
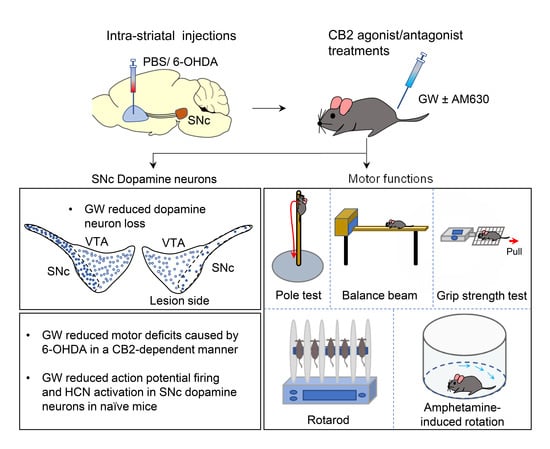
The Neuroprotective Effects of the CB2 Agonist GW842166x in the 6-OHDA Mouse Model of Parkinson’s Disease

 , , , ,
, , , ,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Intra-Striatal 6-OHDA Injection
2.3. Immunohistochemistry (IHC)
2.4. Brain Slicing
2.5. Electrophysiology
2.6. Behavioral Tests
2.6.1. Pole Test
2.6.2. Balance Beam
2.6.3. Grip Strength
2.6.4. Accelerating Rotarod
2.6.5. Spontaneous and Amphetamine-Induced Rotation
2.7. Chemicals
2.8. Statistics
3. Results
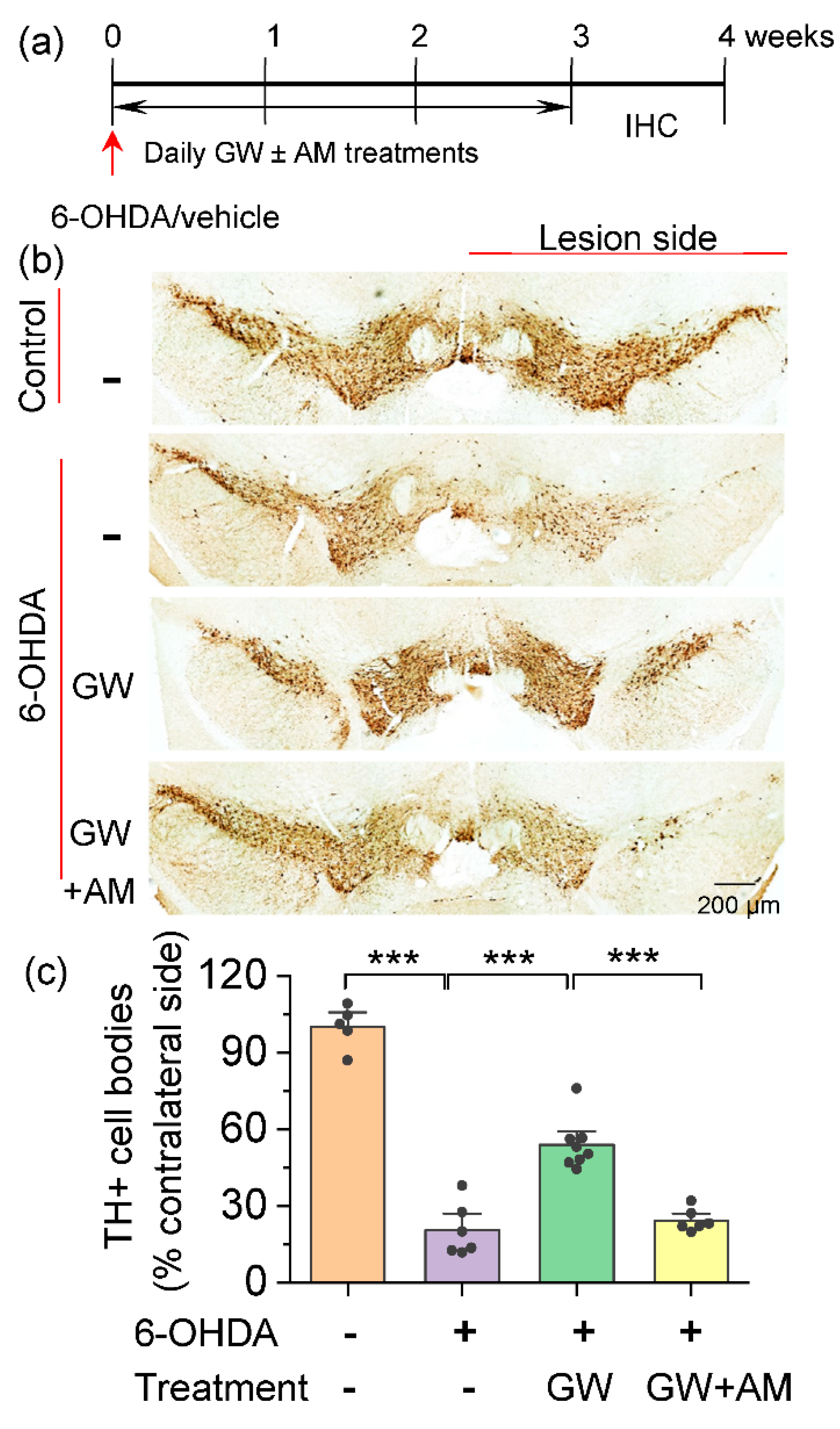
3.1. CB2 Agonism Protected Dopamine Neurons against Degeneration Induced by 6-OHDA

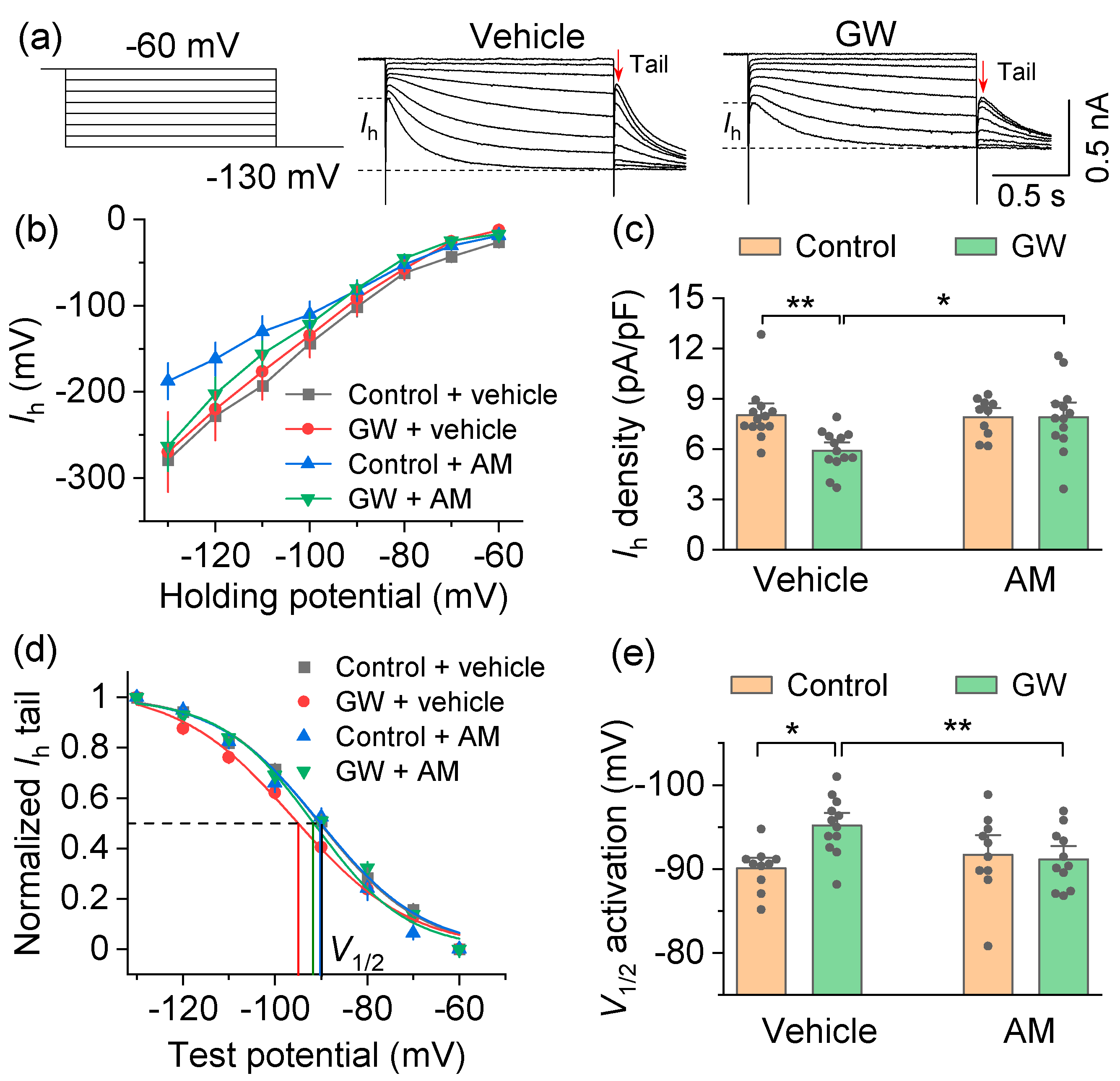
3.2. Mechanisms by Which the CB2 Agonist GW842166x Exerts Neuroprotective Effects
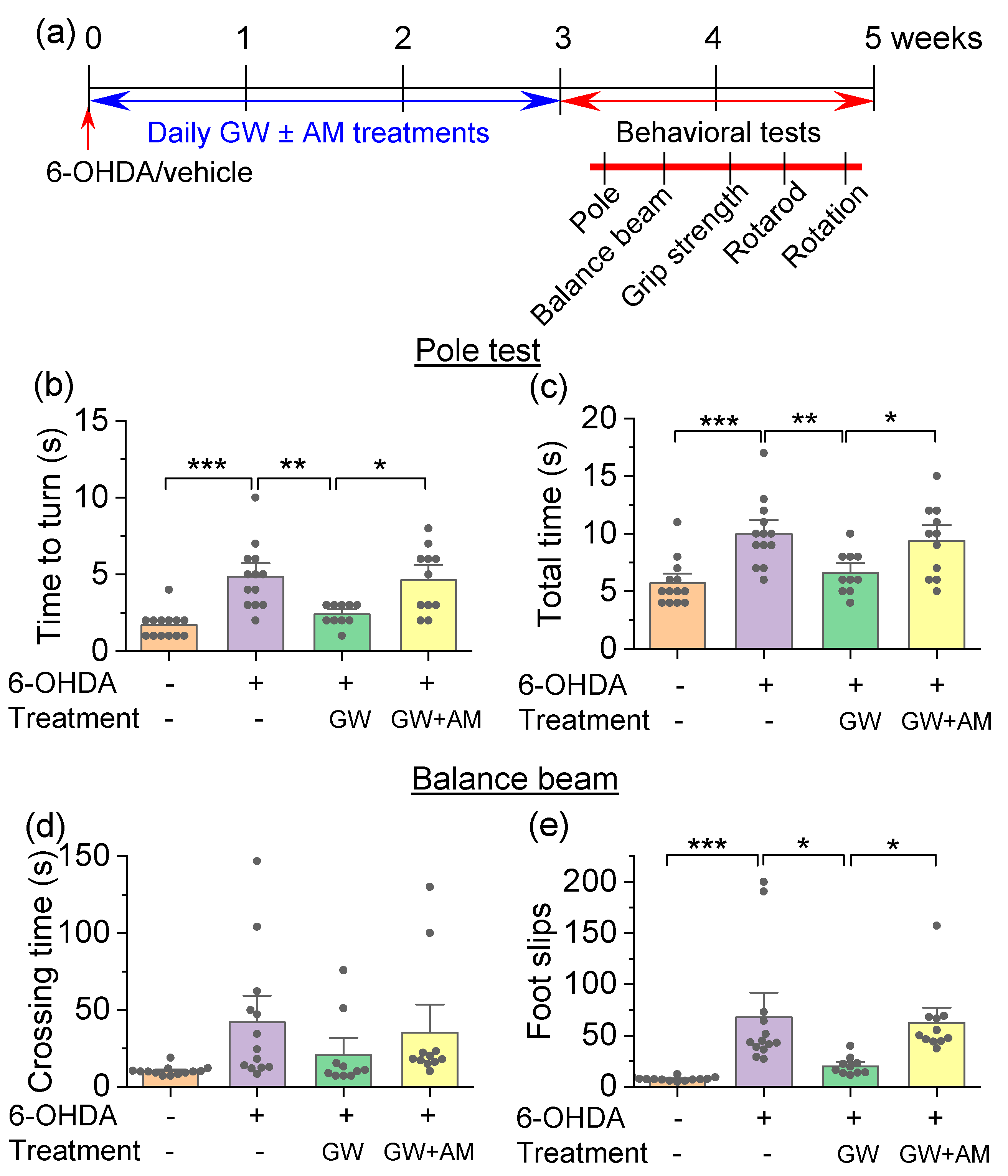
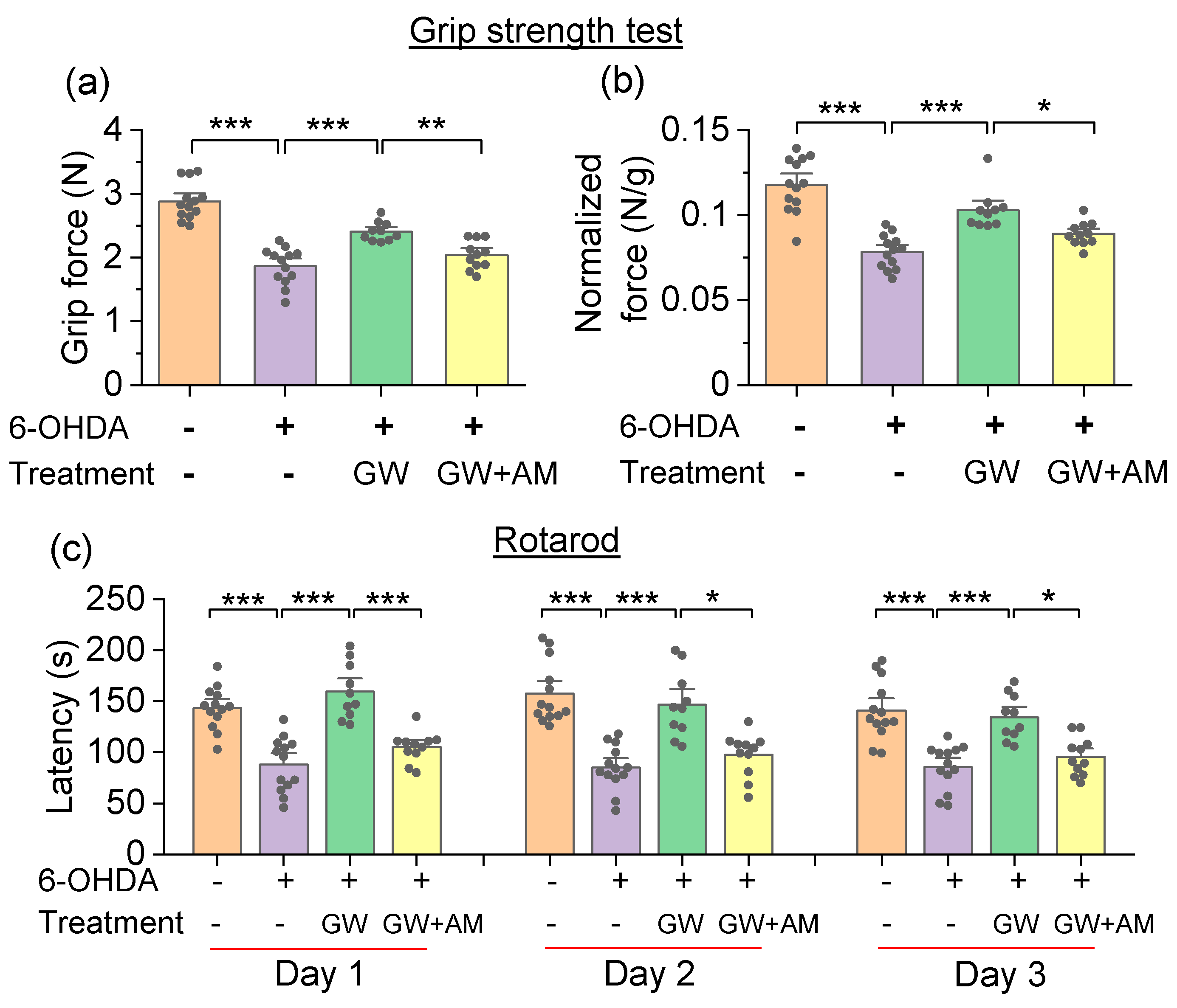
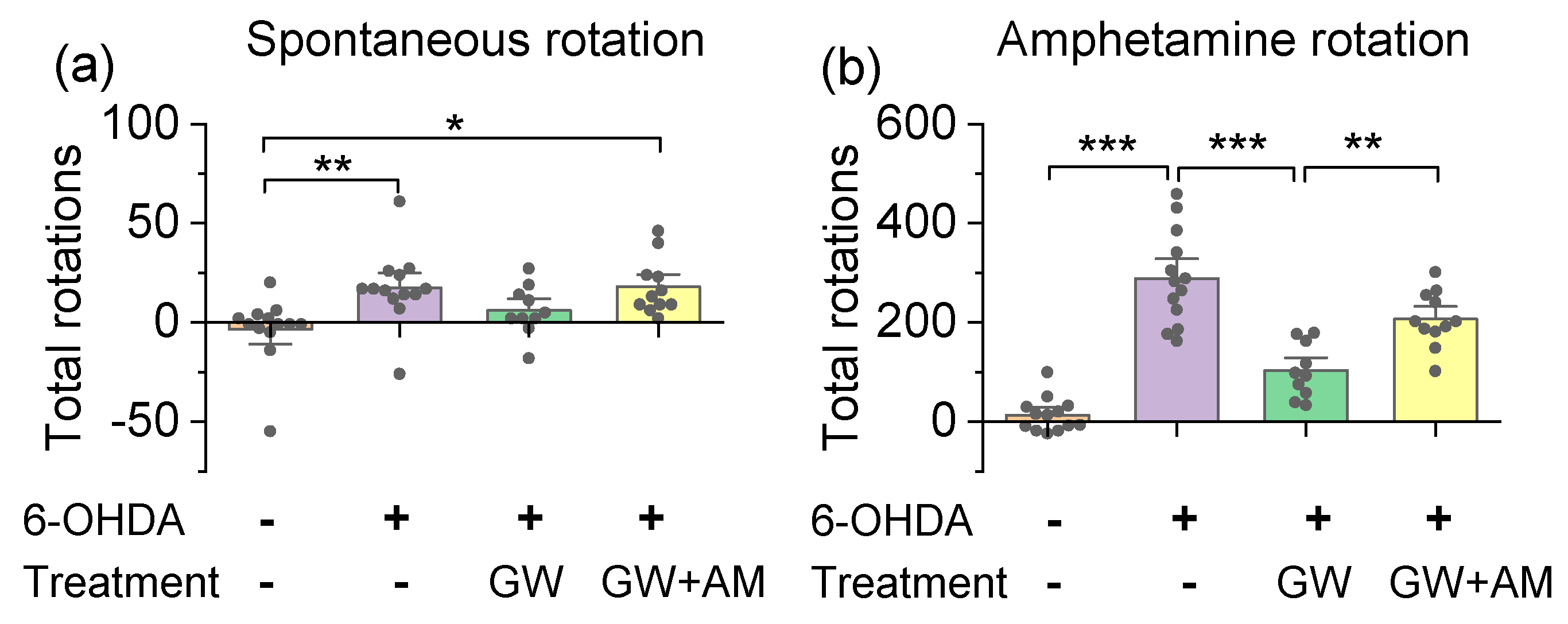
3.3. GW842166x Protected against 6-OHDA-Induced Motor Functions Deficits
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [Green Version]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, P.; Shabbott, B.; Cortes, J.C. Motor control abnormalities in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konta, B.; Frank, W. The treatment of Parkinson’s disease with dopamine agonists. GMS Health Technol. Assess. 2008, 4, Doc05. [Google Scholar]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar] [CrossRef]
- Voon, V.; Napier, T.C.; Frank, M.J.; Sgambato-Faure, V.; Grace, A.A.; Rodriguez-Oroz, M.; Obeso, J.; Bezard, E.; Fernagut, P.O. Impulse control disorders and levodopa-induced dyskinesias in Parkinson’s disease: An update. Lancet Neurol. 2017, 16, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Basavarajappa, B.S.; Shivakumar, M.; Joshi, V.; Subbanna, S. Endocannabinoid system in neurodegenerative disorders. J. Neurochem. 2017, 142, 624–648. