Pathological Functions of LRRK2 in Parkinson’s Disease


Abstract
:1. Introduction
2. LRRK2 in Neurodegeneration
2.1. The Structure of LRRK2 and Regulation of Enzymatic Activities
2.2. Functions of Pathogenic LRRK2 Mutations in Neurodegeneration
2.3. Kinase Substrates of LRRK2 and Their Roles in Neurodegeneration
3. Functions of LRRK2 in Lewy Pathology and Synucleinopathies
3.1. Lewy Pathology in PD
3.2. Roles of LRRK2 in Synucleinopathy
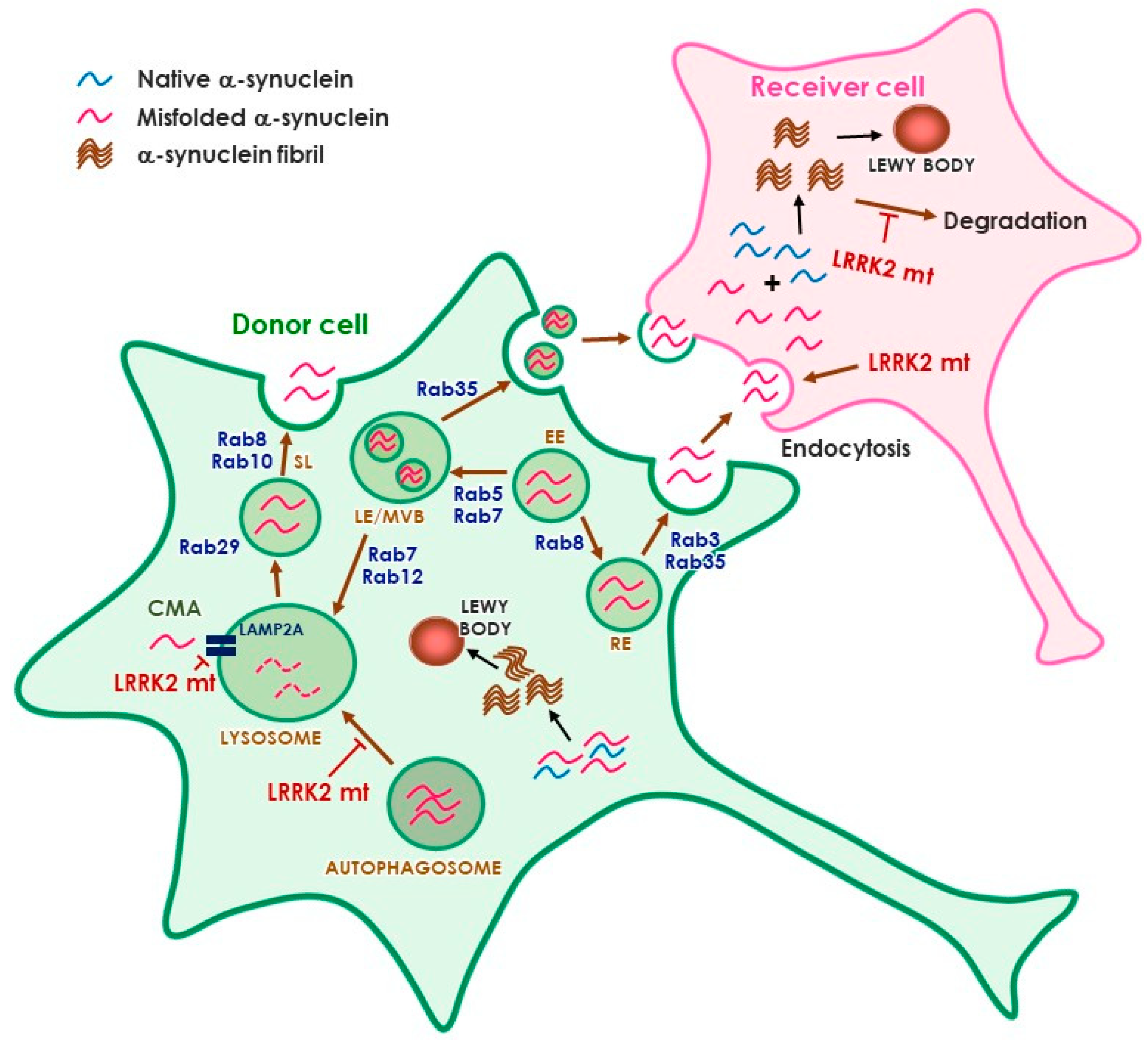
3.3. Roles of LRRK2 in α-Synuclein Propagation
4. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Clarke, C.E. Parkinson’s disease. BMJ 2007, 335, 441–445. [Google Scholar] [CrossRef]
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15 (Suppl. 1), 14–20. [Google Scholar] [CrossRef] [PubMed]
- Ponsen, M.M.; Stoffers, D.; Booij, J.; van Eck-Smit, B.L.; Wolters, E.; Berendse, H.W. Idiopathic hyposmia as a preclinical sign of Parkinson’s disease. Ann. Neurol. 2004, 56, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef] [PubMed]
- Aasly, J.O.; Toft, M.; Fernandez-Mata, I.; Kachergus, J.; Hulihan, M.; White, L.R.; Farrer, M. Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann. Neurol. 2005, 57, 762–765. [Google Scholar] [CrossRef]
- Bras, J.M.; Guerreiro, R.J.; Ribeiro, M.H.; Januario, C.; Morgadinho, A.; Oliveira, C.R.; Cunha, L.; Hardy, J.; Singleton, A. G2019S dardarin substitution is a common cause of Parkinson’s disease in a Portuguese cohort. Mov. Disord. 2005, 20, 1653–1655. [Google Scholar] [CrossRef] [Green Version]
- Infante, J.; Rodriguez, E.; Combarros, O.; Mateo, I.; Fontalba, A.; Pascual, J.; Oterino, A.; Polo, J.M.; Leno, C.; Berciano, J. LRRK2 G2019S is a common mutation in Spanish patients with late-onset Parkinson’s disease. Neurosci. Lett. 2006, 395, 224–226. [Google Scholar] [CrossRef]
- Kachergus, J.; Mata, I.F.; Hulihan, M.; Taylor, J.P.; Lincoln, S.; Aasly, J.; Gibson, J.M.; Ross, O.A.; Lynch, T.; Wiley, J.; et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: Evidence of a common founder across European populations. Am. J. Hum. Genet. 2005, 76, 672–680. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.D.; Dawson, V.L.; Dawson, T.M. Leucine-rich repeat kinase 2 (LRRK2) as a potential therapeutic target in Parkinson’s disease. Trends Pharmacol. Sci. 2012, 33, 365–373. [Google Scholar] [CrossRef] [Green Version]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.; Zhang, S.; Bustos, D.; Kleinheinz, T.; Le Pichon, C.E.; Dominguez, S.L.; Solanoy, H.O.; Drummond, J.; Zhang, X.; Ding, X.; et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci. Transl. Med. 2012, 4, 164ra161. [Google Scholar] [CrossRef] [PubMed]
- Jaleel, M.; Nichols, R.J.; Deak, M.; Campbell, D.G.; Gillardon, F.; Knebel, A.; Alessi, D.R. LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 2007, 405, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.D.; Shin, J.H.; VanKampen, J.; Petrucelli, L.; West, A.B.; Ko, H.S.; Lee, Y.I.; Maguire-Zeiss, K.A.; Bowers, W.J.; Federoff, H.J.; et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 2010, 16, 998–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, G.; Okai, T.; Fujino, G.; Takeda, K.; Ichijo, H.; Katada, T.; Iwatsubo, T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson’s disease. Biochemistry 2007, 46, 1380–1388. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Choi, C.; Andrabi, S.A.; Li, X.; Dikeman, D.; Biskup, S.; Zhang, Z.; Lim, K.L.; Dawson, V.L.; et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007, 16, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Greggio, E.; Zambrano, I.; Kaganovich, A.; Beilina, A.; Taymans, J.M.; Daniels, V.; Lewis, P.; Jain, S.; Ding, J.; Syed, A.; et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 2008, 283, 16906–16914. [Google Scholar] [CrossRef] [Green Version]
- Daniels, V.; Vancraenenbroeck, R.; Law, B.M.; Greggio, E.; Lobbestael, E.; Gao, F.; De Maeyer, M.; Cookson, M.R.; Harvey, K.; Baekelandt, V.; et al. Insight into the mode of action of the LRRK2 Y1699C pathogenic mutant. J. Neurochem. 2011, 116, 304–315. [Google Scholar] [CrossRef] [Green Version]
- Civiero, L.; Vancraenenbroeck, R.; Belluzzi, E.; Beilina, A.; Lobbestael, E.; Reyniers, L.; Gao, F.; Micetic, I.; De Maeyer, M.; Bubacco, L.; et al. Biochemical characterization of highly purified leucine-rich repeat kinases 1 and 2 demonstrates formation of homodimers. PLoS ONE 2012, 7, e43472. [Google Scholar] [CrossRef] [Green Version]
