl. Med. 2018, 16, 169. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Zhou, Q.; Zhou, J.; Huang, Y.; Yan, Y.; Li, W.; Wang, C.; Hu, G.; Lu, Y.; Chen, J. Carboxyl Terminus of Hsp70-Interacting Protein (CHIP) Contributes to Human Glioma Oncogenesis. Cancer Sci. 2011, 102, 959–966. [Google Scholar] [CrossRef]
- Liang, Z.L.; Kim, M.; Huang, S.M.; Lee, H.J.; Kim, J.-M. Expression of Carboxyl Terminus of Hsp70-interacting Protein (CHIP) Indicates Poor Prognosis in Human Gallbladder Carcinoma. Oncol. Lett. 2013, 5, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Saini, S.; Khan, S.; Surendra Lele, S.; Prabhakar, B.S. Restoring Self-Tolerance in Autoimmune Diseases by Enhancing Regulatory T-Cells. Cell. Immunol. 2019, 339, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Barbi, J.; Bu, S.; Yang, H.-Y.; Li, Z.; Gao, Y.; Jinasena, D.; Fu, J.; Lin, F.; Chen, C.; et al. The Ubiquitin Ligase Stub1 Negatively Modulates Regulatory T Cell Suppressive Activity by Promoting Degradation of the Transcription Factor Foxp3. Immunity 2013, 39, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Lan, Q.; Li, Z.; Zhou, X.; Gu, J.; Li, Q.; Wang, J.; Chen, M.; Liu, Y.; Shen, Y.; et al. Critical Role of All-Trans Retinoic Acid in Stabilizing Human Natural Regulatory T Cells under Inflammatory Conditions. Proc. Natl. Acad. Sci. USA 2014, 111, E3432–E3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, Z.; Luo, X.; Wu, B.; Li, B.; Wang, B. Cimetidine Down-Regulates Stability of Foxp3 Protein via Stub1 in Treg Cells. Hum. Vaccines Immunother. 2016, 12, 2512–2518. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhao, M.; Lu, Q. Transcription Factor RFX1 Is Ubiquitinated by E3 Ligase STUB1 in Systemic Lupus Erythematosus. Clin. Immunol. 2016, 169, 1–7. [Google Scholar] [CrossRef]
- Takayama, S.; Bimston, D.N.; Matsuzawa, S.; Freeman, B.C.; Aime-Sempe, C.; Xie, Z.; Morimoto, R.I.; Reed, J.C. BAG-1 Modulates the Chaperone Activity of Hsp70/Hsc70. EMBO J. 1997, 16, 4887–4896. [Google Scholar] [CrossRef] [Green Version]
- Zeiner, M.; Gebauer, M.; Gehring, U. Mammalian Protein RAP46: An Interaction Partner and Modulator of 70 KDa Heat Shock Proteins. EMBO J. 1997, 16, 5483–5490. [Google Scholar] [CrossRef] [Green Version]
- Takayama, S.; Xie, Z.; Reed, J.C. An Evolutionarily Conserved Family of Hsp70/Hsc70 Molecular Chaperone Regulators. J. Biol. Chem. 1999, 274, 781–786. [Google Scholar] [CrossRef] [Green Version]
- Kabbage, M.; Dickman, M.B. The BAG Proteins: A Ubiquitous Family of Chaperone Regulators. Cell. Mol. Life Sci. 2008, 65, 1390–1402. [Google Scholar] [CrossRef]
- Takayama, S.; Reed, J.C. Molecular Chaperone Targeting and Regulation by BAG Family Proteins. Nat. Cell Biol. 2001, 3, E237–E241. [Google Scholar] [CrossRef]
- Mizushima, W.; Sadoshima, J. BAG3 Plays a Central Role in Proteostasis in the Heart. J. Clin. Investig. 2017, 127, 2900–2903. [Google Scholar] [CrossRef] [Green Version]
- Behl, C. Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol. Sci. 2016, 37, 672–688. [Google Scholar] [CrossRef]
- Ryu, S.W.; Stewart, R.; Pectol, D.C.; Ender, N.A.; Wimalarathne, O.; Lee, J.-H.; Zanini, C.P.; Harvey, A.; Huibregtse, J.M.; Mueller, P.; et al. Proteome-Wide Identification of HSP70/HSC70 Chaperone Clients in Human Cells. PLoS Biol. 2020, 18, e3000606. [Google Scholar] [CrossRef] [PubMed]
- Mahmaljy, H.; Yelamanchili, V.S.; Singhal, M. Dilated Cardiomyopathy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Haas, J.; Frese, K.S.; Peil, B.; Kloos, W.; Keller, A.; Nietsch, R.; Feng, Z.; Müller, S.; Kayvanpour, E.; Vogel, B.; et al. Atlas of the Clinical Genetics of Human Dilated Cardiomyopathy. Eur. Heart J. 2015, 36, 1123–1135. [Google Scholar] [CrossRef]
- Franaszczyk, M.; Bilinska, Z.T.; Sobieszczańska-Małek, M.; Michalak, E.; Sleszycka, J.; Sioma, A.; Małek, Ł.A.; Kaczmarska, D.; Walczak, E.; Włodarski, P.; et al. The BAG3 Gene Variants in Polish Patients with Dilated Cardiomyopathy: Four Novel Mutations and a Genotype-Phenotype Correlation. J. Transl. Med. 2014, 12, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez, F.; Cuenca, S.; Bilińska, Z.; Toro, R.; Villard, E.; Barriales-Villa, R.; Ochoa, J.P.; Asselbergs, F.; Sammani, A.; Franaszczyk, M.; et al. Dilated Cardiomyopathy Due to BLC2-Associated Athanogene 3 (BAG3) Mutations. J. Am. Coll. Cardiol. 2018, 72, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.G.; Tawfik, S.; Moravec, C.S.; Pak, T.R.; Kirk, J.A. BAG3 Expression and Sarcomere Localization in the Human Heart Are Linked to HSF-1 and Are Differentially Affected by Sex and Disease. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H2339–H2350. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Wu, N.; Yang, H.; Guo, Y.; Guo, X.; Sun, Y. Different Roles of BAG3 in Cardiac Physiological Hypertrophy and Pathological Remodeling. Transl. Res. J. Lab. Clin. Med. 2021, 233, 47–61. [Google Scholar] [CrossRef]
- Myers, V.D.; Tomar, D.; Madesh, M.; Wang, J.; Song, J.; Zhang, X.; Gupta, M.K.; Tahrir, F.G.; Gordon, J.; McClung, J.M.; et al. Haplo-insufficiency of Bcl2-associated Athanogene 3 in Mice Results in Progressive Left Ventricular Dysfunction, Β-adrenergic Insensitivity, and Increased Apoptosis. J. Cell. Physiol. 2018, 233, 6319–6326. [Google Scholar] [CrossRef]
- Inomata, Y.; Nagasaka, S.; Miyate, K.; Goto, Y.; Hino, C.; Toukairin, C.; Higashio, R.; Ishida, K.; Saino, T.; Hirose, M.; et al. Bcl-2-Associated Athanogene 3 (BAG3) Is an Enhancer of Small Heat Shock Protein Turnover via Activation of Autophagy in the Heart. Biochem. Biophys. Res. Commun. 2018, 496, 1141–1147. [Google Scholar] [CrossRef]