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, J.; Wu, Y.; Wang, D.; Feng, G.; Tang, Y.P.; Teng, Z.; Chen, C. Monoacylglycerol lipase is a therapeutic target for Alzheimer’s disease. Cell Rep. 2012, 2, 1329–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.C.; Bok, E.; Huh, S.H.; Park, J.Y.; Yoon, S.H.; Kim, S.R.; Kim, Y.S.; Maeng, S.; Park, S.H.; Jin, B.K. Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J. Immunol. 2011, 187, 6508–6517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Yang, X.; Ma, Y.; Wu, N.; Liu, Z. The CB1 cannabinoid receptor agonist reduces L-DOPA-induced motor fluctuation and ERK1/2 phosphorylation in 6-OHDA-lesioned rats. Drug Des. Dev. Ther. 2014, 8, 2173–2179. [Google Scholar] [CrossRef] [Green Version]
- Walsh, S.; Gorman, A.M.; Finn, D.P.; Dowd, E. The effects of cannabinoid drugs on abnormal involuntary movements in dyskinetic and non-dyskinetic 6-hydroxydopamine lesioned rats. Brain Res. 2010, 1363, 40–48. [Google Scholar] [CrossRef]
- Chung, E.S.; Bok, E.; Chung, Y.C.; Baik, H.H.; Jin, B.K. Cannabinoids prevent lipopolysaccharide-induced neurodegeneration in the rat substantia nigra in vivo through inhibition of microglial activation and NADPH oxidase. Brain Res. 2012, 1451, 110–116. [Google Scholar] [CrossRef]
- Price, D.A.; Martinez, A.A.; Seillier, A.; Koek, W.; Acosta, Y.; Fernandez, E.; Strong, R.; Lutz, B.; Marsicano, G.; Roberts, J.L.; et al. WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Eur. J. Neurosci. 2009, 29, 2177–2186. [Google Scholar] [CrossRef]
- Sieradzan, K.A.; Fox, S.H.; Hill, M.; Dick, J.P.; Crossman, A.R.; Brotchie, J.M. Cannabinoids reduce levodopa-induced dyskinesia in Parkinson’s disease: A pilot study. Neurology 2001, 57, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H.; Henry, B.; Hill, M.; Crossman, A.; Brotchie, J. Stimulation of cannabinoid receptors reduces levodopa-induced dyskinesia in the MPTP-lesioned nonhuman primate model of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2002, 17, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H.; Kellett, M.; Moore, A.P.; Crossman, A.R.; Brotchie, J.M. Randomised, double-blind, placebo-controlled trial to assess the potential of cannabinoid receptor stimulation in the treatment of dystonia. Mov. Disord. Off. J. Mov. Disord. Soc. 2002, 17, 145–149. [Google Scholar] [CrossRef]