- Guaitoli, G.; Raimondi, F.; Gilsbach, B.K.; Gomez-Llorente, Y.; Deyaert, E.; Renzi, F.; Li, X.; Schaffner, A.; Jagtap, P.K.; Boldt, K.; et al. Structural model of the dimeric Parkinson’s protein LRRK2 reveals a compact architecture involving distant interdomain contacts. Proc. Natl. Acad. Sci. USA 2016, 113, 4357–4366. [Google Scholar] [CrossRef] [Green Version]
- Berger, Z.; Smith, K.A.; Lavoie, M.J. Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 2010, 49, 5511–5523. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Webber, P.J.; West, A.B. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J. Biol. Chem. 2009, 284, 36346–36356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deyaert, E.; Wauters, L.; Guaitoli, G.; Konijnenberg, A.; Leemans, M.; Terheyden, S.; Petrovic, A.; Gallardo, R.; Nederveen-Schippers, L.M.; Athanasopoulos, P.S.; et al. A homologue of the Parkinson’s disease-associated protein LRRK2 undergoes a monomer-dimer transition during GTP turnover. Nat. Commun. 2017, 8, 1008. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Trancikova, A.; Civiero, L.; Glauser, L.; Bubacco, L.; Greggio, E.; Moore, D.J. GTPase activity regulates kinase activity and cellular phenotypes of Parkinson’s disease-associated LRRK2. Hum. Mol. Genet. 2013, 22, 1140–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greggio, E.; Taymans, J.M.; Zhen, E.Y.; Ryder, J.; Vancraenenbroeck, R.; Beilina, A.; Sun, P.; Deng, J.; Jaffe, H.; Baekelandt, V.; et al. The Parkinson’s disease kinase LRRK2 autophosphorylates its GTPase domain at multiple sites. Biochem. Biophys. Res. Commun. 2009, 389, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, P.J.; Smith, A.D.; Sen, S.; Renfrow, M.B.; Mobley, J.A.; West, A.B. Autophosphorylation in the leucine-rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP-binding activities. J. Mol. Biol. 2011, 412, 94–110. [Google Scholar] [CrossRef] [Green Version]
- Gloeckner, C.J.; Boldt, K.; von Zweydorf, F.; Helm, S.; Wiesent, L.; Sarioglu, H.; Ueffing, M. Phosphopeptide analysis reveals two discrete clusters of phosphorylation in the N-terminus and the Roc domain of the Parkinson-disease associated protein kinase LRRK2. J. Proteome. Res. 2010, 9, 1738–1745. [Google Scholar] [CrossRef]
- Xiong, Y.; Yuan, C.; Chen, R.; Dawson, T.M.; Dawson, V.L. ArfGAP1 is a GTPase activating protein for LRRK2: Reciprocal regulation of ArfGAP1 by LRRK2. J. Neurosci. 2012, 32, 3877–3886. [Google Scholar] [CrossRef] [Green Version]
- Stafa, K.; Trancikova, A.; Webber, P.J.; Glauser, L.; West, A.B.; Moore, D.J. GTPase activity and neuronal toxicity of Parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet. 2012, 8, e1002526. [Google Scholar] [CrossRef]
- Dusonchet, J.; Li, H.; Guillily, M.; Liu, M.; Stafa, K.; Derada Troletti, C.; Boon, J.Y.; Saha, S.; Glauser, L.; Mamais, A.; et al. A Parkinson’s disease gene regulatory network identifies the signaling protein RGS2 as a modulator of LRRK2 activity and neuronal toxicity. Hum. Mol. Genet. 2014, 23, 4887–4905. [Google Scholar] [CrossRef] [Green Version]
- Haebig, K.; Gloeckner, C.J.; Miralles, M.G.; Gillardon, F.; Schulte, C.; Riess, O.; Ueffing, M.; Biskup, S.; Bonin, M. ARHGEF7 (Beta-PIX) acts as guanine nucleotide exchange factor for leucine-rich repeat kinase 2. PLoS ONE 2010, 5, e13762. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Lewis, P.A.; Singleton, A.B. LRRK2: Cause, risk, and mechanism. J. Parkinsons Dis. 2013, 3, 85–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloeckner, C.J.; Kinkl, N.; Schumacher, A.; Braun, R.J.; O’Neill, E.; Meitinger, T.; Kolch, W.; Prokisch, H.; Ueffing, M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum. Mol. Genet. 2006, 15, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Bender, S.; Kang, S.; Lin, R.; Glicksman, M.A.; Liu, M. The Parkinson disease-linked LRRK2 protein mutation I2020T stabilizes an active state conformation leading to increased kinase activity. J. Biol. Chem. 2014, 289, 13042–13053. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.W.; Pei, Z.; Jiang, H.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 2006, 9, 1231–1233. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Wu, C.X.; Burlak, C.; Zhang, S.; Sahm, H.; Wang, M.; Zhang, Z.Y.; Vogel, K.W.; Federici, M.; Riddle, S.M.; et al. Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc. Natl. Acad. Sci. USA 2014, 111, 4055–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greggio, E.; Jain, S.; Kingsbury, A.; Bandopadhyay, R.; Lewis, P.; Kaganovich, A.; van der Brug, M.P.; Beilina, A.; Blackinton, J.; Thomas, K.J.; et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006, 23, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Yu, M.; Wang, C.; Xu, Z. Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. J. Neurochem. 2012, 122, 650–658. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, D.; Dowman, J.; Hammond, R.; Leete, T.; Inoue, K.; Abeliovich, A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 2006, 52, 587–593. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, N.; Perrin, J.; Ren, Z.; Lee, B.D.; Zinn, N.; Dawson, V.L.; Tam, D.; Bova, M.; Lang, M.; Drewes, G.; et al. Chemoproteomics-based design of potent LRRK2-selective lead compounds that attenuate Parkinson’s disease-related toxicity in human neurons. ACS Chem. Biol. 2011, 6, 1021–1028. [Google Scholar] [CrossRef]
- Lin, C.H.; Tsai, P.I.; Wu, R.M.; Chien, C.T. LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. J. Neurosci. 2010, 30, 13138–13149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venderova, K.; Kabbach, G.; Abdel-Messih, E.; Zhang, Y.; Parks, R.J.; Imai, Y.; Gehrke, S.; Ngsee, J.; Lavoie, M.J.; Slack, R.S.; et al. Leucine-Rich Repeat Kinase 2 interacts with Parkin, DJ-1 and PINK-1 in a Drosophila melanogaster model of Parkinson’s disease. Hum. Mol. Genet. 2009, 18, 4390–4404. [Google Scholar] [CrossRef] [PubMed]
- Hindle, S.; Afsari, F.; Stark, M.; Middleton, C.A.; Evans, G.J.; Sweeney, S.T.; Elliott, C.J. Dopaminergic expression of the Parkinsonian gene LRRK2-G2019S leads to non-autonomous visual neurodegeneration, accelerated by increased neural demands for energy. Hum. Mol. Genet. 2013, 22, 2129–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, C.H.; Mok, S.Z.; Koh, C.; Ouyang, X.; Fivaz, M.L.; Tan, E.K.; Dawson, V.L.; Dawson, T.M.; Yu, F.; Lim, K.L. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J. Neurosci. 2009, 29, 11257–11262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Hamamichi, S.; Lee, B.D.; Yang, D.; Ray, A.; Caldwell, G.A.; Caldwell, K.A.; Dawson, T.M.; Smith, W.W.; Dawson, V.L. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum. Mol. Genet. 2011, 20, 3933–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.; El Khoury, R.; Wang, W.; Byrd, T.A.; Pehek, E.A.; Thacker, C.; Zhu, X.; Smith, M.A.; Wilson-Delfosse, A.L.; Chen, S.G. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol. Dis. 2010, 40, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsika, E.; Nguyen, A.P.; Dusonchet, J.; Colin, P.; Schneider, B.L.; Moore, D.J. Adenoviral-mediated expression of G2019S LRRK2 induces striatal pathology in a kinase-dependent manner in a rat model of Parkinson’s disease. Neurobiol. Dis. 2015, 77, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Weng, Y.H.; Chien, K.Y.; Lin, K.J.; Yeh, T.H.; Cheng, Y.P.; Lu, C.S.; Wang, H.L. (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 2012, 19, 1623–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, J.S.; Chen, C.Y.; Chen, Y.L.; Weng, Y.H.; Yeh, T.H.; Lu, C.S.; Chang, Y.M.; Wang, H.L. (G2019S) LRRK2 causes early-phase dysfunction of SNpc dopaminergic neurons and impairment of corticostriatal long-term depression in the PD transgenic mouse. Neurobiol. Dis. 2014, 68, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, T.; Mori, S.; Sasaki, Y.; Miyajima, T.; Azuma, S.; Ohta, E.; Obata, F. The I2020T Leucine-rich repeat kinase 2 transgenic mouse exhibits impaired locomotive ability accompanied by dopaminergic neuron abnormalities. Mol. Neurodegener. 2012, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, Y.H.; Chen, C.Y.; Lin, K.J.; Chen, Y.L.; Yeh, T.H.; Hsiao, I.T.; Chen, I.J.; Lu, C.S.; Wang, H.L. (R1441C) LRRK2 induces the degeneration of SN dopaminergic neurons and alters the expression of genes regulating neuronal survival in a transgenic mouse model. Exp. Neurol. 2016, 275, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Neifert, S.; Karuppagounder, S.S.; Liu, Q.; Stankowski, J.N.; Lee, B.D.; Ko, H.S.; Lee, Y.; Grima, J.C.; Mao, X.; et al. Robust kinase- and age-dependent dopaminergic and norepinephrine neurodegeneration in LRRK2 G2019S transgenic mice. Proc. Natl. Acad. Sci. USA 2018, 115, 1635–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giesert, F.; Glasl, L.; Zimprich, A.; Ernst, L.; Piccoli, G.; Stautner, C.; Zerle, J.; Holter, S.M.; Vogt Weisenhorn, D.M.; Wurst, W. The pathogenic LRRK2 R1441C mutation induces specific deficits modeling the prodromal phase of Parkinson’s disease in the mouse. Neurobiol. Dis. 2017, 105, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Mercatelli, D.; Novello, S.; Arcuri, L.; Brugnoli, A.; Vincenzi, F.; Russo, I.; Berti, G.; Mabrouk, O.S.; Kennedy, R.T.; et al. Age-dependent dopamine transporter dysfunction and Serine129 phospho-alpha-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol. Commun. 2017, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Pisani, A.; Martella, G.; Karouani, M.; Yamaguchi, H.; Pothos, E.N.; Shen, J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14622–14627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Patel, J.C.; Wang, J.; Avshalumov, M.V.; Nicholson, C.; Buxbaum, J.D.; Elder, G.A.; Rice, M.E.; Yue, Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J. Neurosci. 2010, 30, 1788–1797. [Google Scholar] [CrossRef]