- Inomata, Y.; Miyate, K.; Goto, Y.; Higashio, R.; Hirose, M.; Sanbe, A. P2625Bcl-2 Associated Athanogene 3 Enhances Alpha-B Crystallin Protein Turnover via Activation of Autophagy in the Heart. Eur. Heart J. 2017, 38. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J.; Stroud, M.J.; Zhang, Z.; Ma, X.; Mu, Y.; et al. Loss-of-Function Mutations in Co-Chaperone BAG3 Destabilize Small HSPs and Cause Cardiomyopathy. J. Clin. Investig. 2017, 127, 3189–3200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judge, L.M.; Perez-Bermejo, J.A.; Truong, A.; Ribeiro, A.J.; Yoo, J.C.; Jensen, C.L.; Mandegar, M.A.; Huebsch, N.; Kaake, R.M.; So, P.-L.; et al. A BAG3 Chaperone Complex Maintains Cardiomyocyte Function during Proteotoxic Stress. JCI Insight 2017, 2, 94623. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Bogomolovas, J.; Zhou, P.S.; Mu, Y.; Ma, X.; Chen, Z.; Zhang, L.; Zhu, M.; Veevers, J.; Ouyang, K.; et al. P209L Mutation in Bag3 Does Not Cause Cardiomyopathy in Mice. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H392–H399. [Google Scholar] [CrossRef]
- Quintana, M.T.; Parry, T.L.; He, J.; Yates, C.C.; Sidorova, T.N.; Murray, K.T.; Bain, J.R.; Newgard, C.B.; Muehlbauer, M.J.; Eaton, S.C.; et al. Cardiomyocyte-Specific Human Bcl2-Associated Anthanogene 3 P209L Expression Induces Mitochondrial Fragmentation, Bcl2-Associated Anthanogene 3 Haploinsufficiency, and Activates P38 Signaling. Am. J. Pathol. 2016, 186, 1989–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, M.S.; Patterson, C. Hold Me Tight: The Role of the HSP Family of Chaperones in Cardiac Disease. Circulation 2010, 122, 1740–1751. [Google Scholar] [CrossRef]
- McDermott-Roe, C.; Lv, W.; Maximova, T.; Wada, S.; Bukowy, J.; Marquez, M.; Lai, S.; Shehu, A.; Benjamin, I.; Geurts, A.; et al. Investigation of a Dilated Cardiomyopathy-Associated Variant in BAG3 Using Genome-Edited IPSC-Derived Cardiomyocytes. JCI Insight 2019, 4, 128799. [Google Scholar] [CrossRef]
- Takayama, S.; Krajewski, S.; Krajewska, M.; Kitada, S.; Zapata, J.M.; Kochel, K.; Knee, D.; Scudiero, D.; Tudor, G.; Miller, G.J.; et al. Expression and Location of Hsp70/Hsc-Binding Anti-Apoptotic Protein BAG-1 and Its Variants in Normal Tissues and Tumor Cell Lines. Cancer Res. 1998, 58, 3116–3131. [Google Scholar]
- Froesch, B.A.; Takayama, S.; Reed, J.C. BAG-1L Protein Enhances Androgen Receptor Function. J. Biol. Chem. 1998, 273, 11660–11666. [Google Scholar] [CrossRef] [Green Version]
- Cato, L.; Neeb, A.; Sharp, A.; Buzón, V.; Ficarro, S.B.; Yang, L.; Muhle-Goll, C.; Kuznik, N.C.; Riisnaes, R.; Nava Rodrigues, D.; et al. Development of Bag-1L as a Therapeutic Target in Androgen Receptor-Dependent Prostate Cancer. eLife 2017, 6, e27159. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.I.; Kuznik, N.C.; Rottenberg, J.T.; Brown, M.; Cato, A.C.B. BAG1L: A Promising Therapeutic Target for Androgen Receptor-Dependent Prostate Cancer. J. Mol. Endocrinol. 2019, 62, R289–R299. [Google Scholar] [CrossRef] [Green Version]
- Townsend, P.A.; Dublin, E.; Hart, I.R.; Kao, R.H.; Hanby, A.M.; Cutress, R.I.; Poulsom, R.; Ryder, K.; Barnes, D.M.; Packham, G. BAG-i Expression in Human Breast Cancer: Interrelationship between BAG-1 RNA, Protein, HSC70 Expression and Clinico-Pathological Data. J. Pathol. 2002, 197, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, E.S.; Reeves, T.; Robson, N.H.; Maishman, T.; Packham, G.; Cutress, R.I. BAG-1 as a Biomarker in Early Breast Cancer Prognosis: A Systematic Review with Meta-Analyses. Br. J. Cancer 2017, 116, 1585–1594. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.-C.; Wang, Z.; Peng, B.; Xia, L.-G.; Lin, L.-W.; Xu, Z.-L. BAG2 Overexpression Correlates with Growth and Poor Prognosis of Esophageal Squamous Cell Carcinoma. Open Life Sci. 2018, 13, 582–588. [Google Scholar] [CrossRef]
- Liu, Y.-S.; Wei, B. Over-Expression of Bcl2-Associated Athanogene 2 in Oral Cancer Promotes Cellular Proliferation and Is Associated with Poor Prognosis. Arch. Oral Biol. 2019, 102, 164–170. [Google Scholar] [CrossRef]
- Sun, L.; Chen, G.; Sun, A.; Wang, Z.; Huang, H.; Gao, Z.; Liang, W.; Liu, C.; Li, K. BAG2 Promotes Proliferation and Metastasis of Gastric Cancer via ERK1/2 Signaling and Partially Regulated by MiR186. Front. Oncol. 2020, 10, 31. [Google Scholar] [CrossRef]
- Wang, H.-Q.; Zhang, H.-Y.; Hao, F.-J.; Meng, X.; Guan, Y.; Du, Z.-X. Induction of BAG2 Protein during Proteasome Inhibitor-Induced Apoptosis in Thyroid Carcinoma Cells. Br. J. Pharmacol. 2008, 155, 655–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marco, M.D.; Turco, M.C.; Marzullo, L. BAG3 in Tumor Resistance to Therapy. Trends Cancer 2020, 6, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Kögel, D.; Linder, B.; Brunschweiger, A.; Chines, S.; Behl, C. At the Crossroads of Apoptosis and Autophagy: Multiple Roles of the Co-Chaperone BAG3 in Stress and Therapy Resistance of Cancer. Cells 2020, 9, 574. [Google Scholar] [CrossRef] [Green Version]
- Terracciano, S.; Lauro, G.; Russo, A.; Vaccaro, M.C.; Vassallo, A.; Marco, M.D.; Ranieri, B.; Rosati, A.; Turco, M.C.; Riccio, R.; et al. Discovery and Synthesis of the First Selective BAG Domain Modulator of BAG3 as an Attractive Candidate for the Development of a New Class of Chemotherapeutics. Chem. Commun. 2018, 54, 7613–7616. [Google Scholar] [CrossRef]
- Rosati, A.; Basile, A.; D’Auria, R.; d’Avenia, M.; De Marco, M.; Falco, A.; Festa, M.; Guerriero, L.; Iorio, V.; Parente, R.; et al. BAG3 Promotes Pancreatic Ductal Adenocarcinoma Growth by Activating Stromal Macrophages. Nat. Commun. 2015, 6, 8695. [Google Scholar] [CrossRef]
- Iorio, V.; Rosati, A.; D’Auria, R.; Marco, M.D.; Marzullo, L.; Basile, A.; Festa, M.; Pascale, M.; Remondelli, P.; Capunzo, M.; et al. Combined Effect of Anti-BAG3 and Anti-PD-1 Treatment on Macrophage Infiltrate, CD8+ T Cell Number and Tumour Growth in Pancreatic Cancer. Gut 2018, 67, 780–782. [Google Scholar] [CrossRef] [Green Version]
- Basile, A.; De Marco, M.; Festa, M.; Falco, A.; Iorio, V.; Guerriero, L.; Eletto, D.; Rea, D.; Arra, C.; Lamolinara, A.; et al. Development of an Anti-BAG3 Humanized Antibody for Treatment of Pancreatic Cancer. Mol. Oncol. 2019, 13, 1388–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolet, C.M.; Craig, E.A. Isolation and Characterization of STI1, a Stress-Inducible Gene from Saccharomyces Cerevisiae. Mol. Cell. Biol. 1989, 9, 3638–3646. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.D.; Schumacher, R.J.; Ross, E.D.; Toft, D.O. Hop Modulates Hsp70/Hsp90 Interactions in Protein Folding. J. Biol. Chem. 1998, 273, 3679–3686. [Google Scholar] [CrossRef] [Green Version]
