
Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development

 ,
,  ,
, 

Abstract
:1. Introduction: Glioblastoma and Its Heterogeneity
2. The Immunosuppressive Microenvironment of Glioblastoma
3. Immunotherapeutic Strategies for Glioblastoma
3.1. Vaccines
3.2. Oncolytic Viruses
3.3. Immune Checkpoint Inhibitors
3.4. Adoptive Cell Therapies: CAR T and NK Therapy
3.5. Resistance to Immunotherapy and Combinatorial Approaches
4. Advanced In Vitro and Animal Tumor Models for Testing Immunotherapeutic Approaches
4.1. CSCs
4.2. Organotypic Tissue Slices
4.3. Organoids
4.4. Animal Models
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma a randomized clinical trial. JAMA-J. Am. Med. Assoc. 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noch, E.K.; Ramakrishna, R.; Magge, R. Challenges in the Treatment of Glioblastoma: Multisystem Mechanisms of Therapeutic Resistance. World Neurosurg. 2018, 116, 505–517. [Google Scholar] [CrossRef]
- Thomas, A.A.; Brennan, C.W.; DeAngelis, L.M.; Omuro, A.M. Emerging therapies for glioblastoma. JAMA Neurol. 2014, 71, 1437–1444. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA-J. Am. Med. Assoc. 2013, 310, 1842–1850. [Google Scholar] [CrossRef]
- Theodorakis, P.E.; Müller, E.A.; Craster, R.V.; Matar, O.K. Physical insights into the blood-brain barrier translocation mechanisms. Phys. Biol. 2017, 14, 041001. [Google Scholar] [CrossRef] [Green Version]
- Fokas, E.; Steinbach, J.P.; Rödel, C. Biology of brain metastases and novel targeted therapies: Time to translate the research. Biochim. Biophys. Acta-Rev. Cancer 2013, 1835, 61–75. [Google Scholar] [CrossRef]
- Herisson, F.; Frodermann, V.; Courties, G.; Rohde, D.; Sun, Y.; Vandoorne, K.; Wojtkiewicz, G.R.; Masson, G.S.; Vinegoni, C.; Kim, J.; et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat. Neurosci. 2018, 21, 1209–1217. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neurooncol. 2020. [Google Scholar] [CrossRef]
- Oberoi, R.K.; Parrish, K.E.; Sio, T.T.; Mittapalli, R.K.; Elmquist, W.F.; Sarkaria, J.N. Strategies to improve delivery of anticancer drugs across the blood–brain barrier to treat glioblastoma. Neuro. Oncol. 2016, 18, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 1203–1217. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018. [Google Scholar] [CrossRef]
- Teng, J.; Da Hora, C.C.; Kantar, R.S.; Nakano, I.; Wakimoto, H.; Batchelor, T.T.; Antonio Chiocca, E.; Badr, C.E.; Tannous, B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro. Oncol. 2017, 19, 820–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otvos, B.; Silver, D.J.; Mulkearns-Hubert, E.E.; Alvarado, A.G.; Turaga, S.M.; Sorensen, M.D.; Rayman, P.; Flavahan, W.A.; Hale, J.S.; Stoltz, K.; et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression. Cell Stem Cell 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hira, V.V.V.; Aderetti, D.A.; van Noorden, C.J.F. Glioma Stem Cell Niches in Human Glioblastoma Are Periarteriolar. J. Histochem. Cytochem. 2018, 66, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Hira, V.V.V.; Breznik, B.; Vittori, M.; Loncq de Jong, A.; Mlakar, J.; Oostra, R.J.; Khurshed, M.; Molenaar, R.J.; Lah, T.; Van Noorden, C.J.F. Similarities Between Stem Cell Niches in Glioblastoma and Bone Marrow: Rays of Hope for Novel Treatment Strategies. J. Histochem. Cytochem. 2019, 68. [Google Scholar] [CrossRef]
