
TREM2 Mediates Microglial Anti-Inflammatory Activations in Alzheimer’s Disease: Lessons Learned from Transcriptomics


Abstract
:1. Introduction
2. Trem2 and Disease-Associated Microglia (DAM)
3. TREM2 and AD Pathology
4. Possible Dark Sides of Trem2
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef]
- Tang, Y.P.; Gershon, E.S. Genetic studies in Alzheimer’s disease. Dialogues Clin. Neurosci. 2003, 5, 17–26. [Google Scholar] [PubMed]
- Villegas-Llerena, C.; Phillips, A.; Garcia-Reitboeck, P.; Hardy, J.; Pocock, J.M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 2016, 36, 74–81. [Google Scholar] [CrossRef]
- Millington, C.; Sonego, S.; Karunaweera, N.; Rangel, A.; Aldrich-Wright, J.R.; Campbell, I.L.; Gyengesi, E.; Munch, G. Chronic neuroinflammation in Alzheimer’s disease: New perspectives on animal models and promising candidate drugs. Biomed Res. Int. 2014, 2014, 309129. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Escott-Price, V.; Bellenguez, C.; Wang, L.S.; Choi, S.H.; Harold, D.; Jones, L.; Holmans, P.; Gerrish, A.; Vedernikov, A.; Richards, A.; et al. Gene-wide analysis detects two new susceptibility genes for Alzheimer’s disease. PLoS ONE 2014, 9, e94661. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amber, S.; Zahid, S. Data integration for functional annotation of regulatory single nucleotide polymorphisms associated with Alzheimer’s disease susceptibility. Gene 2018, 672, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Rezazadeh, M.; Khorrami, A.; Yeghaneh, T.; Talebi, M.; Kiani, S.J.; Heshmati, Y.; Gharesouran, J. Genetic Factors Affecting Late-Onset Alzheimer’s Disease Susceptibility. Neuromolecular Med. 2016, 18, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Sierksma, A.; Lu, A.; Mancuso, R.; Fattorelli, N.; Thrupp, N.; Salta, E.; Zoco, J.; Blum, D.; Buee, L.; De Strooper, B.; et al. Novel Alzheimer risk genes determine the microglia response to amyloid-beta but not to TAU pathology. EMBO Mol. Med. 2020, 12, e10606. [Google Scholar] [CrossRef]
- Mukherjee, S.; Klaus, C.; Pricop-Jeckstadt, M.; Miller, J.A.; Struebing, F.L. A Microglial Signature Directing Human Aging and Neurodegeneration-Related Gene Networks. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Klesney-Tait, J.; Turnbull, I.R.; Colonna, M. The TREM receptor family and signal integration. Nat. Immunol. 2006, 7, 1266–1273. [Google Scholar] [CrossRef]
- Deczkowska, A.; Weiner, A.; Amit, I. The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 2020, 181, 1207–1217. [Google Scholar] [CrossRef]
- Errichiello, E.; Dardiotis, E.; Mannino, F.; Paloneva, J.; Mattina, T.; Zuffardi, O. Phenotypic Expansion in Nasu-Hakola Disease: Immunological Findings in Three Patients and Proposal of a Unifying Pathogenic Hypothesis. Front. Immunol. 2019, 10, 1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paloneva, J.; Autti, T.; Raininko, R.; Partanen, J.; Salonen, O.; Puranen, M.; Hakola, P.; Haltia, M. CNS manifestations of Nasu-Hakola disease: A frontal dementia with bone cysts. Neurology 2001, 56, 1552–1558. [Google Scholar] [CrossRef]
