Comparative Transcriptomics Analyses in Livers of Mice, Humans, and Humanized Mice Define Human-Specific Gene Networks

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. RNA-Seq Analysis for Public Data
2.2. Microarray Analysis for Public GEO Data
2.3. Multiscale Embedded Gene Co-Expression Network Analysis (MEGENA)
2.4. Adenovirus Production
2.5. Animal Experiments
2.6. RNA Extraction and RNA-Seq Analysis
2.7. Human and Mouse Divergence Analysis
2.8. Pathway Enrichment Analysis
2.8.1. Gene Ontology (GO) Enrichment Analysis
2.8.2. Gene Set Enrichment Analysis (GSEA)
2.8.3. Gene Set Variation Analysis (GSVA)
2.9. Data Availability
3. Results
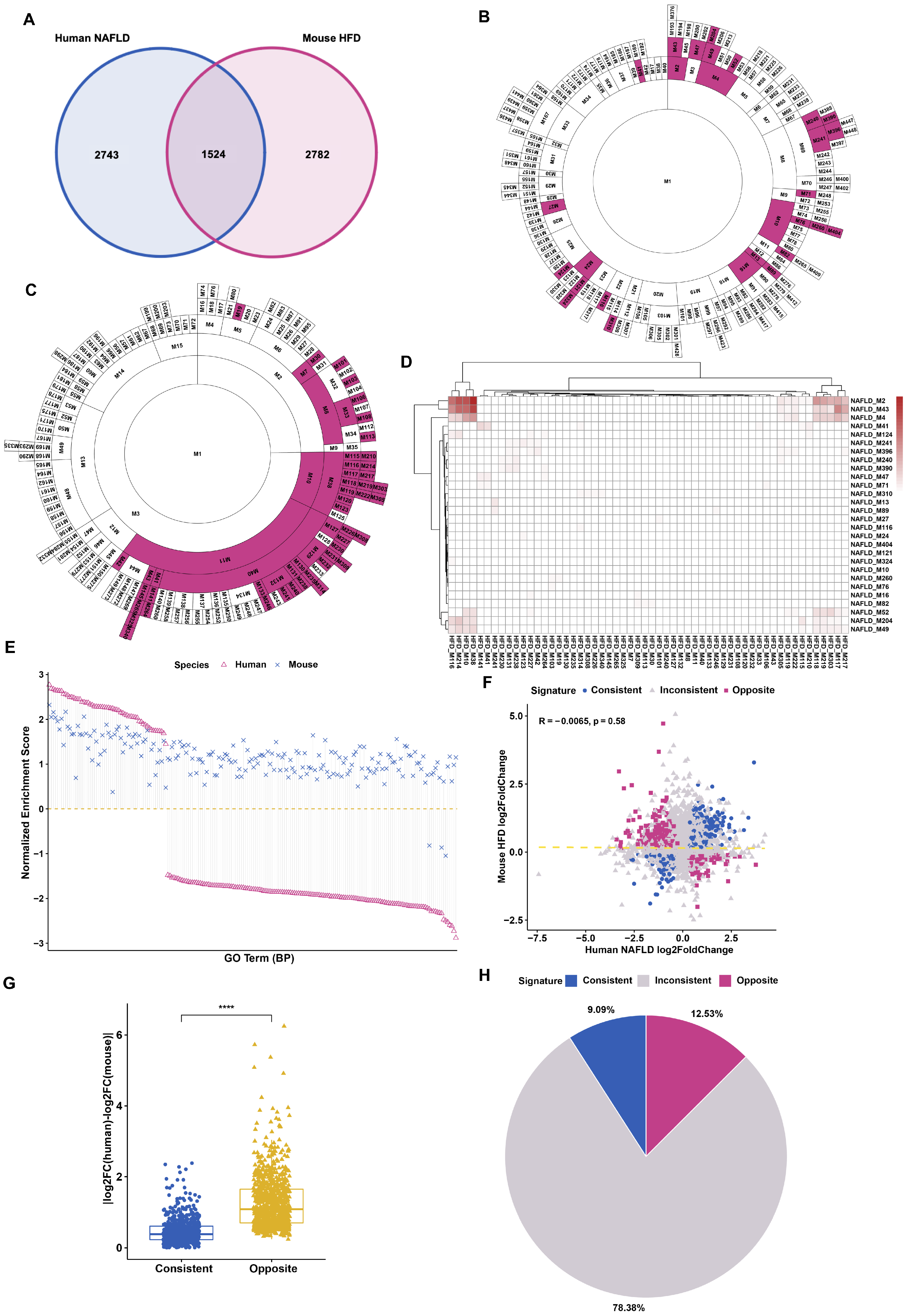
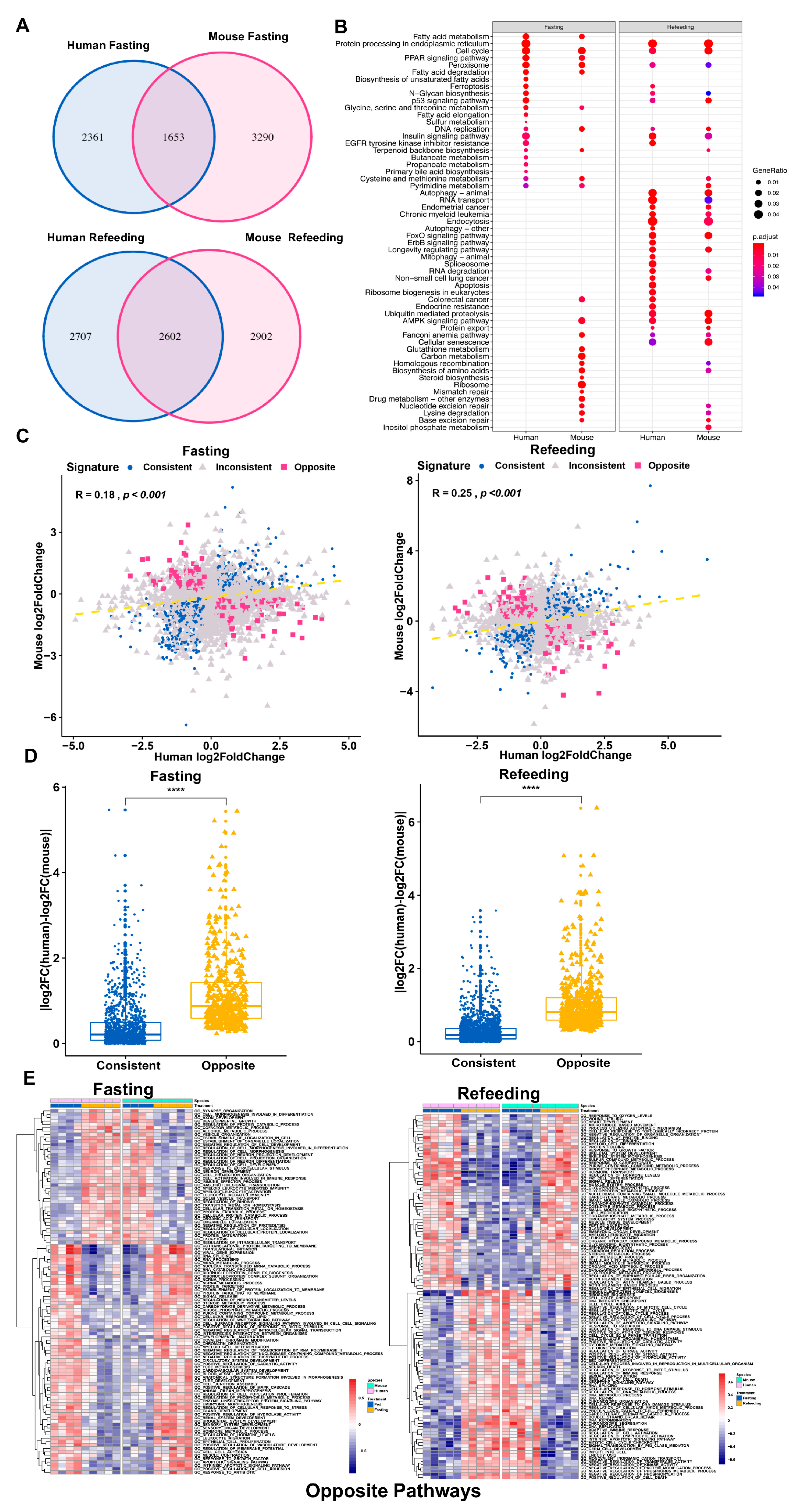
3.1. Differential Gene Regulation in Models of Fatty Liver Disease between Human and Mouse
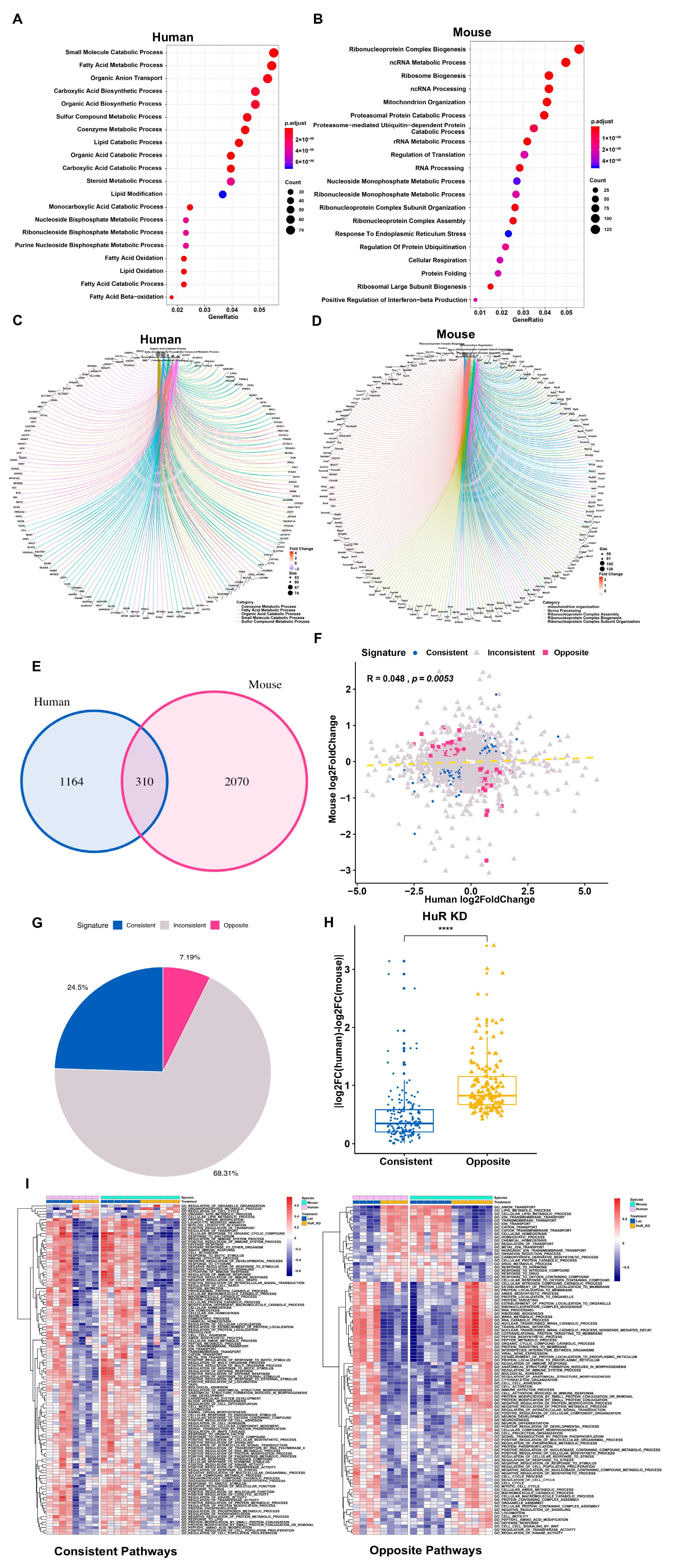
3.2. Distinct Response in Gene Expression between Human and Mouse under Comparable Conditions
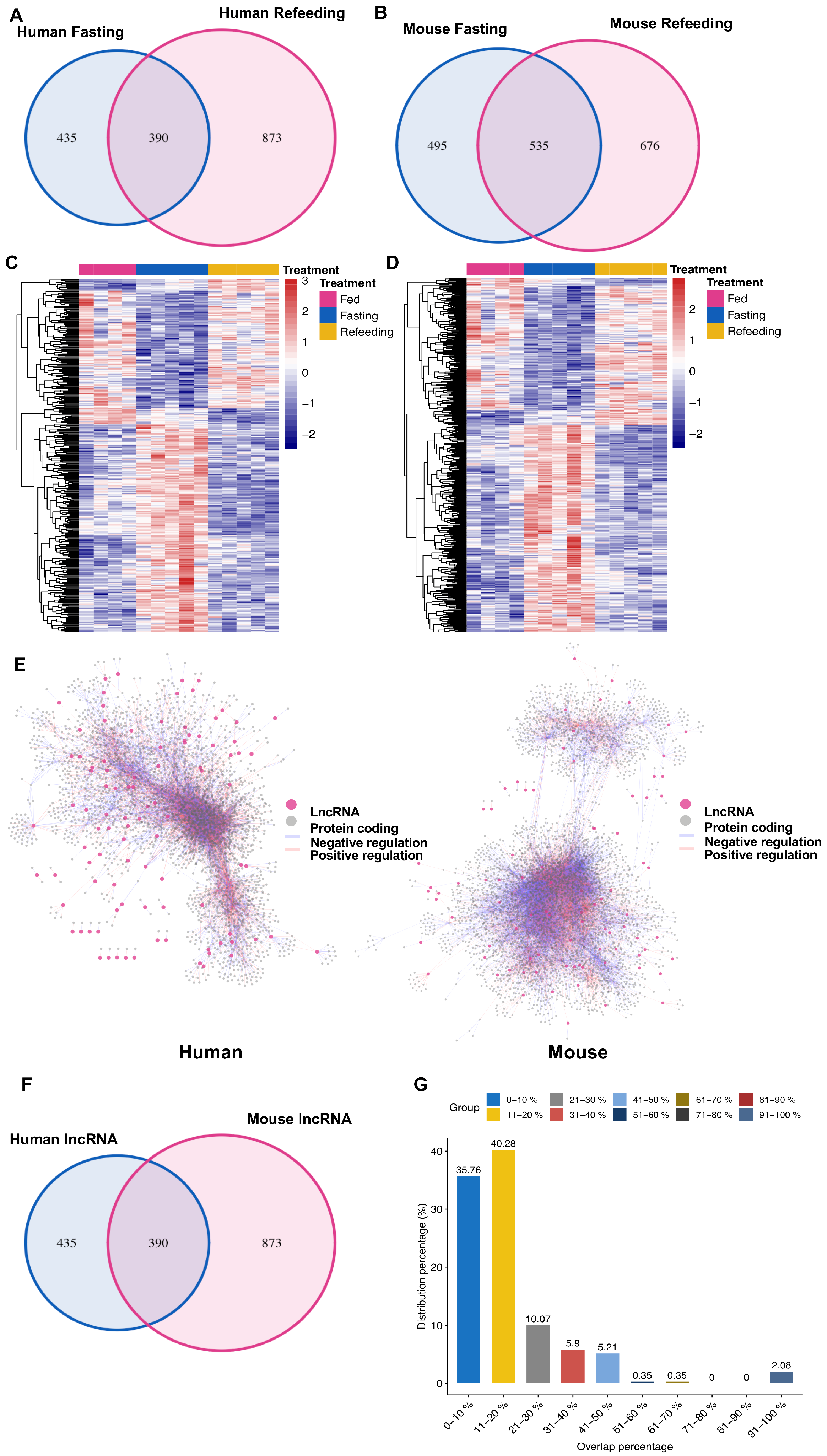
3.3. Distinct Response in Gene Expression between Human and Mouse in the Chimeric Livers
3.4. Nonconserved lncRNAs Might Contribute to the Divergent Gene Regulation between Human and Mouse
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- McKenzie, R.; Fried, M.W.; Sallie, R.; Conjeevaram, H.; Di Bisceglie, A.M.; Park, Y.; Savarese, B.; Kleiner, D.; Tsokos, M.; Luciano, C.; et al. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N. Engl. J. Med. 1995, 333, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Manning, F.J.; Swartz, M. (Eds.) Review of the Fialuridine (FIAU) Clinical Trials; National Academies Press: Washington, DC, USA, 1995. [Google Scholar]
- Xu, D.; Nishimura, T.; Nishimura, S.; Zhang, H.; Zheng, M.; Guo, Y.Y.; Masek, M.; Michie, S.A.; Glenn, J.; Peltz, G. Fialuridine induces acute liver failure in chimeric TK-NOG mice: A model for detecting hepatic drug toxicity prior to human testing. PLoS Med. 2014, 11, e1001628. [Google Scholar] [CrossRef]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schonfels, W.; Herrmann, A.; Bruckner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues from Patients. Gastroenterology 2016, 151, 513–525. [Google Scholar] [CrossRef]
- Mathur, B.; Arif, W.; Patton, M.E.; Faiyaz, R.; Liu, J.; Yeh, J.; Harpavat, S.; Schoonjans, K.; Kalsotra, A.; Wheatley, A.M.; et al. Transcriptomic analysis across liver diseases reveals disease-modulating activation of constitutive androstane receptor in cholestasis. JHEP Rep. 2020, 2, 100140. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Hooiveld, G.; Muller, M.; Kersten, S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS ONE 2009, 4, e6796. [Google Scholar] [CrossRef] [Green Version]
- Song, W.M.; Zhang, B. Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput. Biol. 2015, 11, e1004574. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wang, M.; Song, W.M.; Shen, Q.; McKenzie, A.; Choi, I.; Zhou, X.; Pan, P.Y.; Yue, Z.; et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat. Commun. 2019, 10, 5234. [Google Scholar] [CrossRef]