- Ardebili, S.; Zajc, I.; Gole, B.; Campos, B.; Herold-Mende, C.; Drmota, S.; Lah, T. CD133/prominin1 is prognostic for GBM patient’s survival, but inversely correlated with cysteine cathepsins’ expression in glioblastoma derived spheroids. Radiol. Oncol. 2011, 45, 1–13. [Google Scholar] [CrossRef]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Perus, L.J.M.; Walsh, L.A. Microenvironmental Heterogeneity in Brain Malignancies. Front. Immunol. 2019, 10, 2294. [Google Scholar]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M.V. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Lund, H.; Pieber, M.; Parsa, R.; Han, J.; Grommisch, D.; Ewing, E.; Kular, L.; Needhamsen, M.; Espinosa, A.; Nilsson, E.; et al. Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like cells. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mildner, A.; Schmidt, H.; Nitsche, M.; Merkler, D.; Hanisch, U.K.; Mack, M.; Heikenwalder, M.; Brück, W.; Priller, J.; Prinz, M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007, 10, 1544–1553. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/astrocytes–glioblastoma crosstalk: Crucial molecular mechanisms and microenvironmental factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [Green Version]
- Breznik, B.; Motaln, H.; Turnšek, T.L. Proteases and cytokines as mediators of interactions between cancer and stromal cells in tumours. Biol. Chem. 2016, 398, 709–719. [Google Scholar] [CrossRef] [Green Version]
- O’Hayre, M.; Salanga, C.L.; Handel, T.M.; Allen, S.J. Chemokines and cancer: Migration, intracellular signalling and intercellular communication in the microenvironment. Biochem. J. 2008, 409, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Novak, M.; Koprivnikar Krajnc, M.; Hrastar, B.; Breznik, B.; Majc, B.; Mlinar, M.; Rotter, A.; Porčnik, A.; Mlakar, J.; Stare, K.; et al. CCR5-Mediated Signaling Is Involved in Invasion of Glioblastoma Cells in Its Microenvironment. Int. J. Mol. Sci. 2020, 21, 4199. [Google Scholar] [CrossRef]
- Kranjc, M.K.; Novak, M.; Pestell, R.G.; Lah, T.T. Cytokine CCL5 and receptor CCR5 axis in glioblastoma multiforme. Radiol. Oncol. 2019, 53, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neurooncol. 2020. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Antunes, A.R.P.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife 2020, 9. [Google Scholar] [CrossRef]
- Garris, C.S.; Blaho, V.A.; Hla, T.; Han, M.H. Sphingosine-1-phosphate receptor 1 signalling in T cells: Trafficking and beyond. Immunology 2014, 142, 347–353. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Golán, I.; De La Fuente, L.R.; Costoya, J.A. NK cell-based glioblastoma immunotherapy. Cancers 2018, 10, 522. [Google Scholar] [CrossRef] [Green Version]
- Adhikaree, J.; Moreno-Vicente, J.; Kaur, A.P.; Jackson, A.M.; Patel, P.M. Resistance Mechanisms and Barriers to Successful Immunotherapy for Treating Glioblastoma. Cells 2020, 9, 263. [Google Scholar] [CrossRef] [Green Version]
- Lo, H.C.; Xu, Z.; Kim, I.S.; Pingel, B.; Aguirre, S.; Kodali, S.; Liu, J.; Zhang, W.; Muscarella, A.M.; Hein, S.M.; et al. Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat. Cancer 2020, 1, 709–722. [Google Scholar] [CrossRef]
- Mulder, W.M.C.; Bloemena, E.; Stukart, M.J.; Kummer, J.A.; Wagstaff, J.; Scheper, R.J. T cell receptor-ζ and granzyme B expression in mononuclear cell infiltrates in normal colon mucosa and colon carcinoma. Gut 1997, 40, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Nakagomi, H.; Petersson, M.; Magnusson, I.; Juhlin, C.; Matsuda, M.; Mellstedt, H.; Vivier, J.L.T.; Anderson, P.; Kiessling, R. Decreased Expression of the Signal-transducing ζ Chains in Tumor-infiltrating T-Cells and NK Cells of Patients with Colorectal Carcinoma. Cancer Res. 1993, 53, 5610–5612. [Google Scholar]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013, 121, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Pellegatta, S.; Eoli, M.; Frigerio, S.; Antozzi, C.; Bruzzone, M.G.; Cantini, G.; Nava, S.; Anghileri, E.; Cuppini, L.; Cuccarini, V.; et al. The natural killer cell response and tumor debulking are associated with prolonged survival in recurrent glioblastoma patients receiving dendritic cells loaded with autologous tumor lysates. Oncoimmunology 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, C. IDO inhibitors move center stage in immuno-oncology. Nat. Biotechnol. 