
A LRRK2 GTP Binding Inhibitor, 68, Reduces LPS-Induced Signaling Events and TNF-α Release in Human Lymphoblasts



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Human Lymphoblast Culture and Treatment
2.3. Western Blot and Co-Immunoprecipitation (Co-IP) Assays
2.4. Immunocytochemical Analysis
2.5. TNF-α Measurement
2.6. Data Analysis
3. Results
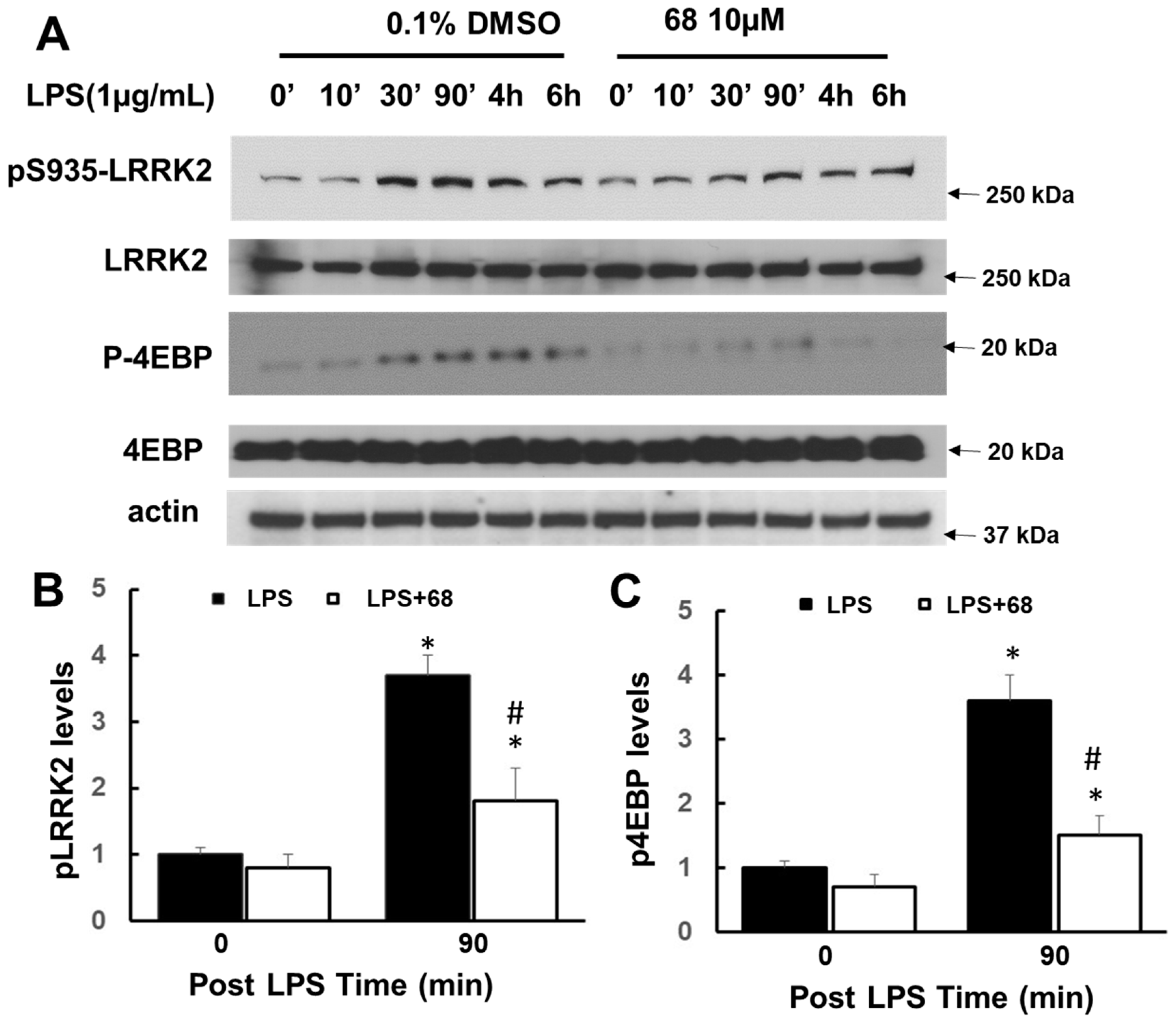
3.1. Inhibition of GTP Binding by 68 Reduced LPS-Induced LRRK2 Kinase Activation
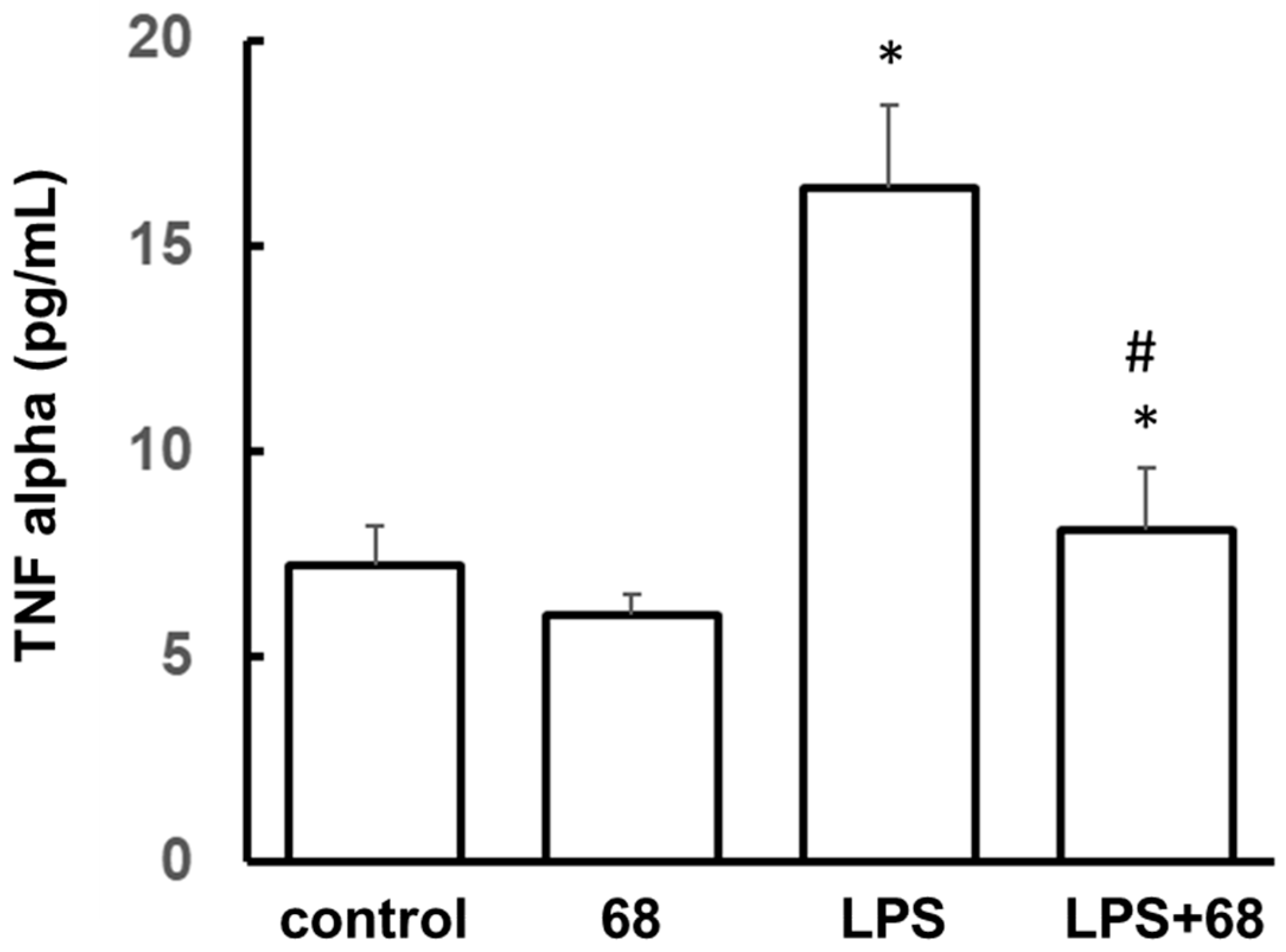
3.2. Compound 68 Reduced LPS-Induced Tumor Necrosis Factor-α (TNF-α) Secretion
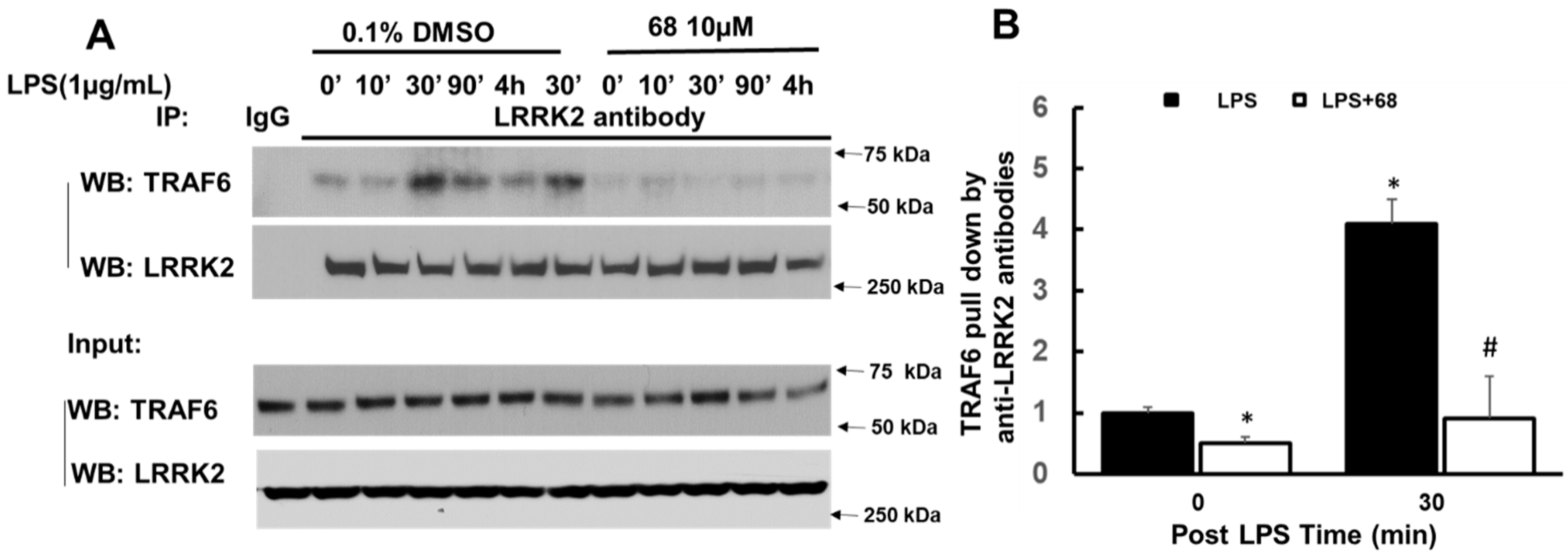
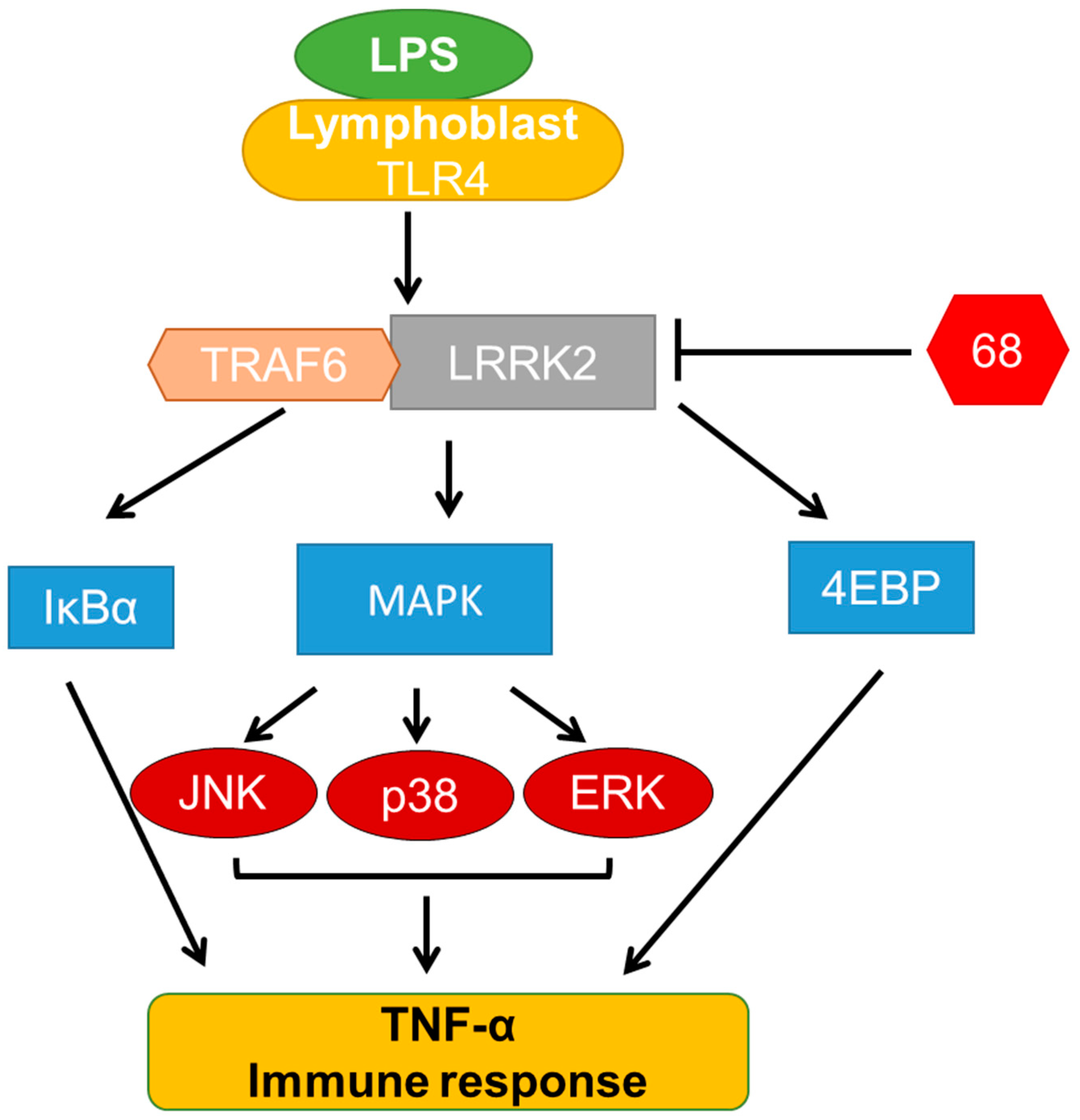
3.3. Compound 68 Reduced LPS-Induced TRAF6/LRRK2 Interaction
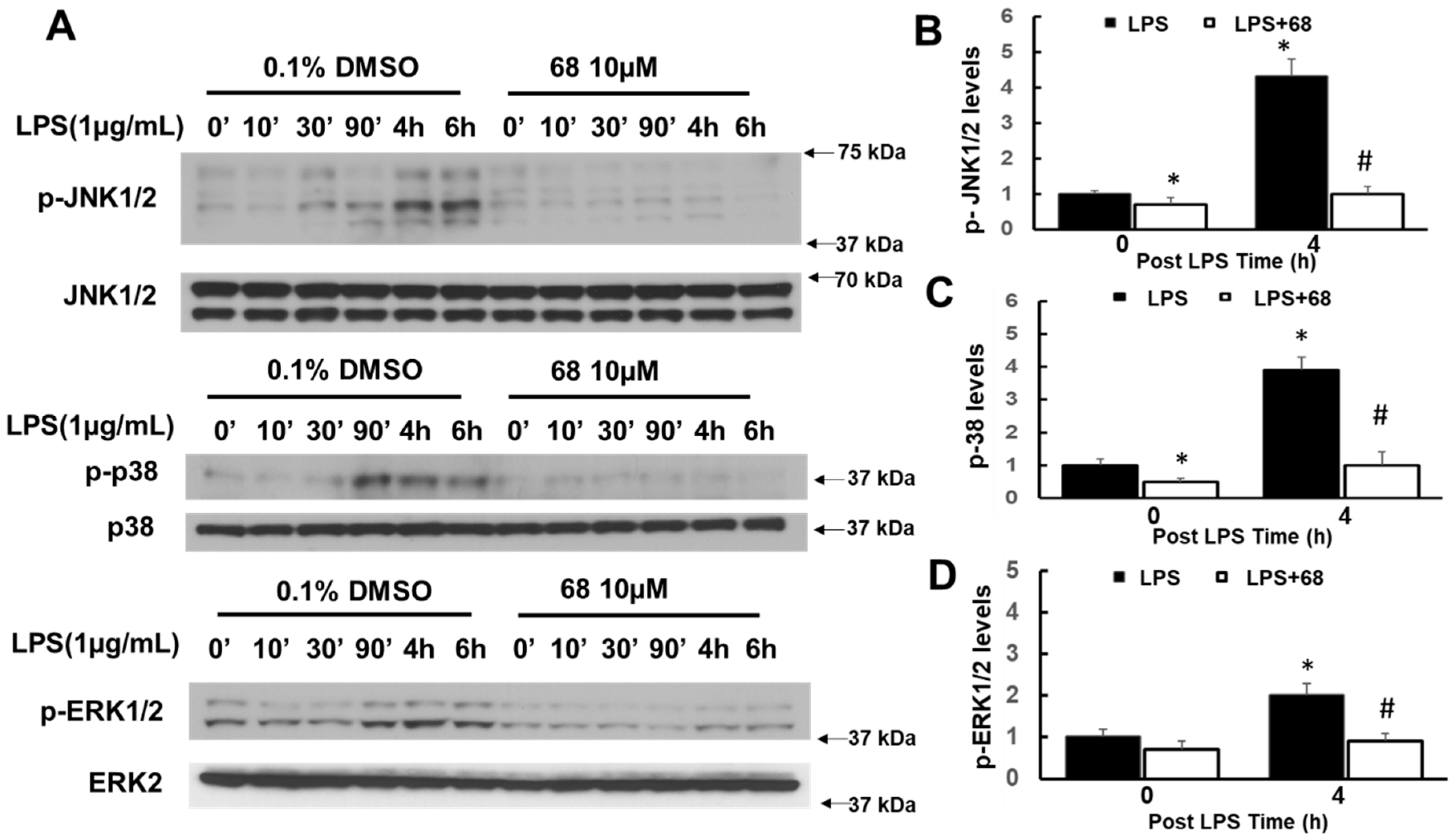
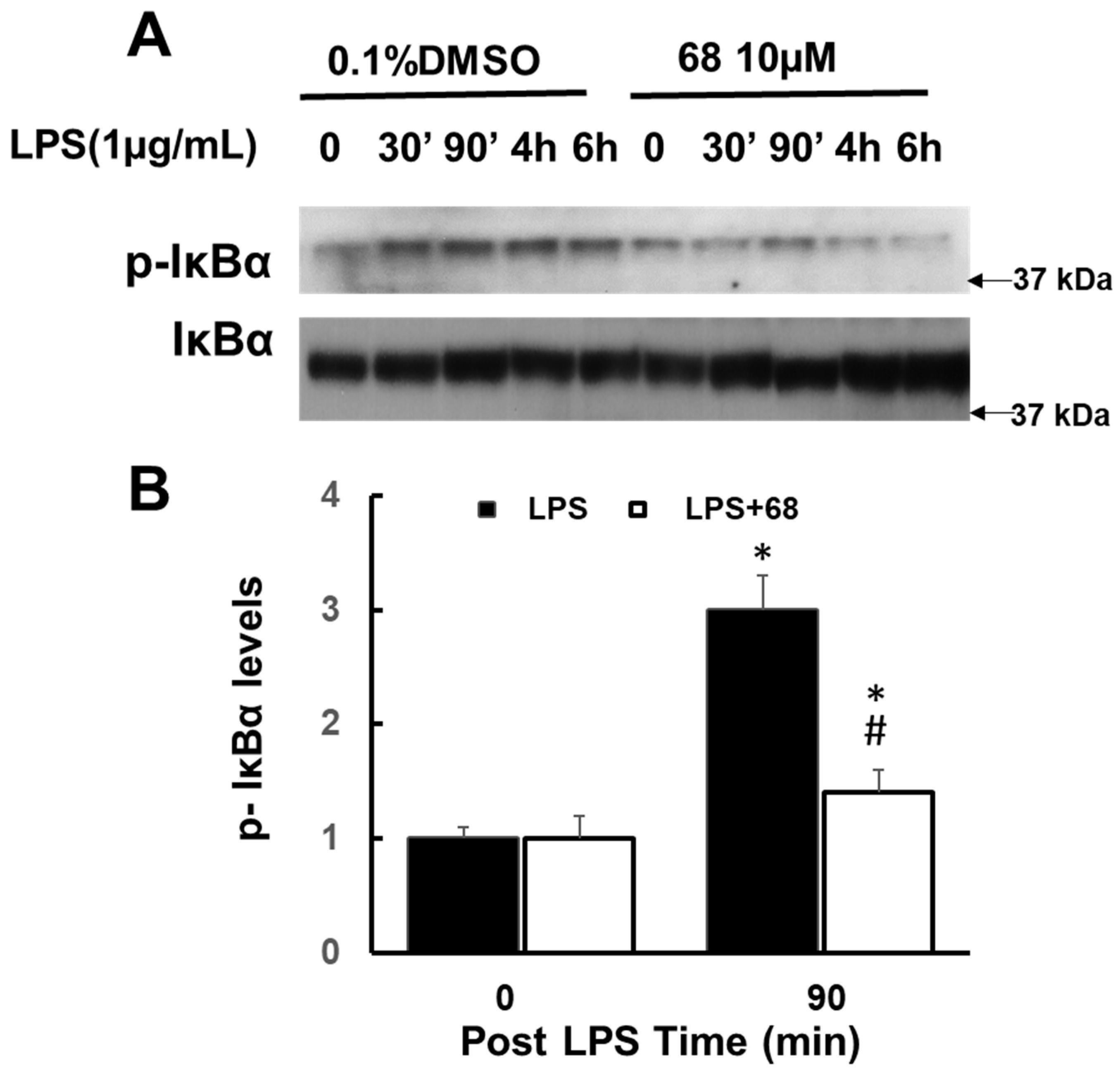
3.4. Compound 68 Attenuated LPS-Induced Activation of TRAF6-Linked MAPK and IkBα Signaling Pathways
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mouradian, M.M. Recent advances in the genetics and pathogenesis of Parkinson disease. Neurology 2002, 58, 179–185. [Google Scholar] [CrossRef]
- Schapira, A.H. Glucocerebrosidase and Parkinson disease: Recent advances. Mol. Cell Neurosci. 2015, 66, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckman, M.G.; Soto-Ortolaza, A.I.; Aasly, J.O.; Abahuni, N.; Annesi, G.; Bacon, J.A.; Bardien, S.; Bozi, M.; Brice, A.; Brighina, L.; et al. Population-specific frequencies for LRRK2 susceptibility variants in the Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium. Mov. Disord. 2013, 28, 1740–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Yang, D.; Sushchky, S.; Liu, Z.; Smith, W.W. Models for LRRK2-Linked Parkinsonism. Parkinsons Dis. 2011, 942412. [Google Scholar] [CrossRef] [Green Version]