- Kleinberger, G.; Brendel, M.; Mracsko, E.; Wefers, B.; Groeneweg, L.; Xiang, X.; Focke, C.; Deussing, M.; Suarez-Calvet, M.; Mazaheri, F.; et al. The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J. 2017, 36, 1837–1853. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 2017, 59, 220.e1–220.e9. [Google Scholar] [CrossRef] [PubMed]
- Prokop, S.; Miller, K.R.; Labra, S.R.; Pitkin, R.M.; Hoxha, K.; Narasimhan, S.; Changolkar, L.; Rosenbloom, A.; Lee, V.M.; Trojanowski, J.Q. Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer’s disease patient brain samples. Acta Neuropathol. 2019, 138, 613–630. [Google Scholar] [CrossRef]
- Bonham, L.W.; Sirkis, D.W.; Yokoyama, J.S. The Transcriptional Landscape of Microglial Genes in Aging and Neurodegenerative Disease. Front. Immunol. 2019, 10, 1170. [Google Scholar] [CrossRef] [Green Version]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef]
- Tabula Muris Consortium; Overall Coordination; Logistical Coordination; Organ Collection and Processing; Library Preparation and Sequencing; Computational Data Analysis; Cell Type Annotation; Writing Group; Supplemental Text Writing Group; Proncipal Investigators. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessard, C.B.; Malnik, S.L.; Zhou, Y.; Ladd, T.B.; Cruz, P.E.; Ran, Y.; Mahan, T.E.; Chakrabaty, P.; Holtzman, D.M.; Ulrich, J.D.; et al. High-affinity interactions and signal transduction between Abeta oligomers and TREM2. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Fitz, N.F.; Wolfe, C.M.; Playso, B.E.; Biedrzycki, R.J.; Lu, Y.; Nam, K.N.; Lefterov, I.; Koldamova, R. Trem2 deficiency differentially affects phenotype and transcriptome of human APOE3 and APOE4 mice. Mol. Neurodegener. 2020, 15, 41. [Google Scholar] [CrossRef]
- Li, C.; Zhao, B.; Lin, C.; Gong, Z.; An, X. TREM2 inhibits inflammatory responses in mouse microglia by suppressing the PI3K/NF-kappaB signaling. Cell Biol. Int. 2019, 43, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Walter, J. The Triggering Receptor Expressed on Myeloid Cells 2: A Molecular Link of Neuroinflammation and Neurodegenerative Diseases. J. Biol. Chem. 2016, 291, 4334–4341. [Google Scholar] [CrossRef] [Green Version]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Finn, M.B.; Wang, Y.; Shen, A.; Mahan, T.E.; Jiang, H.; Stewart, F.R.; Piccio, L.; Colonna, M.; Holtzman, D.M. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 2014, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 92, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.M.; Joshita, S.; Zhou, Y.; Ulland, T.K.; Gilfillan, S.; Colonna, M. Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med. 2018, 215, 745–760. [Google Scholar] [CrossRef] [Green Version]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Remolina Serrano, J.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Wan, Y.; Zhang, Y.D.; Zhou, J.S.; Gao, Q.; Zhu, X.C.; Shi, J.Q.; Lu, H.; Tan, L.; Yu, J.T. TREM2 Overexpression has No Improvement on Neuropathology and Cognitive Impairment in Aging APPswe/PS1dE9 Mice. Mol. Neurobiol. 2017, 54, 855–865. [Google Scholar] [CrossRef]