2015, 33, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3. [Google Scholar] [CrossRef]
- Roy, L.-O.; Poirier, M.-B.; Fortin, D. Transforming growth factor-beta and its implication in the malignancy of gliomas. Target. Oncol. 2015, 10, 1–14. [Google Scholar] [CrossRef]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-β downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro. Oncol. 2010, 12, 7–13. [Google Scholar] [CrossRef]
- Jacobs, J.F.M.; Idema, A.J.; Bol, K.F.; Grotenhuis, J.A.; de Vries, I.J.M.; Wesseling, P.; Adema, G.J. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J. Neuroimmunol. 2010, 225, 195–199. [Google Scholar] [CrossRef]
- El Andaloussi, A.; Lesniak, M.S. CD4+CD25+FoxP3+ T-cell infiltration and heme oxygenase-1 expression correlate with tumor grade in human gliomas. J. Neurooncol. 2007, 83, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Fisher, J.L.; Rahme, G.J.; Hampton, T.H.; Baron, U.; Olek, S.; Schwachula, T.; Rhodes, C.H.; Gui, J.; Tafe, L.J.; et al. Regulatory T cells are not a strong predictor of survival for patients with glioblastoma. Neuro. Oncol. 2015, 17, 801–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Q.; Zhang, X.; Ye, H.X.; Wang, Y.; Du, Z.G.; Yao, Y.; Mao, Y. The prognostic value of Foxp3+ tumor-infiltrating lymphocytes in patients with glioblastoma. J. Neurooncol. 2014, 116, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro. Oncol. 2012, 14, 958–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Vos, K.E.; Abels, E.R.; Zhang, X.; Lai, C.; Carrizosa, E.; Oakley, D.; Prabhakar, S.; Mardini, O.; Crommentuijn, M.H.W.; Skog, J.; et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro. Oncol. 2016, 18, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Stafford, J.H.; Hirai, T.; Deng, L.; Chernikova, S.B.; Urata, K.; West, B.L.; Brown, J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro. Oncol. 2016, 18, 797–806. [Google Scholar] [CrossRef]
- Burger, M.C.; Zhang, C.; Harter, P.N.; Romanski, A.; Strassheimer, F.; Senft, C.; Tonn, T.; Steinbach, J.P.; Wels, W.S. CAR-Engineered NK Cells for the Treatment of Glioblastoma: Turning Innate Effectors Into Precision Tools for Cancer Immunotherapy. Front. Immunol. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Do, A.S.M.S.; Amano, T.; Edwards, L.A.; Zhang, L.; De Peralta-Venturina, M.; Yu, J.S. CD133 mRNA-Loaded Dendritic Cell Vaccination Abrogates Glioma Stem Cell Propagation in Humanized Glioblastoma Mouse Model. Mol. Ther. Oncolytics 2020, 18, 295–303. [Google Scholar] [CrossRef]
- Martikainen, M.; Essand, M. Virus-based immunotherapy of glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Cloughesy, T.F.; Maus, M.V.; Prins, R.M.; Reardon, D.A.; Sonabend, A.M. Emerging immunotherapies for malignant glioma: From immunogenomics to cell therapy. Neuro. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef]