- Cookson, M.R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 2010, 11, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Bardien, S.; Lesage, S.; Brice, A.; Carr, J. Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Parkinsonism Relat. Disord. 2011, 17, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Lewis, P.A.; Singleton, A.B. LRRK2: Cause, Risk, and Mechanism. J. Parkinsons Dis. 2013, 3, 85–103. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Siedlak, S.L.; Smith, M.A.; Perry, G.; Chen, S.G. LRRK2 protein is a component of Lewy bodies. Ann. Neurol. 2006, 60, 617–618. [Google Scholar] [CrossRef]
- Lee, H.; James, W.S.; Cowley, S.A. LRRK2 in peripheral and central nervous system innate immunity: Its link to Parkinson’s disease. Biochem. Soc. Trans. 2017, 45, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Dzamko, N.L. LRRK2 and the Immune System. Adv. Neurobiol. 2017, 14, 123–143. [Google Scholar]
- Brockmann, K.; Apel, A.; Schulte, C.; Schneiderhan-Marra, N.; Pont-Sunyer, C.; Vilas, D.; Ruiz-Martinez, J.; Langkamp, M.; Corvol, J.C.; Cormier, F.; et al. Inflammatory profile in LRRK2-associated prodromal and clinical PD. J. Neuroinflamm. 2016, 13, 122–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Mo, M.; Li, G.; Cen, L.; Wei, L.; Xiao, Y.; Chen, X.; Li, S.; Yang, X.; Qu, S.; et al. The biomarkers of immune dysregulation and inflammation response in Parkinson disease. Transl. Neurodegener. 