- Bichler, Z.; Lim, H.C.; Zeng, L.; Tan, E.K. Non-motor and motor features in LRRK2 transgenic mice. PLoS ONE 2013, 8, e70249. [Google Scholar] [CrossRef] [Green Version]
- Herzig, M.C.; Kolly, C.; Persohn, E.; Theil, D.; Schweizer, T.; Hafner, T.; Stemmelen, C.; Troxler, T.J.; Schmid, P.; Danner, S.; et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet. 2011, 20, 4209–4223. [Google Scholar] [CrossRef] [Green Version]
- Yue, M.; Hinkle, K.M.; Davies, P.; Trushina, E.; Fiesel, F.C.; Christenson, T.A.; Schroeder, A.S.; Zhang, L.; Bowles, E.; Behrouz, B.; et al. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 2015, 78, 172–195. [Google Scholar] [CrossRef] [Green Version]
- Kalogeropulou, A.F.; Zhao, J.; Bolliger, M.F.; Memou, A.; Narasimha, S.; Molitor, T.P.; Wilson, W.H.; Rideout, H.J.; Nichols, R.J. P62/SQSTM1 is a novel leucine-rich repeat kinase 2 (LRRK2) substrate that enhances neuronal toxicity. Biochem. J. 2018, 475, 1271–1293. [Google Scholar] [CrossRef] [Green Version]
- Kiral, F.R.; Kohrs, F.E.; Jin, E.J.; Hiesinger, P.R. Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr. Biol. 2018, 28, R471–R486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Fukuda, M. Small GTPase Rab12 regulates transferrin receptor degradation: Implications for a novel membrane trafficking pathway from recycling endosomes to lysosomes. Cell Logist. 2011, 1, 155–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wei, Z.; Fan, Y.; Huang, W.; Su, Y.; Li, H.; Dong, Z.; Fukuda, M.; Khater, M.; Wu, G. The GTPase Rab43 Controls the Anterograde ER-Golgi Trafficking and Sorting of GPCRs. Cell Rep. 2017, 21, 1089–1101. [Google Scholar] [CrossRef] [Green Version]
- Yun, H.J.; Kim, H.; Ga, I.; Oh, H.; Ho, D.H.; Kim, J.; Seo, H.; Son, I.; Seol, W. An early endosome regulator, Rab5b, is an LRRK2 kinase substrate. J. Biochem. 2015, 157, 485–495. [Google Scholar] [CrossRef]
- Parisiadou, L.; Xie, C.; Cho, H.J.; Lin, X.; Gu, X.L.; Long, C.X.; Lobbestael, E.; Baekelandt, V.; Taymans, J.M.; Sun, L.; et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J. Neurosci. 2009, 29, 13971–13980. [Google Scholar] [CrossRef]
- Gillardon, F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability—A point of convergence in parkinsonian neurodegeneration? J. Neurochem. 2009, 110, 1514–1522. [Google Scholar] [CrossRef]
- Kawakami, F.; Yabata, T.; Ohta, E.; Maekawa, T.; Shimada, N.; Suzuki, M.; Maruyama, H.; Ichikawa, T.; Obata, F. LRRK2 phosphorylates tubulin-associated tau but not the free molecule: LRRK2-mediated regulation of the tau-tubulin association and neurite outgrowth. PLoS ONE 2012, 7, e30834. [Google Scholar] [CrossRef]
- Krumova, P.; Reyniers, L.; Meyer, M.; Lobbestael, E.; Stauffer, D.; Gerrits, B.; Muller, L.; Hoving, S.; Kaupmann, K.; Voshol, J.; et al. Chemical genetic approach identifies microtubule affinity-regulating kinase 1 as a leucine-rich repeat kinase 2 substrate. FASEB J. 2015, 29, 2980–2992. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Liu, H.P.; Lin, W.Y.; Guo, H.; Lu, B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J. Neurosci. 2010, 30, 16959–16969. [Google Scholar] [CrossRef] [Green Version]
- Kanao, T.; Venderova, K.; Park, D.S.; Unterman, T.; Lu, B.; Imai, Y. Activation of FoxO by LRRK2 induces expression of proapoptotic proteins and alters survival of postmitotic dopaminergic neuron in Drosophila. Hum. Mol. Genet. 2010, 19, 3747–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, Y.; Gehrke, S.; Wang, H.Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008, 27, 2432–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, I.; Kim, J.W.; Lee, B.D.; Kang, H.C.; Xu, J.C.; Jia, H.; Stankowski, J.; Kim, M.S.; Zhong, J.; Kumar, M.; et al. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson’s disease. Cell 2014, 157, 472–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matta, S.; Van Kolen, K.; da Cunha, R.; van den Bogaart, G.; Mandemakers, W.; Miskiewicz, K.; De Bock, P.J.; Morais, V.A.; Vilain, S.; Haddad, D.; et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 2012, 75, 1008–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, H.J.; Park, J.; Ho, D.H.; Kim, H.; Kim, C.H.; Oh, H.; Ga, I.; Seo, H.; Chang, S.; Son, I.; et al. LRRK2 phosphorylates Snapin and inhibits interaction of Snapin with SNAP-25. Exp. Mol. Med. 2013, 45, e36. [Google Scholar] [CrossRef] [Green Version]