- Ruan, X.; Li, P.; Chen, Y.; Shi, Y.; Pirooznia, M.; Seifuddin, F.; Suemizu, H.; Ohnishi, Y.; Yoneda, N.; Nishiwaki, M.; et al. In vivo functional analysis of non-conserved human lncRNAs associated with cardiometabolic traits. Nat. Commun. 2020, 11, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, M.; Kawai, K.; Mitsui, T.; Taniguchi, K.; Monnai, M.; Wakui, M.; Ito, M.; Suematsu, M.; Peltz, G.; Nakamura, M.; et al. The reconstituted ‘humanized liver’ in TK-NOG mice is mature and functional. Biochem. Biophys. Res. Commun. 2011, 405, 405–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, K.C.; Floyd, R.R.; Sabir, S.; Jayasekera, D.W.; Leon-Mimila, P.V.; Jones, A.E.; Cortez, A.A.; Shravah, V.; Peterfy, M.; Stiles, L.; et al. Liver Pyruvate Kinase Promotes NAFLD/NASH in both Mice and Humans in a Sex-Specific Manner. Cell Mol. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Suppli, M.P.; Rigbolt, K.T.G.; Veidal, S.S.; Heeboll, S.; Eriksen, P.L.; Demant, M.; Bagger, J.I.; Nielsen, J.C.; Oro, D.; Thrane, S.W.; et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G462–G472. [Google Scholar] [CrossRef] [PubMed]
- Chick, J.M.; Munger, S.C.; Simecek, P.; Huttlin, E.L.; Choi, K.; Gatti, D.M.; Raghupathy, N.; Svenson, K.L.; Churchill, G.A.; Gygi, S.P. Defining the consequences of genetic variation on a proteome-wide scale. Nature 2016, 534, 500–505. [Google Scholar] [CrossRef] [Green Version]