- Lynes, J.P.; Nwankwo, A.K.; Sur, H.P.; Sanchez, V.E.; Sarpong, K.A.; Ariyo, O.I.; Dominah, G.A.; Nduom, E.K. Biomarkers for immunotherapy for treatment of glioblastoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Rampling, R.; Peoples, S.; Mulholland, P.J.; James, A.; Al-Salihi, O.; Twelves, C.J.; McBain, C.; Jefferies, S.; Jackson, A.; Stewart, W.; et al. A cancer research UK first time in human phase i trial of IMA950 (novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin. Cancer Res. 2016, 22, 4776–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 1. [Google Scholar] [CrossRef] [Green Version]
- Eoli, M.; Corbetta, C.; Anghileri, E.; Di Ianni, N.; Milani, M.; Cuccarini, V.; Musio, S.; Paterra, R.; Frigerio, S.; Nava, S.; et al. Expansion of effector and memory T cells is associated with increased survival in recurrent glioblastomas treated with dendritic cell immunotherapy. Neuro-Oncol. Adv. 2019, 1, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A characterization of dendritic cells and their role in immunotherapy in glioblastoma: From preclinical studies to clinical trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef] [Green Version]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef]
- Jiang, H.; Rivera-Molina, Y.; Gomez-Manzano, C.; Clise-Dwyer, K.; Bover, L.; Vence, L.M.; Yuan, Y.; Lang, F.F.; Toniatti, C.; Hossain, M.B.; et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res. 2017, 77, 3894–3907. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, L.A.; Manzanera, A.G.; Bell, S.D.; Cavaliere, R.; McGregor, J.M.; Grecula, J.C.; Newton, H.B.; Lo, S.S.; Badie, B.; Portnow, J.; et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro. Oncol. 2016, 18, 1137–1145. [Google Scholar] [CrossRef] [Green Version]
- Cloughesy, T.F.; Landolfi, J.; Vogelbaum, M.A.; Ostertag, D.; Elder, J.B.; Bloomfield, S.; Carter, B.; Chen, C.C.; Kalkanis, S.N.; Kesari, S.; et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro. Oncol. 2018, 20, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Caffery, B.; Lee, J.S.; Alexander-Bryant, A.A. Vectors for glioblastoma gene therapy: Viral & non-viral delivery strategies. Nanomaterials 2019, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Passaro, C.; Alayo, Q.; De Laura, I.; McNulty, J.; Grauwet, K.; Ito, H.; Bhaskaran, V.; Mineo, M.; Lawler, S.E.; Shah, K.; et al. Arming an oncolytic herpes simplex virus type 1 with a single-chain fragment variable antibody against PD-1 for experimental glioblastoma therapy. Clin. Cancer Res. 2019, 25, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.; Debinski, W. Challenges to successful implementation of the immune checkpoint inhibitors for treatment of glioblastoma. Int. J. Mol. Sci. 2020, 21, 2759. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable Therapeutic Ef fi cacy Utilizing Combinatorial Blockade against IDO, CTLA-4, and PD-L1 in Mice with Brain Tumors. Clin. Cancer Res. 2014, 20. [Google Scholar] [CrossRef] [Green Version]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Khasraw, M.; Reardon, D.A.; Weller, M.; Sampson, J.H. PD-1 Inhibitors: Do they have a Future in the Treatment of Glioblastoma? Clin. Cancer Res. 2020, 26, 5287–5296. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Salinas, R.D.; Durgin, J.S. Rourke, Potential of Glioblastoma—Targeted Chimeric Antigen Receptor (CAR) CAR T Therapy. CNS Drugs. 2020, 34, 127–145. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro. Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e6. [Google Scholar] [CrossRef]
- Jewett, A.; Kos, J.; Kaur, K.; Turnsek, T.L.; Breznik, B.; Senjor, E.; Wong, P.; Nguyen, K.Y.; Ko, M.-W. Multiple defects of Natural Killer cells in cancer patients; Anarchy and dysregulated systemic immunity and immunosuppression in metastatic cancer. Crit. Rev. Immunol. 2020, 40, 93–133. [Google Scholar] [CrossRef]