- Belluzzi, E.; Gonnelli, A.; Cirnaru, M.D.; Marte, A.; Plotegher, N.; Russo, I.; Civiero, L.; Cogo, S.; Carrion, M.P.; Franchin, C.; et al. LRRK2 phosphorylates pre-synaptic N-ethylmaleimide sensitive fusion (NSF) protein enhancing its ATPase activity and SNARE complex disassembling rate. Mol. Neurodegener. 2016, 11, 1. [Google Scholar] [CrossRef]
- Islam, M.S.; Nolte, H.; Jacob, W.; Ziegler, A.B.; Putz, S.; Grosjean, Y.; Szczepanowska, K.; Trifunovic, A.; Braun, T.; Heumann, H.; et al. Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet. 2016, 25, 5365–5382. [Google Scholar] [CrossRef] [Green Version]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. eLife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Jeong, G.R.; Jang, E.H.; Bae, J.R.; Jun, S.; Kang, H.C.; Park, C.H.; Shin, J.H.; Yamamoto, Y.; Tanaka-Yamamoto, K.; Dawson, V.L.; et al. Dysregulated phosphorylation of Rab GTPases by LRRK2 induces neurodegeneration. Mol. Neurodegener. 2018, 13, 8. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef]
- Gao, Y.L.; Wang, N.; Sun, F.R.; Cao, X.P.; Zhang, W.; Yu, J.T. Tau in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef]
- Li, Y.; Liu, W.; Oo, T.F.; Wang, L.; Tang, Y.; Jackson-Lewis, V.; Zhou, C.; Geghman, K.; Bogdanov, M.; Przedborski, S.; et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 2009, 12, 826–828. [Google Scholar] [CrossRef]
- Melrose, H.L.; Dachsel, J.C.; Behrouz, B.; Lincoln, S.J.; Yue, M.; Hinkle, K.M.; Kent, C.B.; Korvatska, E.; Taylor, J.P.; Witten, L.; et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 2010, 40, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Rajput, A.; Dickson, D.W.; Robinson, C.A.; Ross, O.A.; Dachsel, J.C.; Lincoln, S.J.; Cobb, S.A.; Rajput, M.L.; Farrer, M.J. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 2006, 67, 1506–1508. [Google Scholar] [CrossRef]
- Picconi, B.; Piccoli, G.; Calabresi, P. Synaptic dysfunction in Parkinson’s disease. Adv. Exp. Med. Biol. 2012, 970, 553–572. [Google Scholar] [CrossRef]
- Arranz, A.M.; Delbroek, L.; Van Kolen, K.; Guimaraes, M.R.; Mandemakers, W.; Daneels, G.; Matta, S.; Calafate, S.; Shaban, H.; Baatsen, P.; et al. LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J. Cell Sci. 2015, 128, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.; Krainc, D. LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 5576–5581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toma-Fukai, S.; Shimizu, T. Structural Insights into the Regulation Mechanism of Small GTPases by GEFs. Molecules 2019, 24, 3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhekne, H.S.; Yanatori, I.; Gomez, R.C.; Tonelli, F.; Diez, F.; Schule, B.; Steger, M.; Alessi, D.R.; Pfeffer, S.R. A pathway for Parkinson’s Disease LRRK2 kinase to block primary cilia and Sonic hedgehog signaling in the brain. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Madero-Perez, J.; Fdez, E.; Fernandez, B.; Lara Ordonez, A.J.; Blanca Ramirez, M.; Gomez-Suaga, P.; Waschbusch, D.; Lobbestael, E.; Baekelandt, V.; Nairn, A.C.; et al. Parkinson disease-associated mutations in LRRK2 cause centrosomal defects via Rab8a phosphorylation. Mol. Neurodegener. 2018, 13, 3. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, B.; Lara Ordonez, A.J.; Fdez, E.; Mutez, E.; Comptdaer, T.; Leghay, C.; Kreisler, A.; Simonin, C.; Vandewynckel, L.; Defebvre, L.; et al. Centrosomal cohesion deficits as cellular biomarker in lymphoblastoid cell lines from LRRK2 Parkinson’s disease patients. Biochem. J. 2019, 476, 2797–2813. [Google Scholar] [CrossRef] [Green Version]
- Rivero-Rios, P.; Romo-Lozano, M.; Madero-Perez, J.; Thomas, A.P.; Biosa, A.; Greggio, E.; Hilfiker, S. The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A. J. Biol. Chem. 2019, 294, 4738–4758. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, T.; Kuwahara, T.; Sakurai, M.; Komori, T.; Fujimoto, T.; Ito, G.; Yoshimura, S.I.; Harada, A.; Fukuda, M.; Koike, M.; et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9115–E9124. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.C.; Yeh, T.H.; Lai, S.C.; Weng, Y.H.; Huang, Y.C.; Cheng, Y.C.; Chen, R.S.; Huang, Y.Z.; Hung, J.; Chen, C.C.; et al. Increased Rab35 expression is a potential biomarker and implicated in the pathogenesis of Parkinson’s disease. Oncotarget 2016, 7, 54215–54227. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Gronborg, M.; Mobius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef]