- Min-DeBartolo, J.; Schlerman, F.; Akare, S.; Wang, J.; McMahon, J.; Zhan, Y.; Syed, J.; He, W.; Zhang, B.; Martinez, R.V. Thrombospondin-I is a critical modulator in non-alcoholic steatohepatitis (NASH). PLoS ONE 2019, 14, e0226854. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601–614. [Google Scholar] [CrossRef]
- Necsulea, A.; Soumillon, M.; Warnefors, M.; Liechti, A.; Daish, T.; Zeller, U.; Baker, J.C.; Grutzner, F.; Kaessmann, H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014, 505, 635–640. [Google Scholar] [CrossRef]
- Cattley, R.C.; DeLuca, J.; Elcombe, C.; Fenner-Crisp, P.; Lake, B.G.; Marsman, D.S.; Pastoor, T.A.; Popp, J.A.; Robinson, D.E.; Schwetz, B.; et al. Do peroxisome proliferating compounds pose a hepatocarcinogenic hazard to humans? Regul. Toxicol. Pharm. 1998, 27, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Babich, M.A.; Baetcke, K.P.; Cook, J.C.; Corton, J.C.; David, R.M.; DeLuca, J.G.; Lai, D.Y.; McKee, R.H.; Peters, J.M.; et al. PPARalpha agonist-induced rodent tumors: Modes of action and human relevance. Crit. Rev. Toxicol. 2003, 33, 655–780. [Google Scholar] [CrossRef]