- Cheng, J.; Guo, X.; Zhang, T.; Zhong, L.; Bu, G.; Chen, X. TREMs in Alzheimer’s disease: Genetic and clinical investigations. Clin. Chim. Acta 2016, 463, 88–95. [Google Scholar] [CrossRef]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, C.; Qi, R.; Fu, H.; Ma, Q. scREAD: A Single-Cell RNA-Seq Database for Alzheimer’s Disease. iScience 2020, 23, 101769. [Google Scholar] [CrossRef] [PubMed]
- Onuska, K.M. The Dual Role of Microglia in the Progression of Alzheimer’s Disease. J. Neurosci. 2020, 40, 1608–1610. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gomez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef]
- Celarain, N.; Sanchez-Ruiz de Gordoa, J.; Zelaya, M.V.; Roldan, M.; Larumbe, R.; Pulido, L.; Echavarri, C.; Mendioroz, M. TREM2 upregulation correlates with 5-hydroxymethycytosine enrichment in Alzheimer’s disease hippocampus. Clin. Epigenetics 2016, 8, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lue, L.F.; Schmitz, C.T.; Serrano, G.; Sue, L.I.; Beach, T.G.; Walker, D.G. TREM2 Protein Expression Changes Correlate with Alzheimer’s Disease Neurodegenerative Pathologies in Post-Mortem Temporal Cortices. Brain Pathol. 2015, 25, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Strobel, S.; Grunblatt, E.; Riederer, P.; Heinsen, H.; Arzberger, T.; Al-Sarraj, S.; Troakes, C.; Ferrer, I.; Monoranu, C.M. Changes in the expression of genes related to neuroinflammation over the course of sporadic Alzheimer’s disease progression: CX3CL1, TREM2, and PPARgamma. J. Neural Transm. (Vienna) 2015, 122, 1069–1076. [Google Scholar] [CrossRef]
- Srinivasan, K.; Friedman, B.A.; Larson, J.L.; Lauffer, B.E.; Goldstein, L.D.; Appling, L.L.; Borneo, J.; Poon, C.; Ho, T.; Cai, F.; et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun. 2016, 7, 11295. [Google Scholar] [CrossRef]
- Casati, M.; Ferri, E.; Gussago, C.; Mazzola, P.; Abbate, C.; Bellelli, G.; Mari, D.; Cesari, M.; Arosio, B. Increased expression of TREM2 in peripheral cells from mild cognitive impairment patients who progress into Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 805–810. [Google Scholar] [CrossRef]
- Hu, N.; Tan, M.S.; Yu, J.T.; Sun, L.; Tan, L.; Wang, Y.L.; Jiang, T.; Tan, L. Increased expression of TREM2 in peripheral blood of Alzheimer’s disease patients. J. Alzheimers Dis. 2014, 38, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Alsema, A.M.; Jiang, Q.; Kracht, L.; Gerrits, E.; Dubbelaar, M.L.; Miedema, A.; Brouwer, N.; Hol, E.M.; Middeldorp, J.; van Dijk, R.; et al. Profiling Microglia From Alzheimer’s Disease Donors and Non-demented Elderly in Acute Human Postmortem Cortical Tissue. Front. Mol. Neurosci. 2020, 13, 134. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Yin, Z.; Raj, D.; Saiepour, N.; Van Dam, D.; Brouwer, N.; Holtman, I.R.; Eggen, B.J.L.; Moller, T.; Tamm, J.A.; Abdourahman, A.; et al. Immune hyperreactivity of Abeta plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 2017, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.J.; Wu, X.L.; Li, X.G.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.X.; et al. TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Tan, L.; Zhu, X.C.; Zhang, Q.Q.; Cao, L.; Tan, M.S.; Gu, L.Z.; Wang, H.F.; Ding, Z.Z.; Zhang, Y.D.; et al. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.M.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835. [Google Scholar] [CrossRef]
- Smith, A.R.; Smith, R.G.; Condliffe, D.; Hannon, E.; Schalkwyk, L.; Mill, J.; Lunnon, K. Increased DNA methylation near TREM2 is consistently seen in the superior temporal gyrus in Alzheimer’s disease brain. Neurobiol. Aging 2016, 47, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Q.; Li, F.; Chen, X.; Jia, J.; Sun, S.; Zhou, D.; Ma, L.; Jiang, T.; Bai, F.; Xiong, L.; et al. Triggering Receptor Expressed on Myeloid Cells 2, a Novel Regulator of Immunocyte Phenotypes, Confers Neuroprotection by Relieving Neuroinflammation. Anesthesiology 2017, 127, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Dua, P.; Alexandrov, P.N.; Hill, J.M.; Lukiw, W.J. Regulation of TREM2 expression by an NF-small ka, CyrillicB-sensitive miRNA-34a. Neuroreport 2013, 24, 318–323. [Google Scholar] [CrossRef] [Green Version]
- Ye, P.; Xu, D.; Xu, J.; Liu, G.; Huang, S.; Zhang, W.; Zheng, P.; Li, J.; Huang, J. TREM-2 negatively regulates LPS-mediated inflammatory response in rat bone marrow-derived MSCs. Mol. Med. Rep. 2017, 16, 4777–4783. [Google Scholar] [CrossRef]
- Lee, B.; Kim, T.H.; Jun, J.B.; Yoo, D.H.; Woo, J.H.; Choi, S.J.; Lee, Y.H.; Song, G.G.; Sohn, J.; Park-Min, K.H.; et al. Direct inhibition of human RANK+ osteoclast precursors identifies a homeostatic function of IL-1beta. J. Immunol. 2010, 185, 5926–5934. [Google Scholar] [CrossRef] [Green Version]
- Varvel, N.H.; Grathwohl, S.A.; Degenhardt, K.; Resch, C.; Bosch, A.; Jucker, M.; Neher, J.J. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-beta deposition in mouse models of Alzheimer’s disease. J. Exp. Med. 2015, 212, 1803–1809. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinformatics 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Monasor, L.S.; Muller, S.A.; Colombo, A.V.; Tanrioever, G.; Konig, J.; Roth, S.; Liesz, A.; Berghofer, A.; Piechotta, A.; Prestel, M.; et al. Fibrillar A beta triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. Elife 2020, 9, e54083. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Chitu, V.; Biundo, F.; Shlager, G.G.L.; Park, E.S.; Wang, P.; Gulinello, M.E.; Gokhan, S.; Ketchum, H.C.; Saha, K.; DeTure, M.A.; et al. Microglial Homeostasis Requires Balanced CSF-1/CSF-2 Receptor Signaling. Cell Rep. 2020, 30, 3004–3019.e5. [Google Scholar] [CrossRef] [Green Version]