- Scheufler, C.; Brinker, A.; Bourenkov, G.; Pegoraro, S.; Moroder, L.; Bartunik, H.; Hartl, F.U.; Moarefi, I. Structure of TPR Domain-Peptide Complexes: Critical Elements in the Assembly of the Hsp70-Hsp90 Multichaperone Machine. Cell 2000, 101, 199–210. [Google Scholar] [CrossRef]
- Brinker, A.; Scheufler, C.; Von Der Mulbe, F.; Fleckenstein, B.; Herrmann, C.; Jung, G.; Moarefi, I.; Hartl, F.U. Ligand Discrimination by TPR Domains. Relevance and Selectivity of EEVD-Recognition in Hsp70 x Hop x Hsp90 Complexes. J. Biol. Chem. 2002, 277, 19265–19275. [Google Scholar] [CrossRef]
- Richter, K.; Muschler, P.; Hainzl, O.; Reinstein, J.; Buchner, J. Sti1 Is a Non-Competitive Inhibitor of the Hsp90 ATPase: BINDING PREVENTS THE N-TERMINAL DIMERIZATION REACTION DURING THE ATPASE CYCLE. J. Biol. Chem. 2003, 278, 10328–10333. [Google Scholar] [CrossRef] [Green Version]
- Wegele, H.; Wandinger, S.K.; Schmid, A.B.; Reinstein, J.; Buchner, J. Substrate Transfer from the Chaperone Hsp70 to Hsp90. J. Mol. Biol. 2006, 356, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Flom, G.; Behal, R.H.; Rosen, L.; Cole, D.G.; Johnson, J.L. Definition of the Minimal Fragments of Sti1 Required for Dimerization, Interaction with Hsp70 and Hsp90 and in Vivo Functions. Biochem. J. 2007, 404, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Schmid, A.B.; Lagleder, S.; Gräwert, M.A.; Röhl, A.; Hagn, F.; Wandinger, S.K.; Cox, M.B.; Demmer, O.; Richter, K.; Groll, M.; et al. The Architecture of Functional Modules in the Hsp90 Co-Chaperone Sti1/Hop. EMBO J. 2012, 31, 1506–1517. [Google Scholar] [CrossRef] [Green Version]
- Alvira, S.; Cuéllar, J.; Röhl, A.; Yamamoto, S.; Itoh, H.; Alfonso, C.; Rivas, G.; Buchner, J.; Valpuesta, J.M. Structural Characterization of the Substrate Transfer Mechanism in Hsp70/Hsp90 Folding Machinery Mediated by Hop. Nat. Commun. 2014, 5, 5484. [Google Scholar] [CrossRef] [Green Version]
- Röhl, A.; Wengler, D.; Madl, T.; Lagleder, S.; Tippel, F.; Herrmann, M.; Hendrix, J.; Richter, K.; Hack, G.; Schmid, A.B.; et al. Hsp90 Regulates the Dynamics of Its Cochaperone Sti1 and the Transfer of Hsp70 between Modules. Nat. Commun. 2015, 6, 6655. [Google Scholar] [CrossRef] [Green Version]
- Prodromou, C.; Siligardi, G.; O’Brien, R.; Woolfson, D.N.; Regan, L.; Panaretou, B.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Regulation of Hsp90 ATPase Activity by Tetratricopeptide Repeat (TPR)-Domain Co-Chaperones. EMBO J. 1999, 18, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Soroka, J.; Buchner, J. The Hsp90 Chaperone Machinery: Conformational Dynamics and Regulation by Co-Chaperones. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2012, 1823, 624–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-T.; Graf, C.; Mayer, F.J.; Richter, S.M.; Mayer, M.P. Dynamics of the Regulation of Hsp90 by the Co-Chaperone Sti1. EMBO J. 2012, 31, 1518–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, K.; Weidenauer, L.; Luengo, T.M.; Pieters, E.C.; Echeverría, P.C.; Bernasconi, L.; Wider, D.; Sadian, Y.; Koopman, M.B.; Villemin, M.; et al. The Hsp70-Hsp90 Co-Chaperone Hop/Stip1 Shifts the Proteostatic Balance from Folding towards Degradation. Nat. Commun. 2020, 11, 5975. [Google Scholar] [CrossRef]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.-Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.M.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.O.; Cabral, A.L.B.; Lee, K.S.; Juliano, M.A.; et al. Stress-Inducible Protein 1 Is a Cell Surface Ligand for Cellular Prion That Triggers Neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef]
- Lopes, M.H.; Hajj, G.N.M.; Muras, A.G.; Mancini, G.L.; Castro, R.M.P.S.; Ribeiro, K.C.B.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of Cellular Prion and Stress-Inducible Protein 1 Promotes Neuritogenesis and Neuroprotection by Distinct Signaling Pathways. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [Green Version]
- Arantes, C.; Nomizo, R.; Lopes, M.H.; Hajj, G.N.M.; Lima, F.R.S.; Martins, V.R. Prion Protein and Its Ligand Stress Inducible Protein 1 Regulate Astrocyte Development. Glia 2009, 57, 1439–1449. [Google Scholar] [CrossRef]
- Roffé, M.; Beraldo, F.H.; Bester, R.; Nunziante, M.; Bach, C.; Mancini, G.; Gilch, S.; Vorberg, I.; Castilho, B.A.; Martins, V.R.; et al. Prion Protein Interaction with Stress-Inducible Protein 1 Enhances Neuronal Protein Synthesis via MTOR. Proc. Natl. Acad. Sci. USA 2010, 107, 13147–13152. [Google Scholar] [CrossRef] [Green Version]
- Santos, T.G.; Silva, I.R.; Costa-Silva, B.; Lepique, A.P.; Martins, V.R.; Lopes, M.H. Enhanced Neural Progenitor/Stem Cells Self-Renewal via the Interaction of Stress-Inducible Protein 1 with the Prion Protein. Stem Cells 2011, 29, 1126–1136. [Google Scholar] [CrossRef] [Green Version]
- da Fonseca, A.C.C.; Romão, L.; Amaral, R.F.; Assad Kahn, S.; Lobo, D.; Martins, S.; Marcondes de Souza, J.; Moura-Neto, V.; Lima, F.R.S. Microglial Stress Inducible Protein 1 Promotes Proliferation and Migration in Human Glioblastoma Cells. Neuroscience 2012, 200, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Beraldo, F.H.; Soares, I.N.; Goncalves, D.F.; Fan, J.; Thomas, A.A.; Santos, T.G.; Mohammad, A.H.; Roffé, M.; Calder, M.D.; Nikolova, S.; et al. Stress-Inducible Phosphoprotein 1 Has Unique Cochaperone Activity during Development and Regulates Cellular Response to Ischemia via the Prion Protein. FASEB J. 2013, 27, 3594–3607. [Google Scholar] [CrossRef] [Green Version]
- Ostapchenko, V.G.; Beraldo, F.H.; Mohammad, A.H.; Xie, Y.-F.; Hirata, P.H.F.; Magalhaes, A.C.; Lamour, G.; Li, H.; Maciejewski, A.; Belrose, J.C.; et al. The Prion Protein Ligand, Stress-Inducible Phosphoprotein 1, Regulates Amyloid-β Oligomer Toxicity. J. Neurosci. 2013, 33, 16552–16564. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, A.; Ostapchenko, V.G.; Beraldo, F.H.; Prado, V.F.; Prado, M.A.M.; Choy, W.-Y. Domains of STIP1 Responsible for Regulating PrPC-Dependent Amyloid-β Oligomer Toxicity. Biochem. J. 2016, 473, 2119–2130. [Google Scholar] [CrossRef]