- Dhopeshwarkar, A.; Mackie, K. CB2 Cannabinoid receptors as a therapeutic target-what does the future hold? Mol. Pharmacol. 2014, 86, 430–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.Y.; Gao, M.; Liu, Q.R.; Bi, G.H.; Li, X.; Yang, H.J.; Gardner, E.L.; Wu, J.; Xi, Z.X. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E5007–E5015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.C.; Cinquina, V.; Palomo-Garo, C.; Rabano, A.; Fernandez-Ruiz, J. Identification of CB(2) receptors in human nigral neurons that degenerate in Parkinson’s disease. Neurosci. Lett. 2015, 587, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Gao, M.; Shen, H.; Bi, G.H.; Yang, H.J.; Liu, Q.R.; Wu, J.; Gardner, E.L.; Bonci, A.; Xi, Z.X. Expression of functional cannabinoid CB2 receptor in VTA dopamine neurons in rats. Addict. Biol. 2017, 22, 752–765. [Google Scholar] [CrossRef] [Green Version]
- Navarrete, F.; García-Gutiérrez, M.S.; Aracil-Fernández, A.; Lanciego, J.L.; Manzanares, J. Cannabinoid CB1 and CB2 receptors, and monoacylglycerol lipase gene expression alterations in the basal ganglia of patients with Parkinson’s disease. Neurotherapeutics 2018, 15, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid type 2 (CB2) receptors activation protects against oxidative stress and neuroinflammation associated dopaminergic neurodegeneration in rotenone model of Parkinson’s disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Ruiz, J.; Romero, J.; Velasco, G.; Tolon, R.M.; Ramos, J.A.; Guzman, M. Cannabinoid CB2 receptor: A new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007, 28, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, U.; Arbuthnott, G.W. Quantitative recording of rotational behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal dopamine system. Brain Res. 1970, 24, 485–493. [Google Scholar] [CrossRef]