- Yang, Q.; Nagano, T.; Shah, Y.; Cheung, C.; Ito, S.; Gonzalez, F.J. The PPAR alpha-humanized mouse: A model to investigate species differences in liver toxicity mediated by PPAR alpha. Toxicol. Sci. 2008, 101, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Chikada, H.; Ida, K.; Ando, E.; Inagaki, Y.; Sakamoto, A.; Kamiya, A. Establishment and analysis of a mouse model that regulates sex-related differences in liver drug metabolism. Lab. Investig. 2018, 98, 1500–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chukijrungroat, N.; Khamphaya, T.; Weerachayaphorn, J.; Songserm, T.; Saengsirisuwan, V. Hepatic FGF21 mediates sex differences in high-fat high-fructose diet-induced fatty liver. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E203–E212. [Google Scholar] [CrossRef] [Green Version]
- Wilson, E.M.; Bial, J.; Tarlow, B.; Bial, G.; Jensen, B.; Greiner, D.L.; Brehm, M.A.; Grompe, M. Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res. 2014, 13, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zong, C.; Jiang, M.; Hu, H.; Cheng, X.; Ni, J.; Yi, X.; Jiang, B.; Tian, F.; Chang, M.W.; et al. Hepatic HuR modulates lipid homeostasis in response to high-fat diet. Nat. Commun. 2020, 11, 3067. [Google Scholar] [CrossRef]
- Mynatt, R.L.; Noland, R.C.; Elks, C.M.; Vandanmagsar, B.; Bayless, D.S.; Stone, A.C.; Ghosh, S.; Ravussin, E.; Warfel, J.D. The RNA binding protein HuR influences skeletal muscle metabolic flexibility in rodents and humans. Metabolism 2019, 97, 40–49. [Google Scholar] [CrossRef]