- Ambegaokar, S.S.; Jackson, G.R. Functional Genomic Screen and Network Analysis Reveal Novel Modifiers of Tauopathy Dissociated from Tau Phosphorylation. Hum. Mol. Genet. 2011, 20, 4947–4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lackie, R.E.; Marques-Lopes, J.; Ostapchenko, V.G.; Good, S.; Choy, W.-Y.; van Oosten-Hawle, P.; Pasternak, S.H.; Prado, V.F.; Prado, M.A.M. Increased Levels of Stress-Inducible Phosphoprotein-1 Accelerates Amyloid-β Deposition in a Mouse Model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2020, 8, 143. [Google Scholar] [CrossRef]
- Falsone, S.F.; Kungl, A.J.; Rek, A.; Cappai, R.; Zangger, K. The Molecular Chaperone Hsp90 Modulates Intermediate Steps of Amyloid Assembly of the Parkinson-Related Protein Alpha-Synuclein. J. Biol. Chem. 2009, 284, 31190–31199. [Google Scholar] [CrossRef] [Green Version]
- Daturpalli, S.; Waudby, C.A.; Meehan, S.; Jackson, S.E. Hsp90 Inhibits α-Synuclein Aggregation by Interacting with Soluble Oligomers. J. Mol. Biol. 2013, 425, 4614–4628. [Google Scholar] [CrossRef]
- Wolfe, K.J.; Ren, H.Y.; Trepte, P.; Cyr, D.M. The Hsp70/90 Cochaperone, Sti1, Suppresses Proteotoxicity by Regulating Spatial Quality Control of Amyloid-like Proteins. Mol. Biol. Cell 2013, 24, 3588–3602. [Google Scholar] [CrossRef]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A Chaperome Subnetwork Safeguards Proteostasis in Aging and Neurodegenerative Disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Kula-Eversole, E.; Iwanaszko, M.; Hutchison, A.L.; Dinner, A.; Allada, R. Circadian Clocks Function in Concert with Heat Shock Organizing Protein to Modulate Mutant Huntingtin Aggregation and Toxicity. Cell Rep. 2019, 27, 59–70.e4. [Google Scholar] [CrossRef]
- Lin, L.T.-W.; Razzaq, A.; Di Gregorio, S.E.; Hong, S.; Charles, B.; Lopes, M.H.; Beraldo, F.; Prado, V.F.; Prado, M.A.M.; Duennwald, M.L. Hsp90 and Its Co-Chaperone Sti1 Control TDP-43 Misfolding and Toxicity. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21594. [Google Scholar] [CrossRef]
- Walsh, N.; Larkin, A.; Swan, N.; Conlon, K.; Dowling, P.; McDermott, R.; Clynes, M. RNAi Knockdown of Hop (Hsp70/Hsp90 Organising Protein) Decreases Invasion via MMP-2 down Regulation. Cancer Lett. 2011, 306, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Kubota, H.; Yamamoto, S.; Itoh, E.; Abe, Y.; Nakamura, A.; Izumi, Y.; Okada, H.; Iida, M.; Nanjo, H.; Itoh, H.; et al. Increased Expression of Co-Chaperone HOP with HSP90 and HSC70 and Complex Formation in Human Colonic Carcinoma. Cell Stress Chaperones 2010, 15, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A High-Affinity Conformation of Hsp90 Confers Tumour Selectivity on Hsp90 Inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Zhai, E.; Liang, W.; Lin, Y.; Huang, L.; He, X.; Cai, S.; Chen, J.; Zhang, N.; Li, J.; Zhang, Q.; et al. HSP70/HSP90-Organizing Protein Contributes to Gastric Cancer Progression in an Autocrine Fashion and Predicts Poor Survival in Gastric Cancer. Cell. Physiol. Biochem. 2018, 47, 879–892. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-H.; Gong, C.; Guo, F.-J.; Zhou, X.; Zhang, M.-S.; Qiu, H.; Chao, T.-F.; Liu, Y.; Qin, L.; Xiong, H.-H. Knockdown of STIP1 Inhibits the Invasion of CD133-positive Cancer Stem-like Cells of the Osteosarcoma MG63 Cell Line via the PI3K/Akt and ERK1/2 Pathways. Int. J. Mol. Med. 2020, 46, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Chen, J.; Liu, G.; Huang, W.; Wei, X.; Wei, Z.; He, Y. STIP1 Knockdown Suppresses Colorectal Cancer Cell Proliferation, Migration and Invasion by Inhibiting STAT3 Pathway. Chem. Biol. Interact. 2021, 341, 109446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ren, H.; Yang, L.; Zhang, X.; Liang, W.; Wu, H.; Huang, L.; Kang, J.; Xu, J.; Zhai, E.; et al. Aberrant Expression of Stress-Induced Phosphoprotein 1 in Colorectal Cancer and Its Clinicopathologic Significance. Hum. Pathol. 2018, 79, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cao, N.; Mu, L.; Cao, W. Stress Induced Phosphoprotein 1 Promotes Tumor Growth and Metastasis of Melanoma via Modulating JAK2/STAT3 Pathway. Biomed. Pharmacother. 2019, 116, 108962. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-L.; Tsai, C.-N.; Lin, C.-Y.; Chen, H.-W.; Lee, Y.-S.; Chao, A.; Wang, T.-H.; Wang, H.-S.; Lai, C.-H. Secreted Stress-Induced Phosphoprotein 1 Activates the ALK2-SMAD Signaling Pathways and Promotes Cell Proliferation of Ovarian Cancer Cells. Cell Rep. 2012, 2, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlich, R.B.; Kahn, S.A.; Lima, F.R.S.; Muras, A.G.; Martins, R.A.P.; Linden, R.; Chiarini, L.B.; Martins, V.R.; Neto, V.M. STI1 Promotes Glioma Proliferation through MAPK and PI3K Pathways. Glia 2007, 55, 1690–1698. [Google Scholar] [CrossRef]
- Yin, H.; Deng, Z.; Li, X.; Li, Y.; Yin, W.; Zhao, G.; Jiang, D.; Sun, C.; Zhou, Y. Down-Regulation of STIP1 Regulate Apoptosis and Invasion of Glioma Cells via TRAP1/AKT Signaling Pathway. Cancer Genet. 2019, 237, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Baindur-Hudson, S.; Edkins, A.L.; Blatch, G.L. Hsp70/Hsp90 Organising Protein (Hop): Beyond Interactions with Chaperones and Prion Proteins. Subcell. Biochem. 2015, 78, 69–90. [Google Scholar] [CrossRef]
- Yi, F.; Regan, L. A Novel Class of Small Molecule Inhibitors of Hsp90. ACS Chem. Biol. 2008, 3, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Horibe, T.; Kohno, M.; Haramoto, M.; Ohara, K.; Kawakami, K. Designed Hybrid TPR Peptide Targeting Hsp90 as a Novel Anticancer Agent. J. Transl. Med. 2011, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horibe, T.; Torisawa, A.; Kohno, M.; Kawakami, K. Molecular Mechanism of Cytotoxicity Induced by Hsp90-Targeted Antp-TPR Hybrid Peptide in Glioblastoma Cells. Mol. Cancer 2012, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Pimienta, G.; Herbert, K.M.; Regan, L. A Compound That Inhibits the HOP-Hsp90 Complex Formation and Has Unique Killing Effects in Breast Cancer Cell Lines. Mol. Pharm. 2011, 8, 2252–2261. [Google Scholar] [CrossRef]
