




Cells 2023, 12(13), 1799; https://doi.org/10.3390/cells12131799 - 6 Jul 2023
Cited by 8 | Viewed by 2187
Abstract
►
Show Figures
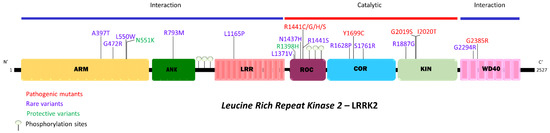
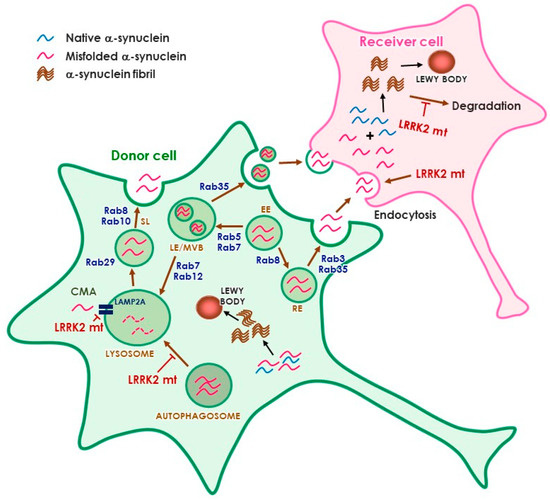
Chronic neuroinflammation plays a crucial role in the progression of several neurodegenerative diseases (NDDs), including Parkinson’s disease (PD) and Alzheimer’s disease (AD). Intriguingly, in the last decade, leucine-rich repeat kinase-2 (LRRK2), a gene mutated in familial and sporadic PD, was revealed
[...] Read more.
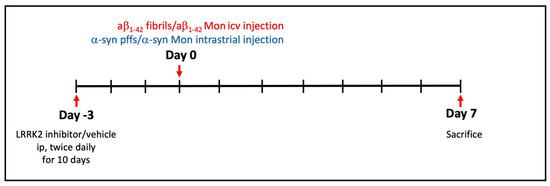
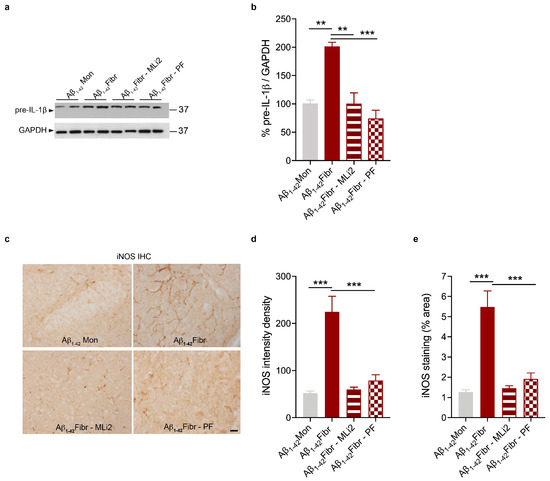
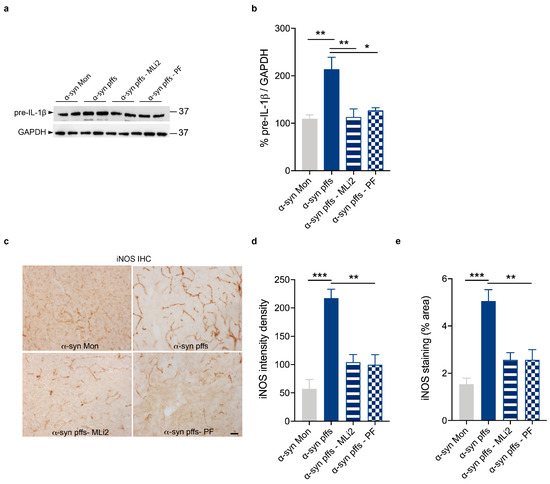
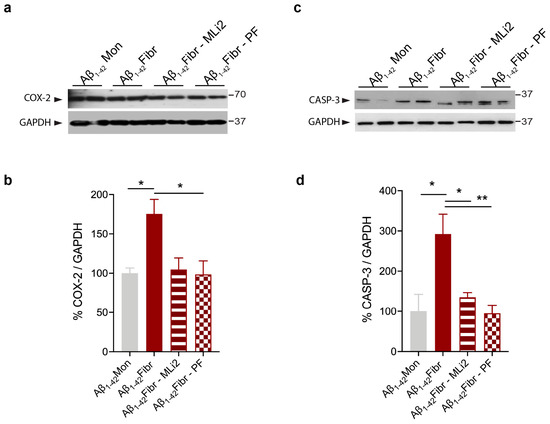