2016, 5, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, N.P.; de Miranda, A.S.; Teixeira, A.L. Insights into Neuroinflammation in Parkinson’s Disease: From Biomarkers to Anti-Inflammatory Based Therapies. BioMed Res. Int. 2015, 628192. [Google Scholar] [CrossRef]
- Dzamko, N.; Geczy, C.L.; Halliday, G.M. Inflammation is genetically implicated in Parkinson’s disease. Neuroscience 2015, 302, 89–102. [Google Scholar] [CrossRef]
- Zhang, F.R.; Huang, W.; Chen, S.M.; Sun, L.D.; Liu, H.; Li, Y.; Cui, Y.; Yan, X.X.; Yang, H.T.; Yang, R.D.; et al. Genomewide association study of leprosy. N. Engl. J. Med. 2009, 361, 2609–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witoelar, A.; Jansen, I.E.; Wang, Y.; Desikan, R.S.; Gibbs, J.R.; Blauwendraat, C.; Thompson, W.K.; Hernandez, D.G.; Djurovic, S.; Schork, A.J.; et al. Genome-wide Pleiotropy Between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 2017, 74, 780–792. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Fang, M.; Jostins, L.; Umicevic, M.M.; Boucher, G.; Anderson, C.A.; Andersen, V.; Cleynen, I.; Cortes, A.; Crins, F.; et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017, 547, 173–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; He, X.; Thomas, J.M.; Yang, D.; Zhong, S.; Xue, F.; Smith, W.W. A novel GTP-binding inhibitor, FX2149, attenuates LRRK2 toxicity in Parkinson’s disease models. PLoS ONE 2015, 10, e0122461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Yang, D.; Zhong, S.; Thomas, J.M.; Xue, F.; Liu, J.; Kong, L.; Voulalas, P.; Hassan, H.E.; Park, J.S.; et al. Novel LRRK2 GTP-binding inhibitors reduced degeneration in Parkinson’s disease cell and mouse models. Hum. Mol. Genet. 