- Ardi, V.C.; Alexander, L.D.; Johnson, V.A.; McAlpine, S.R. Macrocycles That Inhibit the Binding between Heat Shock Protein 90 and TPR-Containing Proteins. ACS Chem. Biol. 2011, 6, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Kou, X.; Jiang, X.; Liu, H.; Wang, X.; Sun, F.; Han, J.; Fan, J.; Feng, G.; Lin, Z.; Jiang, L.; et al. Simvastatin Functions as a Heat Shock Protein 90 Inhibitor against Triple-Negative Breast Cancer. Cancer Sci. 2018, 109, 3272–3284. [Google Scholar] [CrossRef]
- Yamamoto, S.; Subedi, G.P.; Hanashima, S.; Satoh, T.; Otaka, M.; Wakui, H.; Sawada, K.; Yokota, S.; Yamaguchi, Y.; Kubota, H.; et al. ATPase Activity and ATP-Dependent Conformational Change in the Co-Chaperone HSP70/HSP90-Organizing Protein (HOP). J. Biol. Chem. 2014, 289, 9880–9886. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.R. Hsp56: A Novel Heat Shock Protein Associated with Untransformed Steroid Receptor Complexes. J. Biol. Chem. 1990, 265, 22067–22070. [Google Scholar] [CrossRef]
- Smith, D.F.; Faber, L.E.; Toft, D.O. Purification of Unactivated Progesterone Receptor and Identification of Novel Receptor-Associated Proteins. J. Biol. Chem. 1990, 265, 3996–4003. [Google Scholar] [CrossRef]
- Callebaut, I.; Renoir, J.M.; Lebeau, M.C.; Massol, N.; Burny, A.; Baulieu, E.E.; Mornon, J.P. An Immunophilin That Binds M(r) 90,000 Heat Shock Protein: Main Structural Features of a Mammalian P59 Protein. Proc. Natl. Acad. Sci. USA 1992, 89, 6270–6274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, P.K.; Albers, M.W.; Chang, H.; Faber, L.E.; Schreiber, S.L. Association of a 59-Kilodalton Immunophilin with the Glucocorticoid Receptor Complex. Science 1992, 256, 1315–1318. [Google Scholar] [CrossRef] [PubMed]
- Peattie, D.A.; Harding, M.W.; Fleming, M.A.; DeCenzo, M.T.; Lippke, J.A.; Livingston, D.J.; Benasutti, M. Expression and Characterization of Human FKBP52, an Immunophilin That Associates with the 90-KDa Heat Shock Protein and Is a Component of Steroid Receptor Complexes. Proc. Natl. Acad. Sci. USA 1992, 89, 10974–10978. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sullivan, W.P.; Toft, D.O.; Smith, D.F. Differential Interactions of P23 and the TPR-Containing Proteins Hop, Cyp40, FKBP52 and FKBP51 with Hsp90 Mutants. Cell Stress Chaperones 1998, 3, 118–129. [Google Scholar] [CrossRef]
- Smith, D.F. Tetratricopeptide Repeat Cochaperones in Steroid Receptor Complexes. Cell Stress Chaperones 2004, 9, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wochnik, G.M.; Rüegg, J.; Abel, G.A.; Schmidt, U.; Holsboer, F.; Rein, T. FK506-Binding Proteins 51 and 52 Differentially Regulate Dynein Interaction and Nuclear Translocation of the Glucocorticoid Receptor in Mammalian Cells. J. Biol. Chem. 2005, 280, 4609–4616. [Google Scholar] [CrossRef] [Green Version]
- Riggs, D.L.; Cox, M.B.; Tardif, H.L.; Hessling, M.; Buchner, J.; Smith, D.F. Noncatalytic Role of the FKBP52 Peptidyl-Prolyl Isomerase Domain in the Regulation of Steroid Hormone Signaling. Mol. Cell. Biol. 2007, 27, 8658–8669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fries, G.R.; Gassen, N.C.; Rein, T. The FKBP51 Glucocorticoid Receptor Co-Chaperone: Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2017, 18, 2614. [Google Scholar] [CrossRef] [Green Version]
- Zgajnar, N.R.; De Leo, S.A.; Lotufo, C.M.; Erlejman, A.G.; Piwien-Pilipuk, G.; Galigniana, M.D. Biological Actions of the Hsp90-Binding Immunophilins FKBP51 and FKBP52. Biomolecules 2019, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Hähle, A.; Merz, S.; Meyners, C.; Hausch, F. The Many Faces of FKBP51. Biomolecules 2019, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Jinwal, U.K.; Koren, J.; Borysov, S.I.; Schmid, A.B.; Abisambra, J.F.; Blair, L.J.; Johnson, A.G.; Jones, J.R.; Shults, C.L.; O’Leary, J.C.; et al. The Hsp90 Cochaperone, FKBP51, Increases Tau Stability and Polymerizes Microtubules. J. Neurosci. 2010, 30, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C.; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated Neurodegeneration through Chaperone-Mediated Oligomerization of Tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustiniani, J.; Sineus, M.; Sardin, E.; Dounane, O.; Panchal, M.; Sazdovitch, V.; Duyckaerts, C.; Chambraud, B.; Baulieu, E.-E. Decrease of the Immunophilin FKBP52 Accumulation in Human Brains of Alzheimer’s Disease and FTDP-17. J. Alzheimers Dis. JAD 2012, 29, 471–483. [Google Scholar] [CrossRef]
- Chambraud, B.; Sardin, E.; Giustiniani, J.; Dounane, O.; Schumacher, M.; Goedert, M.; Baulieu, E.-E. A Role for FKBP52 in Tau Protein Function. Proc. Natl. Acad. Sci. USA 2010, 107, 2658–2663. [Google Scholar] [CrossRef] [Green Version]
- Jinwal, U.K.; Koren III, J.; Dickey, C.A. Reconstructing the Hsp90/Tau Machine. Curr. Enzym. Inhib. 2013, 9, 41–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustiniani, J.; Chambraud, B.; Sardin, E.; Dounane, O.; Guillemeau, K.; Nakatani, H.; Paquet, D.; Kamah, A.; Landrieu, I.; Lippens, G.; et al. Immunophilin FKBP52 Induces Tau-P301L Filamentous Assembly in Vitro and Modulates Its Activity in a Model of Tauopathy. Proc. Natl. Acad. Sci. USA 2014, 111, 4584–4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustiniani, J.; Guillemeau, K.; Dounane, O.; Sardin, E.; Huvent, I.; Schmitt, A.; Hamdane, M.; Buée, L.; Landrieu, I.; Lippens, G.; et al. The FK506-Binding Protein FKBP52 in Vitro Induces Aggregation of Truncated Tau Forms with Prion-like Behavior. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 3171–3181. [Google Scholar] [CrossRef] [Green Version]
- Meduri, G.; Guillemeau, K.; Dounane, O.; Sazdovitch, V.; Duyckaerts, C.; Chambraud, B.; Baulieu, E.E.; Giustiniani, J. Caspase-Cleaved Tau-D(421) Is Colocalized with the Immunophilin FKBP52 in the Autophagy-Endolysosomal System of Alzheimer’s Disease Neurons. Neurobiol. Aging 2016, 46, 124–137. [Google Scholar] [CrossRef]
- Kamah, A.; Cantrelle, F.X.; Huvent, I.; Giustiniani, J.; Guillemeau, K.; Byrne, C.; Jacquot, Y.; Landrieu, I.; Baulieu, E.E.; Smet, C.; et al. Isomerization and Oligomerization of Truncated and Mutated Tau Forms by FKBP52 Are Independent Processes. J. Mol. Biol. 2016, 428, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Criado-Marrero, M.; Gebru, N.T.; Blazier, D.M.; Gould, L.A.; Baker, J.D.; Beaulieu-Abdelahad, D.; Blair, L.J. Hsp90 Co-Chaperones, FKBP52 and Aha1, Promote Tau Pathogenesis in Aged Wild-Type Mice. Acta Neuropathol. Commun. 2021, 9, 65. [Google Scholar] [CrossRef]