- Kozlowska, A.K.; Tseng, H.C.; Kaur, K.; Topchyan, P.; Inagaki, A.; Bui, V.T.; Kasahara, N.; Cacalano, N.; Jewett, A. Resistance to cytotoxicity and sustained release of interleukin-6 and interleukin-8 in the presence of decreased interferon-γ after differentiation of glioblastoma by human natural killer cells. Cancer Immunol. Immunother. 2016, 65, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef]
- Sharifzad, F.; Mardpour, S.; Mardpour, S.; Fakharian, E.; Taghikhani, A.; Sharifzad, A.; Kiani, S.; Heydarian, Y.; Łos, M.J.; Azizi, Z.; et al. HSP70/IL-2 treated NK cells effectively cross the blood brain barrier and target tumor cells in a rat model of induced glioblastoma multiforme (GBM). Int. J. Mol. Sci. 2020, 21, 2263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016. [Google Scholar] [CrossRef]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015. [Google Scholar] [CrossRef]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [Green Version]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro. Oncol. 2016, 18, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Karachi, A.; Yang, C.; Dastmalchi, F.; Sayour, E.J.; Huang, J.; Azari, H.; Long, Y.; Flores, C.; Mitchell, D.A.; Rahman, M. Neuro-Oncology Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro Oncol. 2019, 21, 730–741. [Google Scholar] [CrossRef]
- Saha, D.; Rabkin, S.D.; Martuza, R.L. Temozolomide antagonizes oncolytic immunovirotherapy in glioblastoma. J. Immunother. Cancer 2020, 8, 1–8. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Cell Biol. 2006. [Google Scholar] [CrossRef]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef]
- Gromeier, M.; Brown, M.C.; Zhang, G.; Lin, X.; Chen, Y.; Wei, Z.; Beaubier, N.; Yan, H.; He, Y.; Desjardins, A.; et al. Very low mutation burden is a feature of inflamed recurrent glioblastomas responsive to cancer immunotherapy. Nat. Commun. 2021, 12, 352. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta-Rev. Cancer 2010, 1805, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.; Hau, A.; Oudin, A.; Golebiewska, A. Glioblastoma Organoids: Pre-Clinical Applications and Challenges in the Context of Immunotherapy Glioblastoma Organoids: Pre-Clinical Applications and Challenges in the Context of Immunotherapy. Front. Oncol. 2020. [Google Scholar] [CrossRef]
- Goodspeed, A.; Heiser, L.M.; Gray, J.W.; Costello, J.C. Tumor-derived cell lines as molecular models of cancer pharmacogenomics. Mol. Cancer Res. 2016, 14, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, K.; Topchyan, P.; Kozlowska, A.K.; Ohanian, N.; Chiang, J.; Maung, P.O.; Park, S.H.; Ko, M.W.; Fang, C.; Nishimura, I.; et al. Super-charged NK cells inhibit growth and progression of stem-like/poorly differentiated oral tumors in vivo in humanized BLT mice; effect on tumor differentiation and response to chemotherapeutic drugs. Oncoimmunology 2018, 7, e1426518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merz, F.; Gaunitz, F.; Dehghani, F.; Renner, C.; Meixensberger, J.; Gutenberg, A.; Giese, A.; Schopow, K.; Hellwig, C.; Schäfer, M.; et al. Organotypic slice cultures of human glioblastoma reveal different susceptibilities to treatments. Neuro. Oncol. 2013, 15, 670–681. [Google Scholar] [CrossRef]