- Kouranti, I.; Sachse, M.; Arouche, N.; Goud, B.; Echard, A. Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Curr. Biol. 2006, 16, 1719–1725. [Google Scholar] [CrossRef] [Green Version]
- Malik, B.R.; Maddison, D.C.; Smith, G.A.; Peters, O.M. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain. 2019, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.M.; Jang, E.H.; Jeong, G.R.; Lee, B.D. LRRK2 and membrane trafficking: Nexus of Parkinson’s disease. BMB Rep. 2019, 52, 533–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J., 3rd; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9879–9884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.W.; Zhang, T.; Jiang, C.; Chen, S.; Guo, M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012, 21, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Giaime, E.; Yamaguchi, H.; Ichimura, T.; Liu, Y.; Si, H.; Cai, H.; Bonventre, J.V.; Shen, J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 2012, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Nuber, S.; Rajsombath, M.; Minakaki, G.; Winkler, J.; Muller, C.P.; Ericsson, M.; Caldarone, B.; Dettmer, U.; Selkoe, D.J. Abrogating Native alpha-Synuclein Tetramers in Mice Causes a L-DOPA-Responsive Motor Syndrome Closely Resembling Parkinson’s Disease. Neuron 2018, 100, 75–90.e75. [Google Scholar] [CrossRef] [Green Version]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Harper, J.D.; Williamson, R.E.; Lansbury, P.T., Jr. Accelerated oligomerization by Parkinson’s disease linked alpha-synuclein mutants. Ann. N. Y. Acad. Sci. 2000, 920, 42–45. [Google Scholar] [CrossRef]
- Leverenz, J.B.; Umar, I.; Wang, Q.; Montine, T.J.; McMillan, P.J.; Tsuang, D.W.; Jin, J.; Pan, C.; Shin, J.; Zhu, D.; et al. Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol. 2007, 17, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Liao, L.; Cheng, D.; Duong, D.M.; Gearing, M.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic identification of novel proteins associated with Lewy bodies. Front. Biosci. 2008, 13, 3850–3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miklossy, J.; Arai, T.; Guo, J.P.; Klegeris, A.; Yu, S.; McGeer, E.G.; McGeer, P.L. LRRK2 expression in normal and pathologic human brain and in human cell lines. J. Neuropathol. Exp. Neurol. 2006, 65, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Siedlak, S.L.; Smith, M.A.; Perry, G.; Chen, S.G. LRRK2 protein is a component of Lewy bodies. Ann. Neurol. 2006, 60, 617–618. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Babar, A.; Siedlak, S.L.; Yang, Q.; Ito, G.; Iwatsubo, T.; Smith, M.A.; Perry, G.; Chen, S.G. LRRK2 in Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 2006, 1, 17. [Google Scholar] [CrossRef] [Green Version]
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain 2004, 127, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Meredith, G.E.; Chen, L.; Zhou, Y.; Xu, J.; Shie, F.S.; Lockhart, P.; Zhang, J. Quantitative proteomic analysis of mitochondrial proteins: Relevance to Lewy body formation and Parkinson’s disease. Brain Res. Mol. Brain. Res. 2005, 134, 119–138. [Google Scholar] [CrossRef]
- Schlossmacher, M.G.; Frosch, M.P.; Gai, W.P.; Medina, M.; Sharma, N.; Forno, L.; Ochiishi, T.; Shimura, H.; Sharon, R.; Hattori, N.; et al. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am. J. Pathol. 2002, 160, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Muqit, M.M.; Stanyer, L.; Healy, D.G.; Abou-Sleiman, P.M.; Hargreaves, I.; Heales, S.; Ganguly, M.; Parsons, L.; Lees, A.J.; et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain 2006, 129, 1720–1731. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Mehta, S.H.; Sethi, K.D. Learning PD from the lark. Mov. Disord. 2011, 26, 2178. [Google Scholar] [CrossRef] [PubMed]