- Criado-Marrero, M.; Gebru, N.T.; Gould, L.A.; Blazier, D.M.; Vidal-Aguiar, Y.; Smith, T.M.; Abdelmaboud, S.S.; Shelton, L.B.; Wang, X.; Dahrendorff, J.; et al. FKBP52 Overexpression Accelerates Hippocampal-Dependent Memory Impairments in a Tau Transgenic Mouse Model. NPJ Aging Mech. Dis. 2021, 7, 9. [Google Scholar] [CrossRef]
- De Leon, J.T.; Iwai, A.; Feau, C.; Garcia, Y.; Balsiger, H.A.; Storer, C.L.; Suro, R.M.; Garza, K.M.; Lee, S.; Sang Kim, Y.; et al. Targeting the Regulation of Androgen Receptor Signaling by the Heat Shock Protein 90 Cochaperone FKBP52 in Prostate Cancer Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11878–11883. [Google Scholar] [CrossRef] [Green Version]
- Ward, B.K.; Mark, P.J.; Ingram, D.M.; Minchin, R.F.; Ratajczak, T. Expression of the Estrogen Receptor-Associated Immunophilins, Cyclophilin 40 and FKBP52, in Breast Cancer. Breast Cancer Res. Treat. 1999, 58, 265–278. [Google Scholar] [CrossRef]
- Periyasamy, S.; Hinds, T.; Shemshedini, L.; Shou, W.; Sanchez, E.R. FKBP51 and Cyp40 Are Positive Regulators of Androgen-Dependent Prostate Cancer Cell Growth and the Targets of FK506 and Cyclosporin A. Oncogene 2010, 29, 1691–1701. [Google Scholar] [CrossRef] [Green Version]
- Maeda, K.; Habara, M.; Kawaguchi, M.; Matsumoto, H.; Hanaki, S.; Masaki, T.; Sato, Y.; Matsuyama, H.; Kunieda, K.; Nakagawa, H.; et al. FKBP51 and FKBP52 Regulate Androgen Receptor Dimerization and Proliferation in Prostate Cancer Cells. Mol. Oncol. 2021. [Google Scholar] [CrossRef]
- Yu, J.; Sun, L.; Hao, T.; Zhang, B.; Chen, X.; Li, H.; Zhang, Z.; Zhu, S.; Quan, C.; Niu, Y.; et al. Restoration of FKBP51 Protein Promotes the Progression of Castration Resistant Prostate Cancer. Ann. Transl. Med. 2019, 7, 729. [Google Scholar] [CrossRef]
- Gao, Y.; Elamin, E.; Zhou, R.; Yan, H.; Liu, S.; Hu, S.; Dong, J.; Wei, M.; Sun, L.; Zhao, Y. FKBP51 Promotes Migration and Invasion of Papillary Thyroid Carcinoma through NF-κB-dependent Epithelial-to-mesenchymal Transition. Oncol. Lett. 2018, 16, 7020–7028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, H.; Li, L.; Fridley, B.L.; Jenkins, G.D.; Kalari, K.R.; Lingle, W.; Petersen, G.; Lou, Z.; Wang, L. FKBP51 Affects Cancer Cell Response to Chemotherapy by Negatively Regulating Akt. Cancer Cell 2009, 16, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Wang, L. FKBP5 as a Selection Biomarker for Gemcitabine and Akt Inhibitors in Treatment of Pancreatic Cancer. PLoS ONE 2012, 7, e36252. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, D.; He, Y.; Li, L.; Liu, S.; Lu, J.; Gui, H.; Wang, Y.; Tao, Y.; Wang, H.; et al. Rapamycin Inhibits AR Signaling Pathway in Prostate Cancer by Interacting with the FK1 Domain of FKBP51. Biochem. Biophys. Rep. 2020, 23, 100778. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Bai, Q.; Zhou, S.; Liu, X.; Liu, H.; Yao, X. Molecular Dynamics Simulation, Binding Free Energy Calculation and Unbinding Pathway Analysis on Selectivity Difference between FKBP51 and FKBP52: Insight into the Molecular Mechanism of Isoform Selectivity. Proteins Struct. Funct. Bioinforma. 2018, 86, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, M.; Dubois-Deruy, E.; Turkieh, A.; Mulder, P.; Peugnet, V.; Chwastyniak, M.; Beseme, O.; Dechaumes, A.; Amouyel, P.; Richard, V.; et al. Desmin Aggrephagy in Rat and Human Ischemic Heart Failure through PKCζ and GSK3β as Upstream Signaling Pathways. Cell Death Discov. 2021, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Tannous, P.; Zhu, H.; Johnstone, J.L.; Shelton, J.M.; Rajasekaran, N.S.; Benjamin, I.J.; Nguyen, L.; Gerard, R.D.; Levine, B.; Rothermel, B.A.; et al. Autophagy Is an Adaptive Response in Desmin-Related Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2008, 105, 9745–9750. [Google Scholar] [CrossRef] [Green Version]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prévost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tomé, F.; Dupret, J.M.; et al. A Missense Mutation in the AlphaB-Crystallin Chaperone Gene Causes a Desmin-Related Myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef]
- Omary, M.B.; Coulombe, P.A.; McLean, W.H.I. Intermediate Filament Proteins and Their Associated Diseases. N. Engl. J. Med. 2004, 351, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Capetanaki, Y.; Papathanasiou, S.; Diokmetzidou, A.; Vatsellas, G.; Tsikitis, M. Desmin Related Disease: A Matter of Cell Survival Failure. Curr. Opin. Cell Biol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schänzer, A.; Rupp, S.; Gräf, S.; Zengeler, D.; Jux, C.; Akintürk, H.; Gulatz, L.; Mazhari, N.; Acker, T.; Van Coster, R.; et al. Dysregulated Autophagy in Restrictive Cardiomyopathy Due to Pro209Leu Mutation in BAG3. Mol. Genet. Metab. 2018, 123, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Li, Y.; Wang, X.; Sun, B.; Fang, W.; Jiang, S.; Liang, C. CRYAB Inhibits Migration and Invasion of Bladder Cancer Cells through the PI3K/AKT and ERK Pathways. Jpn. J. Clin. Oncol. 2020, 50, 254–260. [Google Scholar] [CrossRef]
- Tao, X.; Cheng, L.; Li, Y.; Ci, H.; Xu, J.; Wu, S.; Tao, Y. Expression of CRYAB with the Angiogenesis and Poor Prognosis for Human Gastric Cancer. Medicine 2019, 98, e17799. [Google Scholar] [CrossRef]
- Shi, C.; Yang, X.; Bu, X.; Hou, N.; Chen, P. Alpha B-Crystallin Promotes the Invasion and Metastasis of Colorectal Cancer via Epithelial-Mesenchymal Transition. Biochem. Biophys. Res. Commun. 2017, 489, 369–374. [Google Scholar] [CrossRef]
- Shi, Q.-M.; Luo, J.; Wu, K.; Yin, M.; Gu, Y.-R.; Cheng, X.-G. High Level of AB-Crystallin Contributes to the Progression of Osteosarcoma. Oncotarget 2016, 7, 9007–9016. [Google Scholar] [CrossRef] [Green Version]
- Ousman, S.S.; Tomooka, B.H.; van Noort, J.M.; Wawrousek, E.F.; O’Connor, K.C.; Hafler, D.A.; Sobel, R.A.; Robinson, W.H.; Steinman, L. Protective and Therapeutic Role for AlphaB-Crystallin in Autoimmune Demyelination. Nature 2007, 448, 474–479. [Google Scholar] [CrossRef]