- Von Voigtlander, P.F.; Moore, K.E. Turning behavior of mice with unilateral 6-hydroxydopamine lesions in the striatum: Effects of apomorphine, L-DOPA, amanthadine, amphetamine and other psychomotor stimulants. Neuropharmacology 1973, 12, 451–462. [Google Scholar] [CrossRef]
- Ostenfeld, T.; Price, J.; Albanese, M.; Bullman, J.; Guillard, F.; Meyer, I.; Leeson, R.; Costantin, C.; Ziviani, L.; Nocini, P.F.; et al. A randomized, controlled study to investigate the analgesic efficacy of single doses of the cannabinoid receptor-2 agonist GW842166, ibuprofen or placebo in patients with acute pain following third molar tooth extraction. Clin. J. Pain 2011, 27, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 2009, 62, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ’Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Schumacker, P.T. Calcium, bioenergetics, and neuronal vulnerability in Parkinson’s disease. J. Biol. Chem. 2013, 288, 10736–10741. [Google Scholar] [CrossRef] [Green Version]
- Duda, J.; Potschke, C.; Liss, B. Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 156–178. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Chan, C.S.; Surmeier, D.J. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009, 29, 11011–11019. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.N.; Ilijic, E.; Yang, B.; Sanchez-Padilla, J.; Wokosin, D.; Galtieri, D.; Kondapalli, J.; Schumacker, P.T.; Surmeier, D.J. Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J. Clin. Investig. 2018, 128, 2266–2280. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Gao, F.; Larsen, B.; Gao, M.; Luo, Z.; Chen, D.; Ma, X.; Qiu, S.; Zhou, Y.; Xie, J.; et al. Mechanisms of cannabinoid CB2 receptor-mediated reduction of dopamine neuronal excitability in mouse ventral tegmental area. EBioMedicine 2019, 42, 225–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonito-Oliva, A.; Masini, D.; Fisone, G. A mouse model of non-motor symptoms in Parkinson’s disease: Focus on pharmacological interventions targeting affective dysfunctions. Front. Behav. Neurosci. 