- Spittau, B.; Dokalis, N.; Prinz, M. The Role of TGFbeta Signaling in Microglia Maturation and Activation. Trends Immunol. 2020, 41, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Poliani, P.L.; Wang, Y.; Fontana, E.; Robinette, M.L.; Yamanishi, Y.; Gilfillan, S.; Colonna, M. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest. 2015, 125, 2161–2170. [Google Scholar] [CrossRef] [Green Version]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathan, M.; Keerthikumar, S.; Ang, C.S.; Gangoda, L.; Quek, C.Y.; Williamson, N.A.; Mouradov, D.; Sieber, O.M.; Simpson, R.J.; Salim, A.; et al. FunRich: An open access standalone functional enrichment and interaction network analysis tool. Proteomics 2015, 15, 2597–2601. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron 2017, 94, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Jay, T.R.; von Saucken, V.E.; Landreth, G.E. TREM2 in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Meilandt, W.J.; Ngu, H.; Gogineni, A.; Lalehzadeh, G.; Lee, S.H.; Srinivasan, K.; Imperio, J.; Wu, T.; Weber, M.; Kruse, A.J.; et al. Trem2 Deletion Reduces Late-Stage Amyloid Plaque Accumulation, Elevates the Abeta42:Abeta40 Ratio, and Exacerbates Axonal Dystrophy and Dendritic Spine Loss in the PS2APP Alzheimer’s Mouse Model. J. Neurosci. 2020, 40, 1956–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.Y.; Das, S.; Adiconis, X.; Chen, H.B.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.Y.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Zhang, X.; Dong, W.; Wang, X.; He, S.; Zhang, H.; Wang, X.; Wei, R.; Chen, Y.; Liu, X.; et al. TREM2 suppresses the proinflammatory response to facilitate PRRSV infection via PI3K/NF-kappaB signaling. PLoS Pathog. 2020, 16, e1008543. [Google Scholar] [CrossRef]
- Katzenelenbogen, Y.; Sheban, F.; Yalin, A.; Yofe, I.; Svetlichnyy, D.; Jaitin, D.A.; Bornstein, C.; Moshe, A.; Keren-Shaul, H.; Cohen, M.; et al. Coupled scRNA-Seq and Intracellular Protein Activity Reveal an Immunosuppressive Role of TREM2 in Cancer. Cell 2020, 182, 872–885.e19. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Esaulova, E.; Vermi, W.; Hou, J.; Chen, Y.; Luo, J.; Brioschi, S.; Bugatti, M.; Omodei, A.S.; Ricci, B.; et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 2020, 182, 886–900.e17. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef] [PubMed]



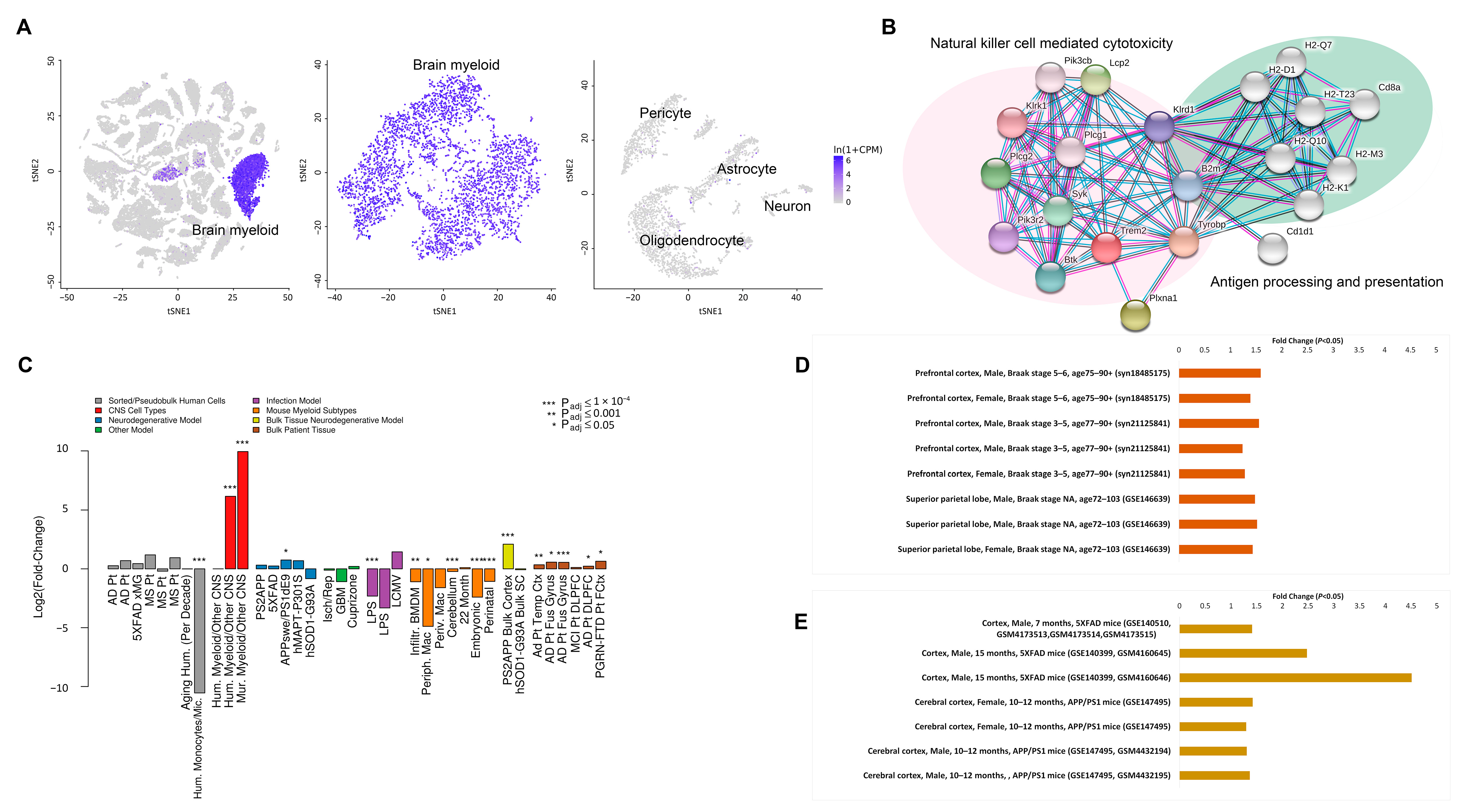{kind=link}
{kind=link}
| Major DAM Markers | Putative or Verified Upstream Regulators | Putative or Verified Functions | Regulation in AD Mouse Models | Regulation in AD Postmortem Samples |
|---|---|---|---|---|
| Cst7 | Trem2, Il10ra (inhibition), Tnf | Inhibiting proteases (papain and Ctsl) activity; possible immune regulation function | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in bulk tissue of temporal cortex (GSE15222) and fusiform gyrus (GSE95587, GSE125583). |