- Wider, C.; Dickson, D.W.; Wszolek, Z.K. Leucine-rich repeat kinase 2 gene-associated disease: Redefining genotype-phenotype correlation. Neurodegener. Dis. 2010, 7, 175–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulopoulos, M.; Cortes, E.; Vonsattel, J.P.; Fahn, S.; Waters, C.; Cote, L.J.; Moskowitz, C.; Honig, L.S.; Clark, L.N.; Marder, K.S.; et al. Clinical and pathological characteristics of LRRK2 G2019S patients with PD. J. Mol. Neurosci. 2012, 47, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalia, L.V.; Lang, A.E.; Hazrati, L.N.; Fujioka, S.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A.; Van Deerlin, V.M.; Trojanowski, J.Q.; Hurtig, H.I.; et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 2015, 72, 100–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agin-Liebes, J.; Cortes, E.; Vonsattel, J.P.; Marder, K.; Alcalay, R.N. Movement disorders rounds: A case of missing pathology in a patient with LRRK2 Parkinson’s disease. Parkinsonism. Relat. Disord. 2020, 74, 76–77. [Google Scholar] [CrossRef]
- Henderson, M.X.; Sengupta, M.; Trojanowski, J.Q.; Lee, V.M.Y. Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 2019, 7, 183. [Google Scholar] [CrossRef] [Green Version]
- Kalia, L.V. Expert comment: “A case of missing pathology in a patient with LRRK2 Parkinson’s disease”. Parkinsonism Relat. Disord. 2020, 74, 78–79. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T., Jr. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef] [Green Version]
- Ding, T.T.; Lee, S.J.; Rochet, J.C.; Lansbury, P.T., Jr. Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 2002, 41, 10209–10217. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef]
- Aasly, J.O.; Johansen, K.K.; Bronstad, G.; Waro, B.J.; Majbour, N.K.; Varghese, S.; Alzahmi, F.; Paleologou, K.E.; Amer, D.A.; Al-Hayani, A.; et al. Elevated levels of cerebrospinal fluid alpha-synuclein oligomers in healthy asymptomatic LRRK2 mutation carriers. Front. Aging Neurosci. 2014, 6, 248. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.; Leung, C.T.; Liu, H.; Pang, S.Y.; Lam, C.S.; Xian, J.; Li, L.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Age-dependent accumulation of oligomeric SNCA/alpha-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (CMA). Autophagy 2020, 16, 347–370. [Google Scholar] [CrossRef] [PubMed]
- Novello, S.; Arcuri, L.; Dovero, S.; Dutheil, N.; Shimshek, D.R.; Bezard, E.; Morari, M. G2019S LRRK2 mutation facilitates alpha-synuclein neuropathology in aged mice. Neurobiol. Dis. 2018, 120, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Parisiadou, L.; Gu, X.L.; Wang, L.; Shim, H.; Sun, L.; Xie, C.; Long, C.X.; Yang, W.J.; Ding, J.; et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 2009, 64, 807–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieri, G.; Brahic, M.; Bousset, L.; Couthouis, J.; Kramer, N.J.; Ma, R.; Nakayama, L.; Monbureau, M.; Defensor, E.; Schule, B.; et al. LRRK2 modifies alpha-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019, 137, 961–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karampetsou, M.; Ardah, M.T.; Semitekolou, M.; Polissidis, A.; Samiotaki, M.; Kalomoiri, M.; Majbour, N.; Xanthou, G.; El-Agnaf, O.M.A.; Vekrellis, K. Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep. 2017, 7, 16533. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef]
- Kondo, K.; Obitsu, S.; Teshima, R. alpha-Synuclein aggregation and transmission are enhanced by leucine-rich repeat kinase 2 in human neuroblastoma SH-SY5Y cells. Biol. Pharm. Bull. 2011, 34, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.J.; Kim, D.K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 kinase regulates alpha-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 2018, 9, 3465. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, T.; Iwatsubo, T. The Emerging Functions of LRRK2 and Rab GTPases in the Endolysosomal System. Front. Neurosci. 2020, 14, 227. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozina, E.; Sadasivan, S.; Jiao, Y.; Dou, Y.; Ma, Z.; Tan, H.; Kodali, K.; Shaw, T.; Peng, J.; Smeyne, R.J. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain 2018, 141, 1753–1769. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; James, W.S.; Cowley, S.A. LRRK2 in peripheral and central nervous system innate immunity: Its link to Parkinson’s disease. Biochem. Soc. Trans. 2017, 45, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Protein | Substrate Phosphorylation | Phospho-Site | Potential Role | Reference | |
|---|---|---|---|---|---|
| In Vitro | In Vivo | ||||
| ArfGAP1 | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ↓ Lrrk2 Knock-out (KO) mouse brain | S155, 246, 284 T189, 216, 292 | GTPase activating protein (GAP) for LRRK2 | [27] |
| β-tubulin | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 D1994A | ND | ND | A component of microtubule (MT) Neurite outgrowth | [67] |