2014, 23, 6212–6222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moehle, M.S.; Webber, P.J.; Tse, T.; Sukar, N.; Standaert, D.G.; DeSilva, T.M.; Cowell, R.M.; West, A.B. LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 2012, 32, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Liu, Z. LRRK2 enhances Nod1/2-mediated inflammatory cytokine production by promoting Rip2 phosphorylation. Protein Cell 2017, 8, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.S.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases-an update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef]
- Kubo, M.; Kamiya, Y.; Nagashima, R.; Maekawa, T.; Eshima, K.; Azuma, S.; Ohta, E.; Obata, F. LRRK2 is expressed in B-2 but not in B-1 B cells, and downregulated by cellular activation. J. Neuroimmunol. 2010, 229, 123–128. [Google Scholar] [CrossRef]
- Kubo, M.; Nagashima, R.; Ohta, E.; Maekawa, T.; Isobe, Y.; Kurihara, M.; Eshima, K.; Iwabuchi, K.; Sasaoka, T.; Azuma, S.; et al. Leucine-rich repeat kinase 2 is a regulator of B cell function, affecting homeostasis, BCR signaling, IgA production, and TI antigen responses. J. Neuroimmunol. 2016, 292, 1–8. [Google Scholar] [CrossRef]
- Smith, W.W.; Pei, Z.; Jiang, H.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 2006, 9, 1231–1233. [Google Scholar] [CrossRef]
- Smith, W.W.; Pei, Z.; Jiang, H.; Moore, D.J.; Liang, Y.; West, A.B.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 18676–18681. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.M.; Li, T.; Yang, W.; Xue, F.; Fishman, P.S.; Smith, W.W. 68 and FX2149 Attenuate Mutant LRRK2-R1441C-Induced Neural Transport Impairment. Front. Aging Neurosci. 2017, 8, 337. [Google Scholar] [CrossRef]