- Parker, J.J.; Lizarraga, M.; Waziri, A.; Foshay, K.M. A human glioblastoma organotypic slice culture model for study of tumor cell migration and patient-specific effects of anti-invasive drugs. J. Vis. Exp. 2017, 2017, 53557. [Google Scholar] [CrossRef]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Bian, S.; Repic, M.; Guo, Z.; Kavirayani, A.; Burkard, T.; Bagley, J.A.; Krauditsch, C.; Knoblich, J.A. Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 2018. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse models for cancer immunotherapy research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Liu, M.; Chan, X.Y.; Tan, S.Y.; Subramaniam, S.; Fan, Y.; Loh, E.; Chang, K.T.E.; Tan, T.C.; Chen, Q. Uncovering the mystery of opposite circadian rhythms between mouse and human leukocytes in humanized mice. Blood 2017, 130, 1995–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, K.M.; Kim, J.; Jin, J.; Kim, M.; Seol, H.J.; Muradov, J.; Yang, H.; Choi, Y.L.; Park, W.Y.; Kong, D.S.; et al. Patient-Specific Orthotopic Glioblastoma Xenograft Models Recapitulate the Histopathology and Biology of Human Glioblastomas In Situ. Cell Rep. 2013, 3, 260–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Shuen, T.W.H.; Toh, T.B.; Chan, X.Y.; Liu, M.; Tan, S.Y.; Fan, Y.; Yang, H.; Lyer, S.G.; Bonney, G.K.; et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 2018, 67, 1845. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, G.; Cheng, L.; Li, Z.; Xiao, Y.; Deng, Q.; Jiang, Y.; Li, B.; Lin, S.; Wang, S.; et al. Establishment of peripheral blood mononuclear cell-derived humanized lung cancer mouse models for studying efficacy of PD-L1/PD-1 targeted immunotherapy. MAbs 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell 2019, 24, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Mehta, G.; Hsiao, A.Y.; Ingram, M.; Luker, G.D.; Takayama, S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Control. Release 2012, 164, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N. Molecular pathology of malignant gliomas. Annu. Rev. Pathol. 2006, 1, 97–117. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Tannock, I.F.; Lee, C.M.; Tunggal, J.K.; Cowan, D.S.M.; Egorin, M.J. Limited penetration of anticancer drugs through tumor tissue: A potential cause of resistance of solid tumors to chemotherapy. Clin. Cancer Res. 2002, 8, 878–884. [Google Scholar]
- Torras, N.; García-Díaz, M.; Fernández-Majada, V.; Martínez, E. Mimicking epithelial tissues in three-dimensional cell culture models. Front. Bioeng. Biotechnol. 2018, 6, 197. [Google Scholar] [CrossRef]
- Cheema, T.A.; Wakimoto, H.; Fecci, P.E.; Ning, J.; Kuroda, T.; Jeyaretna, D.S.; Martuza, R.L.; Rabkin, S.D. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc. Natl. Acad. Sci. USA 2013, 110, 12006–12011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Seo, Y.D.; Sullivan, K.M.; Pillarisetty, V.G. Establishment of slice cultures as a tool to study the cancer immune microenvironment. Methods Mol. Biol. 2019, 1884, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Izpisua Belmonte, J.C. Organoids—Preclinical models of human disease. N. Engl. J. Med. 2019, 380, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van Den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Lin, M.; Rao, M.; Thompson, H.; Hirai, K.; Choi, M.; Georgakis, G.V.; Sasson, A.R.; Bucobo, J.C.; Tzimas, D.; et al. Development of Patient-Derived Gastric Cancer Organoids from Endoscopic Biopsies and Surgical Tissues. Ann. Surg. Oncol. 2018, 25, 2767–2775. [Google Scholar] [CrossRef]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.M.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopper, O.; de Witte, C.J.; Lõhmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2016, 22, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Passier, R.; Orlova, V.; Mummery, C. Complex Tissue and Disease Modeling using hiPSCs. Cell Stem Cell 2016, 18, 309–321. [Google Scholar] [CrossRef] [Green Version]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e5. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.; Ming, G.; Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR-T cell testing. Nat. Protoc. 2020, 15, 4000–4033. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [Green Version]