2014, 8, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, P.; Vickstrom, C.R.; Liu, X.; Hu, Y.; Yu, L.; Yu, H.G.; Liu, Q.S. HCN2 channels in the ventral tegmental area regulate behavioral responses to chronic stress. eLife 2018, 7, e32420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, J.T.; Lee, B.R.; Chong, P.; Soler-Llavina, G.; Cobbs, C.; Koch, C.; Zeng, H.; Lein, E. Preparation of acute brain slices using an optimized N-Methyl-D-Glucamine protective recovery method. J. Vis. Exp. 2018, 132, 53825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, K.; Kabuto, H.; Makino, H.; Ogawa, N. Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods 1997, 73, 45–48. [Google Scholar] [CrossRef]
- Castro, B.; Kuang, S. Evaluation of muscle performance in mice by treadmill exhaustion test and whole-limb grip strength assay. Bio. Protoc. 2017, 7, e2237. [Google Scholar] [CrossRef] [PubMed]
- Mandillo, S.; Tucci, V.; Holter, S.M.; Meziane, H.; Banchaabouchi, M.A.; Kallnik, M.; Lad, H.V.; Nolan, P.M.; Ouagazzal, A.M.; Coghill, E.L.; et al. Reliability, robustness, and reproducibility in mouse behavioral phenotyping: A cross-laboratory study. Physiol. Genom. 2008, 34, 243–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björklund, A.; Dunnett, S.B. The amphetamine induced rotation test: A re-assessment of its use as a tool to monitor motor impairment and functional recovery in rodent models of Parkinson’s disease. J. Parkinson’s Dis. 2019, 9, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, S.J.; DiLella, A.G.; Lis, E.V.; Jones, K.; Levine, D.M.; Stone, D.J.; Hess, J.F. Molecular profiling of a 6-hydroxydopamine model of Parkinson’s disease. Neurochem. Res. 2010, 35, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Giblin, G.M.; O’Shaughnessy, C.T.; Naylor, A.; Mitchell, W.L.; Eatherton, A.J.; Slingsby, B.P.; Rawlings, D.A.; Goldsmith, P.; Brown, A.J.; Haslam, C.P.; et al. Discovery of 2-[(2,4-dichlorophenyl)amino]-N-[(tetrahydro- 2H-pyran-4-yl)methyl]-4-(trifluoromethyl)- 5-pyrimidinecarboxamide, a selective CB2 receptor agonist for the treatment of inflammatory pain. J. Med. Chem. 2007, 50, 2597–2600. [Google Scholar] [CrossRef]
- Ross, R.A.; Brockie, H.C.; Stevenson, L.A.; Murphy, V.L.; Templeton, F.; Makriyannis, A.; Pertwee, R.G. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br. J. Pharmacol. 1999, 126, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Philippart, F.; Destreel, G.; Merino-Sepulveda, P.; Henny, P.; Engel, D.; Seutin, V. Differential somatic Ca2+ channel profile in midbrain dopaminergic neurons. J. Neurosci. 2016, 36, 7234–7245. [Google Scholar] [CrossRef] [Green Version]
- German, D.C.; Manaye, K.F.; Sonsalla, P.K.; Brooks, B.A. Midbrain dopaminergic cell loss in Parkinson’s disease and MPTP-induced parkinsonism: Sparing of calbindin-D28k-containing cells. Ann. N. Y. Acad. Sci. 1992, 648, 42–62. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N. Engl. J. Med. 1988, 318, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Ibsen, M.S.; Connor, M.; Glass, M. Cannabinoid CB1 and CB2 receptor signaling and bias. Cannabis Cannabinoid Res. 2017, 2, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biglan, K.M.; Oakes, D.; Lang, A.E.; Hauser, R.A.; Hodgeman, K.; Greco, B.; Lowell, J.; Rockhill, R.; Shoulson, I.; Venuto, C.; et al. A novel design of a Phase III trial of isradipine in early Parkinson disease (STEADY-PD III). Ann. Clin. Transl. Neurol. 2017, 4, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. Handb. Exp. Pharmacol. 2009, 191, 111–136. [Google Scholar] [CrossRef]
- Wainger, B.J.; DeGennaro, M.; Santoro, B.; Siegelbaum, S.A.; Tibbs, G.R. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 2001, 411, 805–810. [Google Scholar] [CrossRef]