- Thomas, J.M.; Wang, X.; Guo, G.; Li, T.; Dai, B.; Nucifora, L.G.; Nucifora, F.C., Jr.; Liu, Z.; Xue, F.; Liu, C.; et al. GTP-binding inhibitors increase LRRK2-linked ubiquitination and Lewy body-like inclusions. J. Cell Physiol. 2020, 235, 7309–7320. [Google Scholar] [CrossRef]
- Lee, B.D.; Shin, J.H.; Van Kampen, J.; Petrucelli, L.; West, A.B.; Ko, H.S.; Lee, Y.I.; Maguire-Zeiss, K.A.; Bowers, W.J.; Federoff, H.J.; et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 2010, 16, 998–1000. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Li, T.; Liu, Z.; Arbez, N.; Yan, J.; Moran, T.H.; Ross, C.A.; Smith, W.W. LRRK2 kinase activity mediates toxic interactions between genetic mutation and oxidative stress in a Drosophila model: Suppression by curcumin. Neurobiol. Dis. 2012, 47, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Dzamko, N.; Prescott, A.; Davies, P.; Liu, Q.; Yang, Q.; Lee, J.D.; Patricelli, M.P.; Nomanbhoy, T.K.; Alessi, D.R.; et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205. [Google Scholar] [CrossRef] [Green Version]
- Greggio, E.; Jain, S.; Kingsbury, A.; Bandopadhyay, R.; Lewis, P.; Kaganovich, A.; van der Brug, M.P.; Beilina, A.; Blackinton, J.; Thomas, K.J.; et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006, 23, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, J.M.; Yang, K.; Marsman, A.; Pradhan, S.; Wang, M.; Ward, R.E.; Bonekamp, S.; Ambinder, W.B.; Higgs, C.P.; Kim, P.K.; et al. A multimodal approach to studying the relationship between peripheral glutathione, brain glutamate, and cognition in health and in schizophrenia. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Foster, S.J.; McCormick, L.M.; Ntolosi, B.A.; Campbell, D. Production of TNF-α by LPS-stimulated murine, rat and human blood and its pharmacological modulation. Agents Actions 1993, 38, C77–C79. [Google Scholar] [CrossRef]
- Wang, Y.R.; Yang, F.; Liu, J.; Zhou, Y. Effect of sophoridine and TLR4/MD-2 blocking agent on pathway of LPS-induced RAW264. 7 macrophage TLR4-NF-kappaB-TNF-α. Zhongguo Zhong Yao Za Zhi 2012, 37, 3107–3111. [Google Scholar]
- Santpere, G.; Ferrer, I. LRRK2 and neurodegeneration. Acta Neuropathol. 2009, 117, 227–246. [Google Scholar] [CrossRef]
- Jin, H.G.; Yamashita, H.; Nakamura, T.; Fukuba, H.; Takahashi, T.; Hiji, M.; Kohriyama, T.; Matsumoto, M. Synphilin-1 transgenic mice exhibit mild motor impairments. Neurosci. Lett. 2008, 445, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Takagawa, T.; Kitani, A.; Fuss, I.; Levine, B.; Brant, S.R.; Peter, I.; Tajima, M.; Nakamura, S.; Strober, W. An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci. Transl. Med. 2018, 10, eaan8162. [Google Scholar] [CrossRef] [Green Version]
- Han, K.A.; Yoo, L.; Sung, J.Y.; Chung, S.A.; Um, J.W.; Kim, H.; Seol, W.; Chung, K.C. Leucine-Rich Repeat Kinase 2 (LRRK2) Stimulates IL-1β-Mediated Inflammatory Signaling through Phosphorylation of RCAN1. Front Cell Neurosci. 2017, 11, 125. [Google Scholar] [CrossRef] [Green Version]