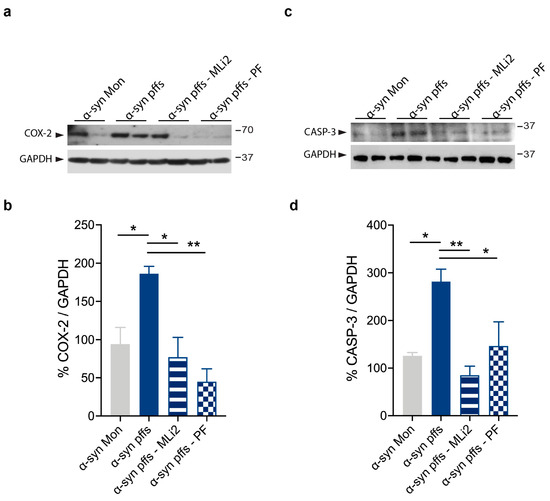
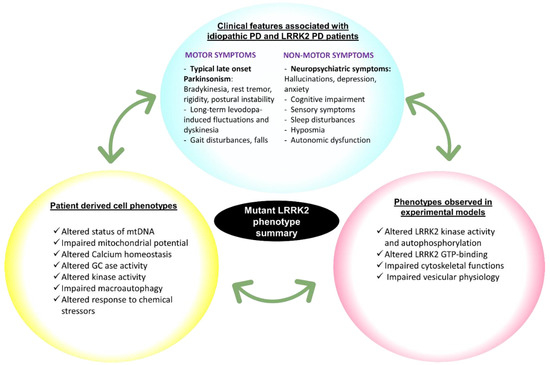
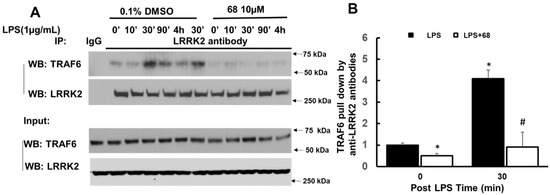
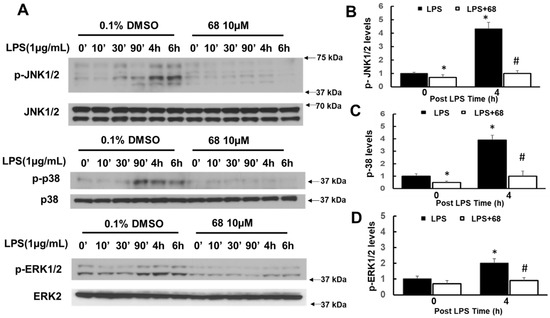
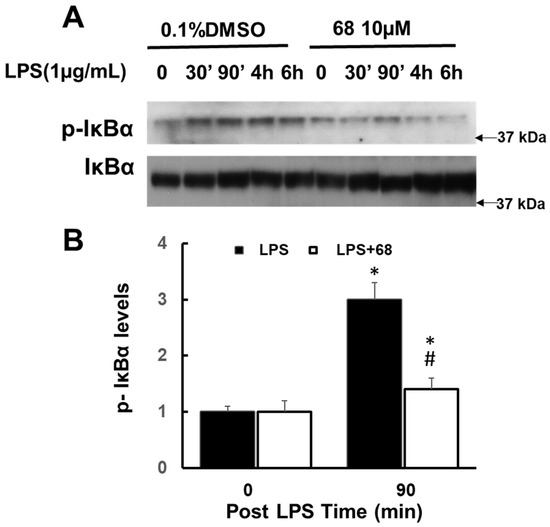
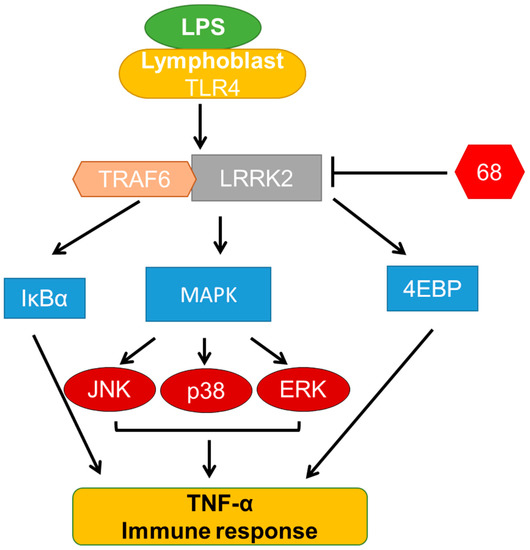
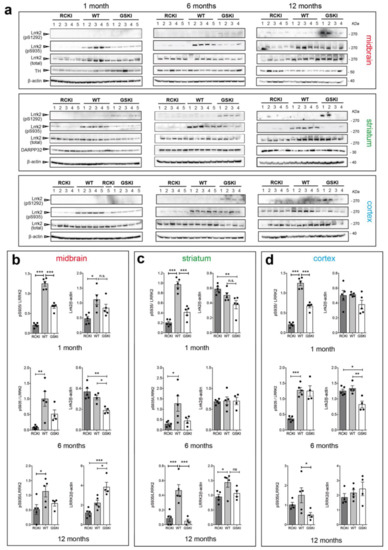
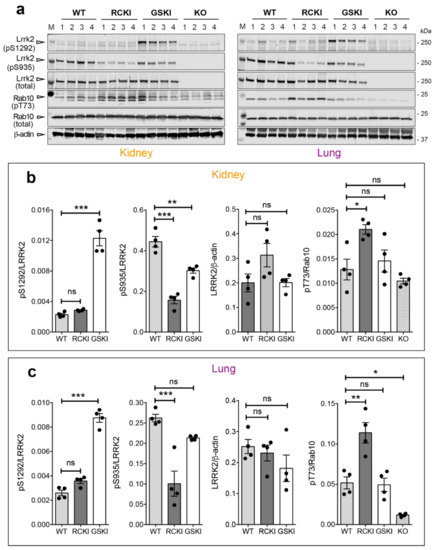
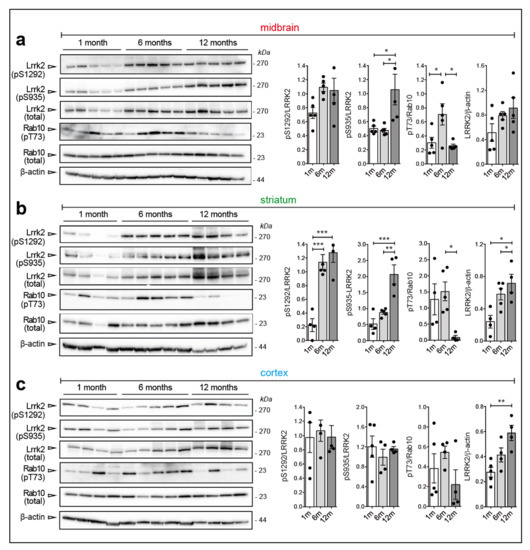
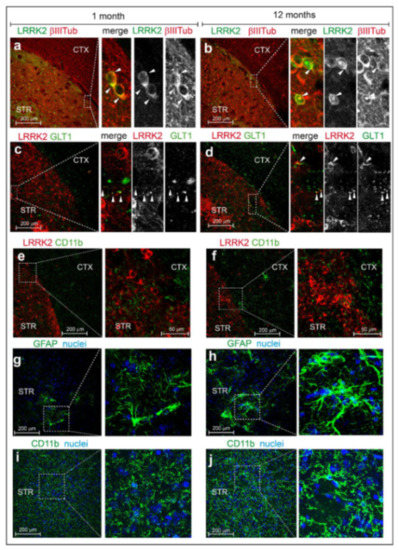
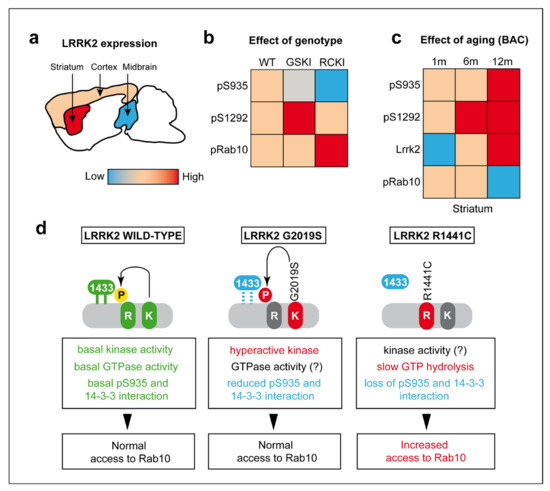
Chronic neuroinflammation plays a crucial role in the progression of several neurodegenerative diseases (NDDs), including Parkinson’s disease (PD) and Alzheimer’s disease (AD). Intriguingly, in the last decade, leucine-rich repeat kinase-2 (LRRK2), a gene mutated in familial and sporadic PD, was revealed as a key mediator of neuroinflammation. Therefore, the anti-inflammatory properties of LRRK2 inhibitors have started to be considered as a disease-modifying treatment for PD; however, to date, there is little evidence on the beneficial effects of targeting LRRK2-related neuroinflammation in preclinical models. In this study, we further validated LRRK2 kinase modulation as a pharmacological intervention in preclinical models of AD- and PD-related neuroinflammation. Specifically, we reported that LRRK2 kinase inhibition with MLi2 and PF-06447475 (PF) molecules attenuated neuroinflammation, gliosis and cytotoxicity in mice with intracerebral injection of Aβ1-42 fibrils or α-syn preformed fibrils (pffs). Moreover, for the first time in vivo, we showed that LRRK2 kinase activity participates in AD-related neuroinflammation and therefore might contribute to AD pathogenesis. Overall, our findings added evidence on the anti-inflammatory effects of LRRK2 kinase inhibition in preclinical models and indicate that targeting LRRK2 activity could be a disease-modifying treatment for NDDs with an inflammatory component.
Full article

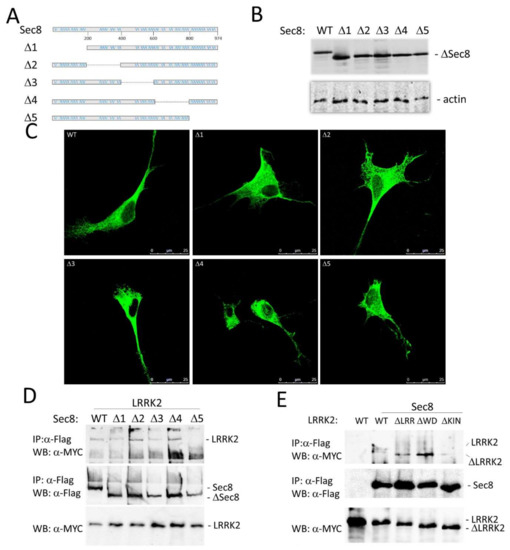
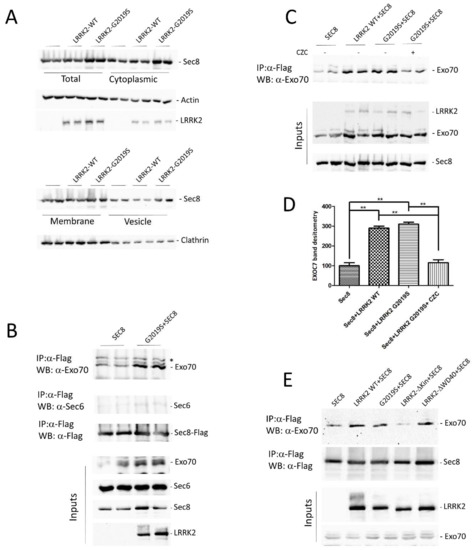
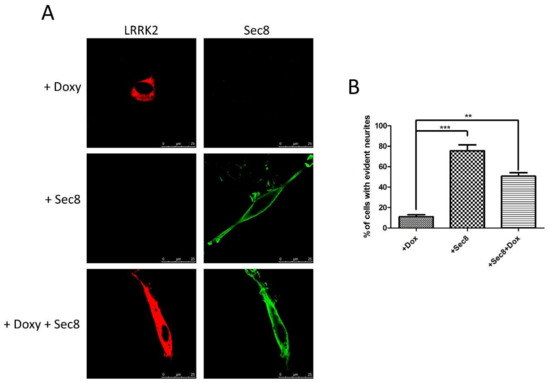
Figure 1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}