- Franz, O.; Liss, B.; Neu, A.; Roeper, J. Single-cell mRNA expression of HCN1 correlates with a fast gating phenotype of hyperpolarization-activated cyclic nucleotide-gated ion channels (Ih) in central neurons. Eur. J. Neurosci. 2000, 12, 2685–2693. [Google Scholar] [CrossRef] [PubMed]
- Santoro, B.; Chen, S.; Luthi, A.; Pavlidis, P.; Shumyatsky, G.P.; Tibbs, G.R.; Siegelbaum, S.A. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J. Neurosci. 2000, 20, 5264–5275. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, S.; Siegelbaum, S.A. Regulation of hyperpolarization-activated HCN channel gating and cAMP modulation due to interactions of COOH terminus and core transmembrane regions. J. Gen. Physiol. 2001, 118, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Jackson-Lewis, V.; Blesa, J.; Przedborski, S. Animal models of Parkinson’s disease. Park. Relat. Disord. 2012, 18 (Suppl. S1), S183–S185. [Google Scholar] [CrossRef]
- Konnova, E.A.; Swanberg, M. Animal models of Parkinson’s disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar] [CrossRef]
- Shi, J.; Cai, Q.; Zhang, J.; He, X.; Liu, Y.; Zhu, R.; Jin, L. AM1241 alleviates MPTP-induced Parkinson’s disease and promotes the regeneration of DA neurons in PD mice. Oncotarget 2017, 8, 67837–67850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, C.; Palomo-Garo, C.; García-Arencibia, M.; Ramos, J.A.; Pertwee, R.G.; Fernández-Ruiz, J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Δ(9)-THCV in animal models of Parkinson’s disease. Br. J. Pharmacol. 2011, 163, 1495–1506. [Google Scholar] [CrossRef] [Green Version]
- Heuer, A.; Smith, G.A.; Lelos, M.J.; Lane, E.L.; Dunnett, S.B. Unilateral nigrostriatal 6-hydroxydopamine lesions in mice I: Motor impairments identify extent of dopamine depletion at three different lesion sites. Behav. Brain Res. 2012, 228, 30–43. [Google Scholar] [CrossRef]