| Trem2 (Stage 2) | Trem2, miRNA-34a (inhibition), LPS (inhibition), Tgfb1 (inhibition), Il4, Csf1, Csf2, PU.1 | Receptor of amyloid-beta protein 42; immune signaling receptor for microglial activation, proliferation, migration, apoptosis, and expression of both pro-inflammation and anti-inflammation factors | Upregulation in sorted microglia of APP/PS1 (GSE74615). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in bulk tissue of temporal cortex (GSE15222) and fusiform gyrus (GSE95587, GSE125583). |
| Apoe (Stage 2) | Trem2, Hif1a (Csf2, Il5, Stat1, Tgfb1, Tnf), App (Ifng, Ngf, Tgfb1, Tnf), Map2k1 (Jak/Stat, Tgfb1, Chemokine, Il15), Stat1, Tgfb1, Tnf (inhibition) | Inhibiting apoptosis; accelerating chemotaxis; Il12 signaling and production in macrophage; quantity of neuroglia; lipid transport in CNS; innate and adaptive immune responses | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). | Upregulation in sorted microglia (GSE125050), and purified microglial nucleus (snRNAseq, syn18485175). |
| Ctsd/Ctsb (Stage 1) | App (Ifng, Ngf, Tgfb1, Tnf), Ifng, Tgfb1, Tnf, Nfe2l2, Tp53 | Lysosomal proteases; playing roles in APP processing or degradation | Upregulation in sorted microglia of APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in bulk tissue of temporal cortex (GSE15222). |
| Csf1 | Hif1a (Csf2, Il5, Stat1, Tgfb1, Tnf), Egr1, Cebpa, Csf2 (Trem1), App (Ifng, Ngf, Tgfb1, Tnf), Csf1r, Il1b, Ifng, Tgfb1, Tnf | Inhibiting apoptosis; accelerating chemotaxis; tyrosine phosphorylation of protein; inhibition of cells; interaction of leukemia cell line; quantity of neuroglia; major cytokine for survival, proliferation and differentiation of myeloid cells | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in bulk tissue of fusiform gyrus (GSE95587, GSE125583). |
| Lpl (Stage 2) | Cebpa, Ifng (inhibition), Nfe2l2, Tgfb1 (inhibition), Tnf (inhibition) | Inhibiting disorder of lipid metabolism; key enzyme in triglyceride metabolism | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Downregulation in bulk tissue of temporal cortex (GSE15222) and fusiform gyrus (GSE95587, GSE125583). |
| Spp1 | Egr1, Cebpa, miRNA-135, IgE (Il8), Csf2, Zfp36 (Il8), Akt1 (Interferon, Jak/Stat, Trem1), Mavs, Pdx1, Igf1, Il1b, Il2, Tgfb1, Tnf, Ifng, PU.1 | Accelerating chemotaxis; tyrosine phosphorylation of protein; interaction of leukemia cell line; cytokine for enhancing pro-inflammatory activation | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in purified microglial nucleus (snRNAseq, syn18485175). Upregulation in bulk tissue of temporal cortex (GSE15222) and fusiform gyrus (GSE95587, GSE125583). |
| Cd9 (Stage 2) | Il5 (Cebpd, Il1), Cxcl12, Il2 (inhibition), Il5 | Tyrosine phosphorylation of protein; chemotaxis of phagocytes; involved in platelet activation and aggregation | Upregulation in sorted microglia of APP/PS1 (GSE74615) and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in bulk tissue of fusiform gyrus (GSE95587, GSE125583). |