- van Noort, J.M.; Bsibsi, M.; Gerritsen, W.H.; van der Valk, P.; Bajramovic, J.J.; Steinman, L.; Amor, S. Alphab-Crystallin Is a Target for Adaptive Immune Responses and a Trigger of Innate Responses in Preactive Multiple Sclerosis Lesions. J. Neuropathol. Exp. Neurol. 2010, 69, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Kuerten, S.; Lanz, T.V.; Lingampalli, N.; Lahey, L.J.; Kleinschnitz, C.; Mäurer, M.; Schroeter, M.; Braune, S.; Ziemssen, T.; Ho, P.P.; et al. Autoantibodies against Central Nervous System Antigens in a Subset of B Cell–Dominant Multiple Sclerosis Patients. Proc. Natl. Acad. Sci. USA 2020, 117, 21512–21518. [Google Scholar] [CrossRef]
- Noort, J.M.; van Bsibsi, M.; Nacken, P.J.; Verbeek, R.; Venneker, E.H.G. Therapeutic Intervention in Multiple Sclerosis with Alpha B-Crystallin: A Randomized Controlled Phase IIa Trial. PLoS ONE 2015, 10, e0143366. [Google Scholar] [CrossRef] [Green Version]
- van Noort, J.M.; van Sechel, A.C.; Bajramovic, J.J.; Ouagmiri, M.E.; Polman, C.H.; Lassmann, H.; Ravid, R. The Small Heat-Shock Protein AB-Crystallin as Candidate Autoantigen in Multiple Sclerosis. Nature 1995, 375, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Skowyra, D.; Elledge, S.J.; Harper, J.W.; Hieter, P. SGT1 Encodes an Essential Component of the Yeast Kinetochore Assembly Pathway and a Novel Subunit of the SCF Ubiquitin Ligase Complex. Mol. Cell 1999, 4, 21–33. [Google Scholar] [CrossRef]
- Niikura, Y.; Kitagawa, K. Identification of a Novel Splice Variant: Human SGT1B (SUGT1B). DNA Seq. 2003, 14, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-T.; Jacob, J.; Michowski, W.; Nowotny, M.; Kuznicki, J.; Chazin, W.J. Human Sgt1 Binds HSP90 through the CHORD-Sgt1 Domain and Not the Tetratricopeptide Repeat Domain. J. Biol. Chem. 2004, 279, 16511–16517. [Google Scholar] [CrossRef] [Green Version]
- Steensgaard, P.; Garrè, M.; Muradore, I.; Transidico, P.; Nigg, E.A.; Kitagawa, K.; Earnshaw, W.C.; Faretta, M.; Musacchio, A. Sgt1 Is Required for Human Kinetochore Assembly. EMBO Rep. 2004, 5, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo-Brenni, M.C.; Thomas, S.; Bouck, D.C.; Kaplan, K.B. Sgt1p and Skp1p Modulate the Assembly and Turnover of CBF3 Complexes Required for Proper Kinetochore Function. Mol. Biol. Cell 2004, 15, 3366–3378. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.E.; Kaplan, K.B. Hsp90–Sgt1 and Skp1 Target Human Mis12 Complexes to Ensure Efficient Formation of Kinetochore–Microtubule Binding Sites. J. Cell Biol. 2010, 189, 261–274. [Google Scholar] [CrossRef]
- Zabka, M.; Leśniak, W.; Prus, W.; Kuźnicki, J.; Filipek, A. Sgt1 Has Co-Chaperone Properties and Is up-Regulated by Heat Shock. Biochem. Biophys. Res. Commun. 2008, 370, 179–183. [Google Scholar] [CrossRef]
- Spiechowicz, M.; Bernstein, H.-G.; Dobrowolny, H.; Leśniak, W.; Mawrin, C.; Bogerts, B.; Kuźnicki, J.; Filipek, A. Density of Sgt1-Immunopositive Neurons Is Decreased in the Cerebral Cortex of Alzheimer’s Disease Brain. Neurochem. Int. 2006, 49, 487–493. [Google Scholar] [CrossRef]
- Bohush, A.; Niewiadomska, G.; Weis, S.; Filipek, A. HSP90 and Its Novel Co-Chaperones, SGT1 and CHP-1, in Brain of Patients with Parkinson’s Disease and Dementia with Lewy Bodies. J. Park. Dis. 2019, 9, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.-B.; Shao, Y.-M.; Miao, S.; Wang, L. The Diversity of the DnaJ/Hsp40 Family, the Crucial Partners for Hsp70 Chaperones. Cell. Mol. Life Sci. CMLS 2006, 63, 2560–2570. [Google Scholar] [CrossRef]
- Gorenberg, E.L.; Chandra, S.S. The Role of Co-Chaperones in Synaptic Proteostasis and Neurodegenerative Disease. Front. Neurosci. 2017, 11, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.M.; Prodromou, N.V.; Webb, T.R.; Gallo, J.-M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The Ataxia Protein Sacsin Is a Functional Co-Chaperone That Protects against Polyglutamine-Expanded Ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef] [Green Version]
- Panayi, G.S.; Corrigall, V.M. Immunoglobulin Heavy-Chain-Binding Protein (BiP): A Stress Protein That Has the Potential to Be a Novel Therapy for Rheumatoid Arthritis. Biochem. Soc. Trans. 2014, 42, 1752–1755. [Google Scholar] [CrossRef] [PubMed]
- Naseri, N.; Sharma, M.; Velinov, M. Autosomal Dominant Neuronal Ceroid Lipofuscinosis: Clinical Features and Molecular Basis. Clin. Genet. 2021, 99, 111–118. [Google Scholar] [CrossRef]
- Ichhaporia, V.P.; Hendershot, L.M. Role of the HSP70 Co-Chaperone SIL1 in Health and Disease. Int. J. Mol. Sci. 2021, 22, 1564. [Google Scholar] [CrossRef]
- Synofzik, M.; Schüle, R.; Schulze, M.; Gburek-Augustat, J.; Schweizer, R.; Schirmacher, A.; Krägeloh-Mann, I.; Gonzalez, M.; Young, P.; Züchner, S.; et al. Phenotype and Frequency of STUB1 Mutations: Next-Generation Screenings in Caucasian Ataxia and Spastic Paraplegia Cohorts. Orphanet J. Rare Dis. 2014, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Benitez, B.A.; Alvarado, D.; Cai, Y.; Mayo, K.; Chakraverty, S.; Norton, J.; Morris, J.C.; Sands, M.S.; Goate, A.; Cruchaga, C. Exome-Sequencing Confirms DNAJC5 Mutations as Cause of Adult Neuronal Ceroid-Lipofuscinosis. PLoS ONE 2011, 6, e26741. [Google Scholar] [CrossRef] [Green Version]
- Nosková, L.; Stránecký, V.; Hartmannová, H.; Přistoupilová, A.; Barešová, V.; Ivánek, R.; Hůlková, H.; Jahnová, H.; van der Zee, J.; Staropoli, J.F.; et al. Mutations in DNAJC5, Encoding Cysteine-String Protein Alpha, Cause Autosomal-Dominant Adult-Onset Neuronal Ceroid Lipofuscinosis. Am. J. Hum. Genet. 2011, 89, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkovic, S.F.; Staropoli, J.F.; Carpenter, S.; Oliver, K.L.; Kmoch, S.; Anderson, G.W.; Damiano, J.A.; Hildebrand, M.S.; Sims, K.B.; Cotman, S.L.; et al. Diagnosis and Misdiagnosis of Adult Neuronal Ceroid Lipofuscinosis (Kufs Disease). Neurology 2016, 87, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Chacón, R.; Wölfel, M.; Nishimune, H.; Tabares, L.; Schmitz, F.; Castellano-Muñoz, M.; Rosenmund, C.; Montesinos, M.L.; Sanes, J.R.; Schneggenburger, R.; et al. The Synaptic Vesicle Protein CSP Alpha Prevents Presynaptic Degeneration. Neuron 2004, 42, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.C.; Swayne, L.A.; Chen, L.; Feng, Z.-P.; Wacker, J.L.; Muchowski, P.J.; Zamponi, G.W.; Braun, J.E.A. Cysteine String Protein (CSP) Inhibition of N-Type Calcium Channels Is Blocked by Mutant Huntingtin. J. Biol. Chem. 2003, 278, 53072–53081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network Organization of the Huntingtin Proteomic Interactome in Mammalian Brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantej, J.; Polasik, K.; Piotrowska, E.; Tukaj, S. Autoantibodies to Heat Shock Proteins 60, 70, and 90 in Patients with Rheumatoid Arthritis. Cell Stress Chaperones 2019, 24, 283–287. [Google Scholar] [CrossRef]