| 4E-BP-1 | ↑ dLRRK WT, Y1383C, I1915T ↑ hLRRK2 WT, I2020T ↓ dLRRK 3KD | ↑ hLRRK2WT, I2020T in 293T cells ↓ hLRRK2 3KD in 293T cells | T37/46 | Cap-dependent protein translation Survival under starvation, oxidative, and unfolded protein stress | [72] |
| Endophilin A | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ↑ hLRRK2 WT, G2019S in CHO cells and Drosophila ↓ hLRRK2 KD in CHO cells and Drosophila | S75 | Regulation of membrane curvature Synaptic vesicle endocytosis | [74] |
| FoxO1 | ↑ dLRRK ↑ hLRRK2 WT ↓ hLRRK2 3KD | ↑ hLRRK2 WT, G2019S in 293T cells ↓ hLRRK2 3KD in 293T cells ↓ dLRRK null in Drosophila | S319 | Transcriptional regulation of pro-apoptotic genes | [71] |
| Futsch | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ND | ND | Microtubule-association protein (MAP), regulation of MT dynamics Negative regulator of synaptic functions | [70] |
| MARK1 | ↑ hLRRK2 G2019S | ↑ hLRRK2 WT, G2019S in HEK-293 cells ↓ hLRRK2 KD in HEK-293T cells | ND | Regulation of MT stability through phosphorylation of MAPs | [69] |
| Moesin/Ezrin/Radixin | ↑ hLRRK2 WT, G2019S | ↑ hLRRK2 WT, G2019S in HEK-293 cells | T558 | Actin cytoskeleton rearrangement, neurite outgrowth, neuronal morphogenesis | [12,66] |
| NSF | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ND | T645 | SNARE complex dissociation, synaptic vesicle endocytosis | [76] |
| P62/SQSTM1 | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ↑ hLRRK2 WT, G2019S, N1437, R1441G, Y1699C in HEK-293 cells ↓ hLRRK2 KD in HEK-293 cells | T138 | Autophagy | [60] |
| Rab1a/b/c | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ↑ hLRRK2 WT, G2019S in HEK-293 cells ↓ hLRRK2 KD in HEK-293 cells | T75 | Endoplasmic reticulum (ER)-Golgi trafficking | [61,78,80] |
| Rab3a/b/c/d | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ↑ hLRRK2 WT, R1441G, Y1699C, G2019S in HEK-293 cells | T94 | Exocytosis, neurotransmitter release | [62,79,80] |
| Rab5a/b/c | ↑ hLRRK2 WT | ↑ hLRRK2 WT, R1441G, Y1699C, G2019S in HEK-293 cells | T6 | Early and recycling endosomal trafficking | [61,65,78,79] |
| Rab8a/b | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ↑ hLRRK2 WT, R1441C/G/H, Y1699C, I2020T, G2019S in HEK-293 cells | T72 | Post-Golgi trafficking, ciliogenesis | [61,78,79,80] |
| Rab10 | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ↑ hLRRK2 WT, R1441C/G/H, Y1699C, I2020T, G2019S in HEK-293 cells | T73 | Exocytosis, trans-Golgi/recycling endosome trafficking to plasma membrane | [62,78,79] |
| Rab12 | ↑ hLRRK2 WT, G2019S | ↑ hLRRK2 WT, R1441G, Y1699C, G2019S in HEK-293 cells | S106 | Recycling of endosomes and lysosomes, ciliogenesis | [63,64,78,79] |
| Rab29 | ↑ hLRRK2 WT | ↑ hLRRK2 WT, R1441G, Y1699C, G2019S in HEK-293 cells | S72 | Endolysosomal sorting/degradation | [61,78,79] |
| Rab35 | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ↑ hLRRK2 WT, R1441G, Y1699C, G2019S in HEK-293 cells | T72 | Recycling endosomal trafficking, exosome secretion | [61,79,80] |
| Rab43 | ND | ↑ hLRRK2 WT, R1441G, Y1699C, G2019S in HEK-293 cells | T82 | Anterograde ER-Golgi trafficking | [79] |
| RGS2 | ↑ hLRRK2 WT, G2019S, I2020T ↓ hLRRK2 KD | ↑ hLRRK2 WT, G2019S in HEK293 cells cells ↓ hLRRK2 KD in ES derived human DA cells | ND | GAP for LRRK2 | [29] |
| RPS15 | ↑ hLRRK2 WT, G2019S, I2020T ↓ hLRRK2 KD | ↑ hLRRK2 WT, G2019S in ES derived human DA cells ↓ hLRRK2 KD in ES derived human DA cells | T136 | Bulk protein translation | [73] |
| Snapin | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ND | T117 | Synaptic vesicle trafficking | [75] |
| Synaptojanin-1 | ↑ hLRRK2 WT, G2019S ↓ hLRRK2 KD | ND | T1343,1348,1452,1503 | Clathrin uncoating, down-regulation of actin polymerization, modulation of dynamin activity | [77] |
| Tau | ↑ hLRRK2 WT, G2019S, I2020T | ↑ hLRRK2 WT in SH-SY5Y cells ↓ hLRRK2 RNAi in SH-SY5Y cells | T181 | Modulation of microtubule dynamics Neurite outgrowth | [68] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, G.R.; Lee, B.D. Pathological Functions of LRRK2 in Parkinson’s Disease. Cells 2020, 9, 2565. https://doi.org/10.3390/cells9122565
Jeong GR, Lee BD. Pathological Functions of LRRK2 in Parkinson’s Disease. Cells. 2020; 9(12):2565. https://doi.org/10.3390/cells9122565
Chicago/Turabian StyleJeong, Ga Ram, and Byoung Dae Lee. 2020. "Pathological Functions of LRRK2 in Parkinson’s Disease" Cells 9, no. 12: 2565. https://doi.org/10.3390/cells9122565
APA StyleJeong, G. R., & Lee, B. D. (2020). Pathological Functions of LRRK2 in Parkinson’s Disease. Cells, 9(12), 2565. https://doi.org/10.3390/cells9122565