- Gambardella, C.; Pignatelli, A.; Belluzzi, O. The h-current in the substantia Nigra pars compacta neurons: A re-examination. PLoS ONE 2012, 7, e52329. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Baltazar, D.; Zavala-Flores, L.M.; Villanueva-Olivo, A. The 6-hydroxydopamine model and parkinsonian pathophysiology: Novel findings in an older model. Neurologia 2017, 32, 533–539. [Google Scholar] [CrossRef]
- Concannon, R.M.; Okine, B.N.; Finn, D.P.; Dowd, E. Differential upregulation of the cannabinoid CB(2) receptor in neurotoxic and inflammation-driven rat models of Parkinson’s disease. Exp. Neurol. 2015, 269, 133–141. [Google Scholar] [CrossRef]
- Han, Q.W.; Yuan, Y.H.; Chen, N.H. The therapeutic role of cannabinoid receptors and its agonists or antagonists in Parkinson’s disease. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2020, 96, 109745. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Liu, X.; Chen, B.; Vickstrom, C.R.; Friedman, V.; Kelly, T.J.; Bai, X.; Zhao, L.; Hillard, C.J.; Liu, Q.-S. The Neuroprotective Effects of the CB2 Agonist GW842166x in the 6-OHDA Mouse Model of Parkinson’s Disease. Cells 2021, 10, 3548. https://doi.org/10.3390/cells10123548
Yu H, Liu X, Chen B, Vickstrom CR, Friedman V, Kelly TJ, Bai X, Zhao L, Hillard CJ, Liu Q-S. The Neuroprotective Effects of the CB2 Agonist GW842166x in the 6-OHDA Mouse Model of Parkinson’s Disease. Cells. 2021; 10(12):3548. https://doi.org/10.3390/cells10123548
Chicago/Turabian StyleYu, Hao, Xiaojie Liu, Bixuan Chen, Casey R. Vickstrom, Vladislav Friedman, Thomas J. Kelly, Xiaowen Bai, Li Zhao, Cecilia J. Hillard, and Qing-Song Liu. 2021. "The Neuroprotective Effects of the CB2 Agonist GW842166x in the 6-OHDA Mouse Model of Parkinson’s Disease" Cells 10, no. 12: 3548. https://doi.org/10.3390/cells10123548
APA StyleYu, H., Liu, X., Chen, B., Vickstrom, C. R., Friedman, V., Kelly, T. J., Bai, X., Zhao, L., Hillard, C. J., & Liu, Q. -S. (2021). The Neuroprotective Effects of the CB2 Agonist GW842166x in the 6-OHDA Mouse Model of Parkinson’s Disease. Cells, 10(12), 3548. https://doi.org/10.3390/cells10123548