- Siang, D.T.C.; Lim, Y.C.; Kyaw, A.M.M.; Win, K.N.; Chia, S.Y.; Degirmenci, U.; Hu, X.; Tan, B.C.; Walet, A.C.E.; Sun, L.; et al. The RNA-binding protein HuR is a negative regulator in adipogenesis. Nat. Commun. 2020, 11, 213. [Google Scholar] [CrossRef]
- Halees, A.S.; El-Badrawi, R.; Khabar, K.S. ARED Organism: Expansion of ARED reveals AU-rich element cluster variations between human and mouse. Nucleic Acids Res. 2008, 36, D137–D140. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, C.; Li, P.; Ruan, X.; Ma, Y.; Kawai, K.; Suemizu, H.; Cao, H. Comparative Transcriptomics Analyses in Livers of Mice, Humans, and Humanized Mice Define Human-Specific Gene Networks. Cells 2020, 9, 2566. https://doi.org/10.3390/cells9122566
Jiang C, Li P, Ruan X, Ma Y, Kawai K, Suemizu H, Cao H. Comparative Transcriptomics Analyses in Livers of Mice, Humans, and Humanized Mice Define Human-Specific Gene Networks. Cells. 2020; 9(12):2566. https://doi.org/10.3390/cells9122566
Chicago/Turabian StyleJiang, Chengfei, Ping Li, Xiangbo Ruan, Yonghe Ma, Kenji Kawai, Hiroshi Suemizu, and Haiming Cao. 2020. "Comparative Transcriptomics Analyses in Livers of Mice, Humans, and Humanized Mice Define Human-Specific Gene Networks" Cells 9, no. 12: 2566. https://doi.org/10.3390/cells9122566
APA StyleJiang, C., Li, P., Ruan, X., Ma, Y., Kawai, K., Suemizu, H., & Cao, H. (2020). Comparative Transcriptomics Analyses in Livers of Mice, Humans, and Humanized Mice Define Human-Specific Gene Networks. Cells, 9(12), 2566. https://doi.org/10.3390/cells9122566