- Tukaj, S.; Kotlarz, A.; Jóźwik, A.; Smoleńska, Z.; Bryl, E.; Witkowski, J.M.; Lipińska, B. Cytokines of the Th1 and Th2 Type in Sera of Rheumatoid Arthritis Patients; Correlations with Anti-Hsp40 Immune Response and Diagnostic Markers. Acta Biochim. Pol. 2010, 57. [Google Scholar] [CrossRef] [Green Version]
- Tukaj, S.; Kotlarz, A.; Jozwik, A.; Smolenska, Z.; Bryl, E.; Witkowski, J.M.; Lipinska, B. Hsp40 Proteins Modulate Humoral and Cellular Immune Response in Rheumatoid Arthritis Patients. Cell Stress Chaperones 2010, 15, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Kotlarz, A.; Tukaj, S.; Krzewski, K.; Brycka, E.; Lipinska, B. Human Hsp40 Proteins, DNAJA1 and DNAJA2, as Potential Targets of the Immune Response Triggered by Bacterial DnaJ in Rheumatoid Arthritis. Cell Stress Chaperones 2013, 18, 653–659. [Google Scholar] [CrossRef] [Green Version]
- Koffeman, E.C.; Genovese, M.; Amox, D.; Keogh, E.; Santana, E.; Matteson, E.L.; Kavanaugh, A.; Molitor, J.A.; Schiff, M.H.; Posever, J.O.; et al. Epitope-Specific Immunotherapy of Rheumatoid Arthritis: Clinical Responsiveness Occurs with Immune Deviation and Relies on the Expression of a Cluster of Molecules Associated with T Cell Tolerance in a Double-Blind, Placebo-Controlled, Pilot Phase II Trial. Arthritis Rheum. 2009, 60, 3207–3216. [Google Scholar] [CrossRef] [PubMed]
- Gestwicki, J.E.; Shao, H. Inhibitors and Chemical Probes for Molecular Chaperone Networks. J. Biol. Chem. 2019, 294, 2151–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, J.; Blagg, B.S.J. The Right Tool for the Job: An Overview of Hsp90 Inhibitors. Adv. Exp. Med. Biol. 2020, 1243, 135–146. [Google Scholar] [CrossRef]
- Banerjee, M.; Hatial, I.; Keegan, B.M.; Blagg, B.S.J. Assay Design and Development Strategies for Finding Hsp90 Inhibitors and Their Role in Human Diseases. Pharmacol. Ther. 2021, 221, 107747. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Barua, S.; Huang, M.Y.; Park, J.; Yenari, M.A.; Lee, J.E. Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury. Cells 2020, 9, 2020. [Google Scholar] [CrossRef]
- Deniset, J.F.; Pierce, G.N. Heat Shock Proteins: Mediators of Atherosclerotic Development. Curr. Drug Targets 2015, 16, 816–826. [Google Scholar] [CrossRef]
- Der Sarkissian, S.; Aceros, H.; Williams, P.-M.; Scalabrini, C.; Borie, M.; Noiseux, N. Heat Shock Protein 90 Inhibition and Multi-Target Approach to Maximize Cardioprotection in Ischaemic Injury. Br. J. Pharmacol. 2020, 177, 3378–3388. [Google Scholar] [CrossRef]
- Costa, T.E.M.M.; Raghavendra, N.M.; Penido, C. Natural Heat Shock Protein 90 Inhibitors in Cancer and Inflammation. Eur. J. Med. Chem. 2020, 189, 112063. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N. Heat Shock Protein 90 Inhibition in the Inflamed Lungs. Cell Stress Chaperones 2020, 25, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Serwetnyk, M.A.; Blagg, B.S.J. The Disruption of Protein-Protein Interactions with Co-Chaperones and Client Substrates as a Strategy towards Hsp90 Inhibition. Acta Pharm. Sin. B 2021, 11, 1446–1468. [Google Scholar] [CrossRef]
- Bickel, D.; Gohlke, H. C-Terminal Modulators of Heat Shock Protein of 90 kDa (HSP90): State of Development and Modes of Action. Bioorg. Med. Chem. 2019, 27, 115080. [Google Scholar] [CrossRef]
- Enthammer, M.; Papadakis, E.S.; Gachet, M.S.; Deutsch, M.; Schwaiger, S.; Koziel, K.; Ashraf, M.I.; Khalid, S.; Wolber, G.; Packham, G.; et al. Isolation of a Novel Thioflavin S–Derived Compound That Inhibits BAG-1–Mediated Protein Interactions and Targets BRAF Inhibitor–Resistant Cell Lines. Mol. Cancer Ther. 2013, 12, 2400–2414. [Google Scholar] [CrossRef] [Green Version]
- Kilbas, P.O.; Akcay, I.M.; Doganay, G.D.; Arisan, E.D. Bag-1 Silencing Enhanced Chemotherapeutic Drug-Induced Apoptosis in MCF-7 Breast Cancer Cells Affecting PI3K/Akt/MTOR and MAPK Signaling Pathways. Mol. Biol. Rep. 2019, 46, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ruan, Q.; Han, S.; Xi, L.; Jiang, W.; Jiang, H.; Ostrov, D.A.; Cai, J. Discovery of Structure-Based Small Molecular Inhibitor of AB-Crystallin against Basal-like/Triple-Negative Breast Cancer Development in Vitro and in Vivo. Breast Cancer Res. Treat. 2014, 145, 45–59. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altinok, S.; Sanchez-Hodge, R.; Stewart, M.; Smith, K.; Schisler, J.C. With or without You: Co-Chaperones Mediate Health and Disease by Modifying Chaperone Function and Protein Triage. Cells 2021, 10, 3121. https://doi.org/10.3390/cells10113121
Altinok S, Sanchez-Hodge R, Stewart M, Smith K, Schisler JC. With or without You: Co-Chaperones Mediate Health and Disease by Modifying Chaperone Function and Protein Triage. Cells. 2021; 10(11):3121. https://doi.org/10.3390/cells10113121
Chicago/Turabian StyleAltinok, Selin, Rebekah Sanchez-Hodge, Mariah Stewart, Kaitlan Smith, and Jonathan C. Schisler. 2021. "With or without You: Co-Chaperones Mediate Health and Disease by Modifying Chaperone Function and Protein Triage" Cells 10, no. 11: 3121. https://doi.org/10.3390/cells10113121
APA StyleAltinok, S., Sanchez-Hodge, R., Stewart, M., Smith, K., & Schisler, J. C. (2021). With or without You: Co-Chaperones Mediate Health and Disease by Modifying Chaperone Function and Protein Triage. Cells, 10(11), 3121. https://doi.org/10.3390/cells10113121