- Zumwalde, N.A.; Haag, J.D.; Sharma, D.; Mirrielees, J.A.; Wilke, L.G.; Gould, M.N.; Gumperz, J.E. Analysis of immune cells from human mammary ductal epithelial organoids reveals Vδ2+ T cells that efficiently target breast carcinoma cells in the presence of bisphosphonate. Cancer Prev. Res. 2016, 9, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ootani, A.; Kuo, C. An air-liquid interface culture system for 3D organoid culture of diverse primary gastrointestinal tissues. Methods Mol. Biol. 2016. [Google Scholar] [CrossRef]
- Zhu, Z.; Mesci, P.; Bernatchez, J.A.; Gimple, R.C.; Wang, X.; Schafer, S.T.; Wettersten, H.I.; Beck, S.; Clark, A.E.; Wu, Q.; et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin αvβ5 Axis. Cell Stem Cell 2020, 26, 187–204.e10. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Fakurnejad, S.; Sayegh, E.T.; Clark, A.J.; Ivan, M.E.; Sun, M.Z.; Safaee, M.; Bloch, O.; James, C.D.; Parsa, A.T. Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med. 2014, 12, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in Cancer: Cancer Stem Cells versus Clonal Evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Sun, R.; Shi, B.; Wang, Y.; Di, S.; Luo, H.; Sun, Y.; Li, Z.; Zhou, M.; Jiang, H. Antitumor efficacy of chimeric antigen receptor T cells against EGFRvIII-expressing glioblastoma in C57BL/6 mice. Biomed. Pharmacother. 2019, 113, 108734. [Google Scholar] [CrossRef]
- Lenting, K.; Verhaak, R.; ter Laan, M.; Wesseling, P.; Leenders, W. Glioma: Experimental models and reality. Acta Neuropathol. 2017, 133, 263–282. [Google Scholar] [CrossRef] [Green Version]
- Huszthy, P.C.; Daphu, I.; Niclou, S.P.; Stieber, D.; Nigro, J.M.; Sakariassen, P.O.; Miletic, H.; Thorsen, F.; Bjerkvig, R. In vivo models of primary brain tumors: Pitfalls and perspectives. Neuro. Oncol. 2012, 14, 979–993. [Google Scholar] [CrossRef] [Green Version]
- Federspiel, M.J.; Bates, P.; Young, J.A.T.; Varmus, H.E.; Hughes, S.H. A system for tissue-specific gene targeting: Transgenic mice susceptible to subgroup A avian leukosis virus-based retroviral vectors. Proc. Natl. Acad. Sci. USA 1994, 91, 11241–11245. [Google Scholar] [CrossRef] [Green Version]
- Mathews, S.; Branch Woods, A.; Katano, I.; Makarov, E.; Thomas, M.B.; Gendelman, H.E.; Poluektova, L.Y.; Ito, M.; Gorantla, S. Human Interleukin-34 facilitates microglia-like cell differentiation and persistent HIV-1 infection in humanized mice. Mol. Neurodegener. 2019, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; De Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018. [Google Scholar] [CrossRef] [Green Version]
- Capasso, A.; Lang, J.; Pitts, T.M.; Jordan, K.R.; Lieu, C.H.; Davis, S.L.; Diamond, J.R.; Kopetz, S.; Barbee, J.; Peterson, J.; et al. Characterization of immune responses to anti-PD-1 mono and combination immunotherapy in hematopoietic humanized mice implanted with tumor xenografts. J. Immunother. Cancer 2019, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Kozlowska, A.K.; Topchyan, P.; Ko, M.W.; Ohanian, N.; Chiang, J.; Cook, J.; Maung, P.O.; Park, S.H.; Cacalano, N.; et al. Probiotic-treated super-charged NK cells efficiently clear poorly differentiated pancreatic tumors in Hu-BLT mice. Cancers 2020, 12, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, N.; Flutter, B.; Sanchez