- Wallings, R.L.; Tansey, M.G. LRRK2 regulation of immune-pathways and inflammatory disease. Biochem. Soc. Trans. 2019, 47, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Kozina, E.; Sadasivan, S.; Jiao, Y.; Dou, Y.; Ma, Z.; Tan, H.; Kodali, K.; Shaw, T.; Peng, J.; Smeyne, R.J. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain 2018, 141, 1753–1769. [Google Scholar] [CrossRef]
- Daher, J.P.; Volpicelli-Daley, L.A.; Blackburn, J.P.; Moehle, M.S.; West, A.B. Abrogation of α-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc. Natl. Acad. Sci. USA 2014, 111, 9289–9294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillardon, F.; Schmid, R.; Draheim, H. Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 2012, 208, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Tian, X.; Zhang, J.; Wang, Z.; Chen, G. Roles of TRAF6 in Central Nervous System. Curr. Neuropharmacol. 2018, 16, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- West, A.B.; Moore, D.J.; Choi, C.; Andrabi, S.A.; Li, X.; Dikeman, D.; Biskup, S.; Zhang, Z.; Lim, K.L.; Dawson, V.L.; et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007, 16, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Thomas, J.M.; Li, T.; Lee, Y.; Liu, Z.; Smith, W.W. The Drosophila hep pathway mediates Lrrk2-induced neurodegeneration. Biochem. Cell Biol. 2018, 96, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Kuss, M.; Adamopoulou, E.; Kahle, P.J. Interferon-γ induces leucine-rich repeat kinase LRRK2 via extracellular signal-regulated kinase ERK5 in macrophages. J. Neurochem. 