| Ccl6 (Stage 2) | Ifng, Tnf, Myc | Accelerating chemotaxis; promoting innate immune activation | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Not detected. |
| Itgax (Cd11c) (Stage 2) | Csf2, Ifng, Irf7, Stat1, Tgfb1 (inhibition), Tnf | Monocyte adhesion and chemotaxis | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in bulk tissue of fusiform gyrus (GSE95587, GSE125583). |
| Tyrobp (Dap12) (Stage 1) | Trem2, Ifng (inhibition) | Quantity of neuroglia; a ligand binding by multiple receptors, e.g., Trem1 and Trem2 | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), and APP/PS1 (GSE74615). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in bulk tissue of temporal cortex (GSE15222) and fusiform gyrus (GSE95587, GSE125583). |
| Igf1 | Tp53, Estrogen, Tnf (inhibition) | Regulator of cellular proliferation; inducer of PI3K-AKT signaling pathway | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Downregulation in bulk tissue of temporal cortex (GSE15222) and fusiform gyrus (GSE95587, GSE125583). |
| Clec7a (Stage 2) | Il1, Csf2, Ifng (inhibition) | Tyrosine phosphorylation of protein; enhancer for pro-inflammatory activation | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in bulk tissue of fusiform gyrus (GSE95587, GSE125583). |
| Axl (Stage 2) | Hif1a (Csf2, Il5, Stat1, Tgfb1, Tnf), App (Ifng, Ngf, Tgfb1, Tnf), Cxcl12, Stat1, Tgfb1 | Inhibiting apoptosis; accelerating chemotaxis; Il15 production; chemotaxis of phagocytes | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in bulk tissue of temporal cortex (GSE15222) and fusiform gyrus (GSE125583). |
| Cd63 | Trem2 | Cell surface receptor involved in activation of Akt, Fak/Ptk2, and Mapk; promoting cell survival, adhesion, and migration | Upregulation in sorted microglia of APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Upregulation in bulk tissue of fusiform gyrus (GSE125583). |
| Ank | Hif1a, Tgfb1 | Attaching integral membrane proteins to cytoskeletal elements | Upregulation in sorted microglia of PS2APP (GSE75431), 5XFAD (GSE65067), APP/PS1 (GSE74615), and Tau-P301S (GSE93180). Upregulation in bulk cortex of PS2APP (GSE75357). | Not changed. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, F.; Du, H. TREM2 Mediates Microglial Anti-Inflammatory Activations in Alzheimer’s Disease: Lessons Learned from Transcriptomics. Cells 2021, 10, 321. https://doi.org/10.3390/cells10020321
Xue F, Du H. TREM2 Mediates Microglial Anti-Inflammatory Activations in Alzheimer’s Disease: Lessons Learned from Transcriptomics. Cells. 2021; 10(2):321. https://doi.org/10.3390/cells10020321
Chicago/Turabian StyleXue, Feng, and Heng Du. 2021. "TREM2 Mediates Microglial Anti-Inflammatory Activations in Alzheimer’s Disease: Lessons Learned from Transcriptomics" Cells 10, no. 2: 321. https://doi.org/10.3390/cells10020321
APA StyleXue, F., & Du, H. (2021). TREM2 Mediates Microglial Anti-Inflammatory Activations in Alzheimer’s Disease: Lessons Learned from Transcriptomics. Cells, 10(2), 321. https://doi.org/10.3390/cells10020321