Rodriguez, R.; Sharif-Paghaleh, E.; Barber, L.D.; Lombardi, G.; Nestle, F.O. Xenogeneic Graft-versus-Host-Disease in NOD-scid IL-2Rγnull Mice Display a T-Effector Memory Phenotype. PLoS ONE 2012, 7, e44219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Frankenberger, B.; Schendel, D.J. NOD/scid IL-2Rg nullmice: A preclinical model system to evaluate human dendritic cell-based vaccine strategies in vivo. J. Transl. Med. 2012, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, V.; Oyer, J.L.; Igarashi, R.Y.; Gitto, S.B.; Copik, A.J.; Altomare, D.A. Anti-ovarian tumor response of donor peripheral blood mononuclear cells is due to infiltrating cytotoxic NK cells. Oncotarget 2016, 7, 7318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashizawa, T.; Iizuka, A.; Nonomura, C.; Kondou, R.; Maeda, C.; Miyata, H.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; et al. Antitumor effect of Programmed Death-1 (PD-1) blockade in humanized the NOG-MHC double knockout mouse. Clin. Cancer Res. 2017, 23, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]



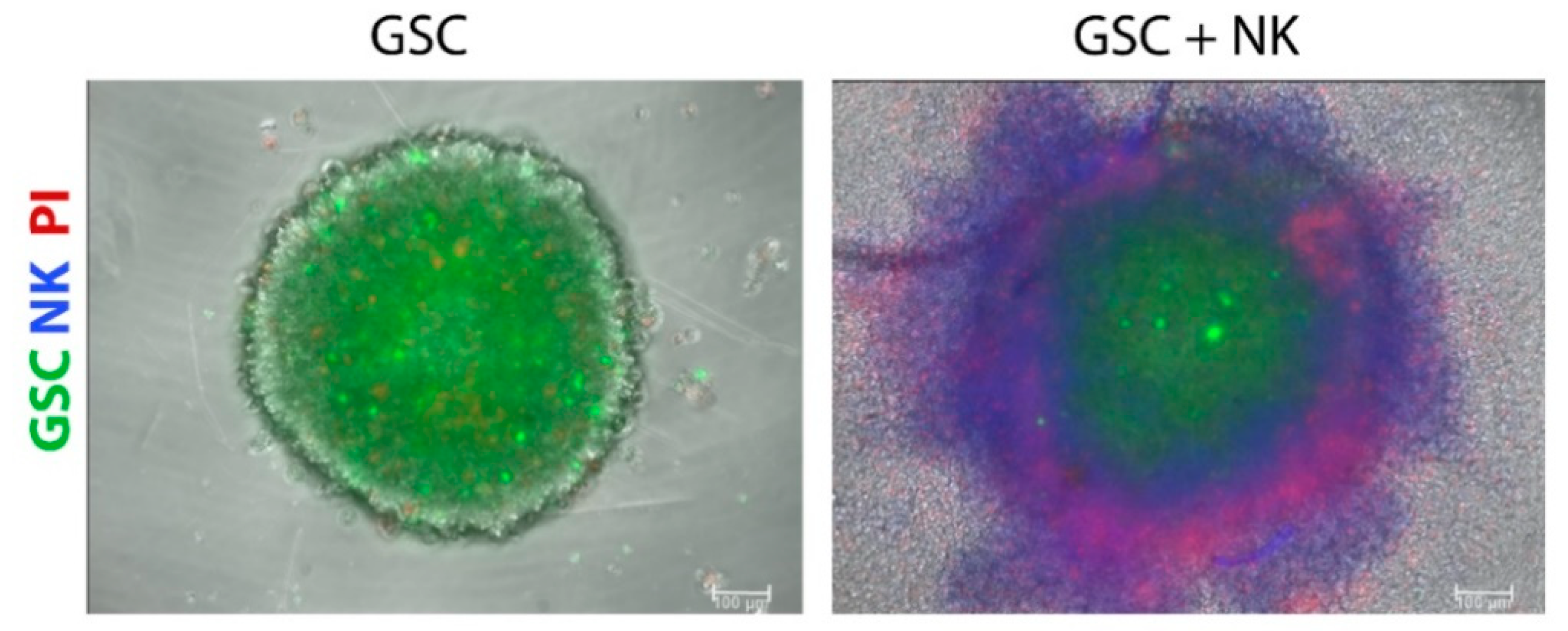{kind=link}
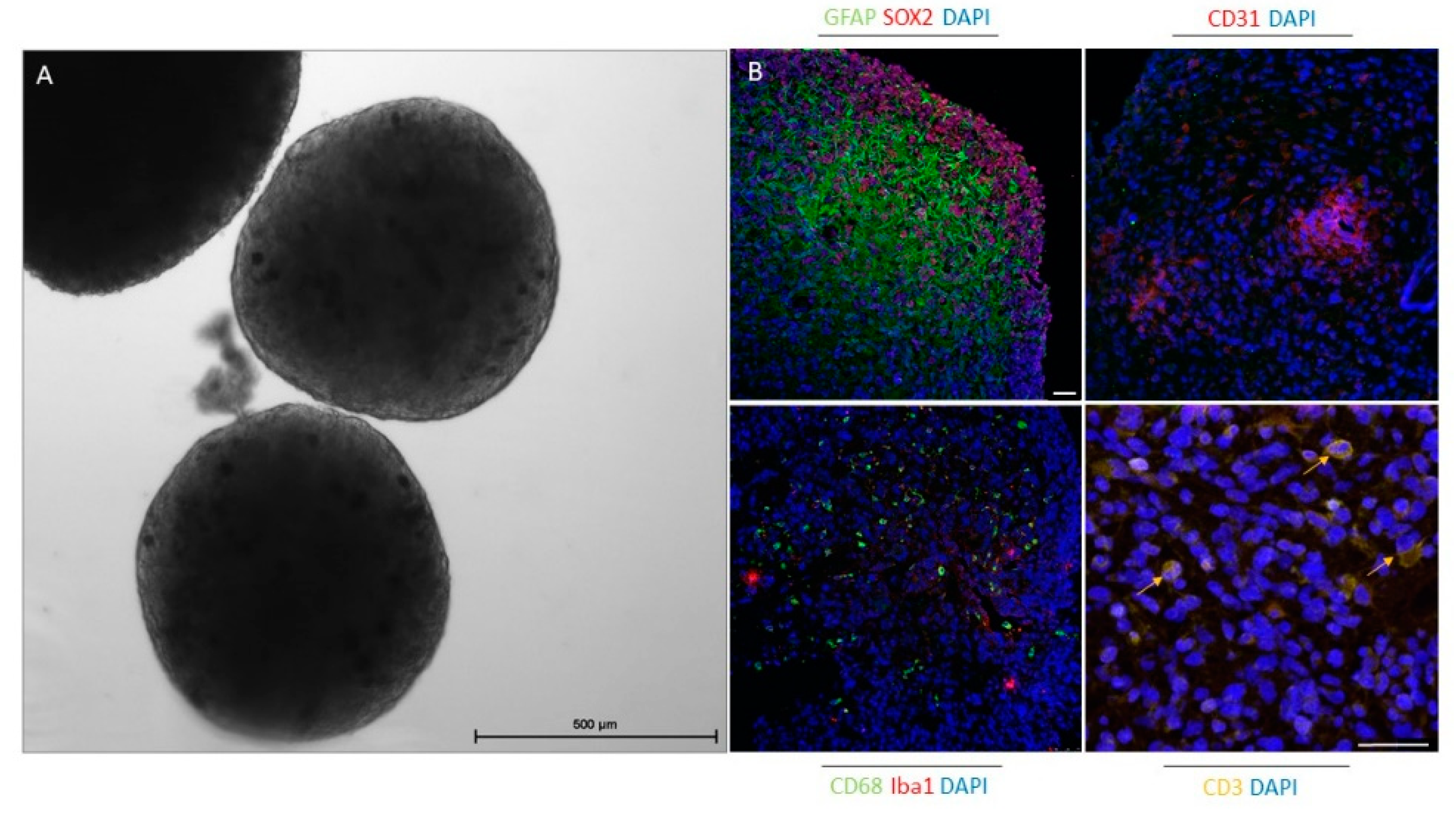{kind=link}
| Tumor Model | Description | Advantages | Disadvantages | References |
|---|---|---|---|---|
| In Vitro | ||||
| Tumor cell lines | established tumor cell lines, grown as monolayers in serum-containing media |
|
| [111] |
| Cancer stem cells | patient-derived tumor cells grown in serum-free and growth factor-supplemented media as tumorspheres |
|
| [112] |
| Cell co-cultures | 2D or 3D co-cultures of tumor and non-tumor cells, such as immune cells and stromal cells |
|
| [113] |
| Organotypic tissue slice cultures | precision-cut slices of tumor tissue, mounted onto porous membranes for mechanical support, and cultured in a controlled conditions |
|
| [114,115] |
| Patient-derived organoids | 3D in vitro tissue constructs composed of multiple cell types, patient-based from resected tumors |
|
| [116,117] |
| Genetically- engineered cerebral organoids | 3D in vitro tissue constructs created by using genetic manipulations to induce tumorigenesis in cerebral organoids |
|
| [118,119] |
| In Vivo | ||||
| Syngeneic mouse model | derived by transplanting mouse tumor cell lines or CSCs into strain-matched mice |
|
| [110,120] |
| Genetically engineered mouse tumor model | created by introducing genetic modifications that result in spontaneous tumor development |
|
| [121] |
| Patient-derived xenografts | derived by transplanting human tumor explants into immunodeficient mice |
|
| [90,122] |
| Humanized mouse tumor model | generated by the engraftment of human cancer cell lines or human PDX tumors into mice with a reconstituted human immune response |
|
| [110,123,124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majc, B.; Novak, M.; Kopitar-Jerala, N.; Jewett, A.; Breznik, B. Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development. Cells 2021, 10, 265. https://doi.org/10.3390/cells10020265
Majc B, Novak M, Kopitar-Jerala N, Jewett A, Breznik B. Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development. Cells. 2021; 10(2):265. https://doi.org/10.3390/cells10020265
Chicago/Turabian StyleMajc, Bernarda, Metka Novak, Nataša Kopitar-Jerala, Anahid Jewett, and Barbara Breznik. 2021. "Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development" Cells 10, no. 2: 265. https://doi.org/10.3390/cells10020265
APA StyleMajc, B., Novak, M., Kopitar-Jerala, N., Jewett, A., & Breznik, B. (2021). Immunotherapy of Glioblastoma: Current Strategies and Challenges in Tumor Model Development. Cells, 10(2), 265. https://doi.org/10.3390/cells10020265