2014, 129, 980–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzamko, N.; Inesta-Vaquera, F.; Zhang, J.; Xie, C.; Cai, H.; Arthur, S.; Tan, L.; Choi, H.; Gray, N.; Cohen, P.; et al. The IkappaB Kinase Family Phosphorylates the Parkinson’s Disease Kinase LRRK2 at Ser935 and Ser910 during Toll-Like Receptor Signaling. PLoS ONE 2012, 7, e39132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, I.; Berti, G.; Plotegher, N.; Bernardo, G.; Filograna, R.; Bubacco, L.; Greggio, E. Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-κB p50 signaling in cultured microglia cells. J. Neuroinflamm. 2015, 12, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Ning, B.; Kong, L.; Dai, B.; He, X.; Thomas, J.M.; Sawa, A.; Ross, C.A.; Smith, W.W. A LRRK2 GTP Binding Inhibitor, 68, Reduces LPS-Induced Signaling Events and TNF-α Release in Human Lymphoblasts. Cells 2021, 10, 480. https://doi.org/10.3390/cells10020480
Li T, Ning B, Kong L, Dai B, He X, Thomas JM, Sawa A, Ross CA, Smith WW. A LRRK2 GTP Binding Inhibitor, 68, Reduces LPS-Induced Signaling Events and TNF-α Release in Human Lymphoblasts. Cells. 2021; 10(2):480. https://doi.org/10.3390/cells10020480
Chicago/Turabian StyleLi, Tianxia, Bo Ning, Lingbo Kong, Bingling Dai, Xiaofei He, Joseph M. Thomas, Akira Sawa, Christopher A. Ross, and Wanli W. Smith. 2021. "A LRRK2 GTP Binding Inhibitor, 68, Reduces LPS-Induced Signaling Events and TNF-α Release in Human Lymphoblasts" Cells 10, no. 2: 480. https://doi.org/10.3390/cells10020480
APA StyleLi, T., Ning, B., Kong, L., Dai, B., He, X., Thomas, J. M., Sawa, A., Ross, C. A., & Smith, W. W. (2021). A LRRK2 GTP Binding Inhibitor, 68, Reduces LPS-Induced Signaling Events and TNF-α Release in Human Lymphoblasts. Cells, 10(2), 480. https://doi.org/10.3390/cells10020480