1. Introduction
Apical periodontitis (AP) is an inflammatory lesion in the periodontal tissues surrounding the root apices. This disease is quite common, with a prevalence of 6.3% of all teeth [
1]. The pathogenesis of AP following tooth necrosis originates from the associated infection which causes necrosis in the tooth pulp. The bacteria or their products (commonly Lipopolysaccharide, LPS) egress into the periapex and stimulate a complex immune response of the host. The microbial factors and host defense forces destroy the periapical tissues and form AP. Some of the cellular elements involved in AP include polymorphonuclear cells (PMN), lymphocytes, plasma cells, macrophages, and osteoclasts. Important cytokines include IL-1, 6,8, TNFα/β, and IFNα/β. Matrix metalloproteinases (MMPs) are involved in tissue degradation. It is accepted to divide the immune reaction in AP to the acute initial phase and the chronic phase. During the initial stage, pro-inflammatory cytokines are secreted and bone resorption is at its peak. During the chronic phase, the pro-inflammatory cytokines are downregulated, reducing bone resorption and the tissue reaches an equilibrium. However, this equilibrium can be disturbed, and the progression of the disease occurs in leaps, with periods of stability in-between. Since the microbes are located within the root canal, which is beyond the reach of the body’s defenses, AP is not self-healing [
2]. Due to the increase in human lifespan, elderly patients are frequently seen at the dental clinic, although not much is known about how age effects the development of AP. Our hypothesis was that since AP is a disease with a strong involvement of the immune system, age-related immune changes may affect the progression of AP.
Immune-system aging, termed immunosenescence, is a complex process involving all arms of the immune system and many different cell types. In the innate immune system, older people present a reduction in cytokine signaling, production of peroxide and nitric oxide, and the phagocytic function of neutrophils. Human natural killer (NK) cells exhibit reduced activity (cytokine secretion and cytotoxicity) as well as diminished migration, and the ability of macrophages to phagocytose apoptotic cells is diminished. The uptake of antigens by DCs is reduced, decreasing consequent T cell stimulation. The adaptive immune system is substantially affected by aging and presents a decrease in the variance of the T cell receptors (TCRs), diminished levels of naïve T and B lymphocytes, and a decrease in antibody avidity in older people [
3].
Another related process which occurs with old age is inflamm-aging, a chronic persistent low-grade inflammation with elevated levels of cytokines such as C-Reactive Protein (CRP), IL-6, IL-1β, and TNF. These two processes (immunosenescence and inflamm-aging) lead, on the one hand, to immune stimulation at the basal level, and on the other hand, to a decreased ability of the immune system to preform specific roles. Inflamm-aging and immunosenescence progress in parallel and form a vicious cycle. Increased production of inflammatory mediators characteristic of inflamm-aging contributes to decreases in the adaptive immune response and to immunosenescence. In contrast, the decrease in the adaptive immune response reinforces the stimulation of the innate immune response, leading to inflamm-aging. The outcome of this situation is an increased frequency of infections, cancer, and chronic diseases in the elderly [
4].
Although not much work has previously been done on the progression of AP with age, a previous study on humans observed lower levels of macrophages in apical lesions in adult compared to young patients [
5]. The aim of the current study was to investigate how age affects the progression of AP in a mouse model. For this purpose, we chose to use a mouse model of induced apical periodontitis for our investigation [
6,
7,
8] and compared different age groups with the same genetic and environmental conditions.
In our study we followed the histopathological sequence of AP progression starting from root canal infection (% of necrotic canals, bacteria localization, and apical foramen volume), and ending in formation of a periapical (PA) lesion (cellular density and cellular composition of the lesion), with subsequent bone resorption (osteoclast numbers and lesion volume) [
2].
A better understanding of how age impacts AP progression may produce information to help provide an endodontic treatment plan more tailored to older patients.
2. Methods
2.1. Animal Procedures
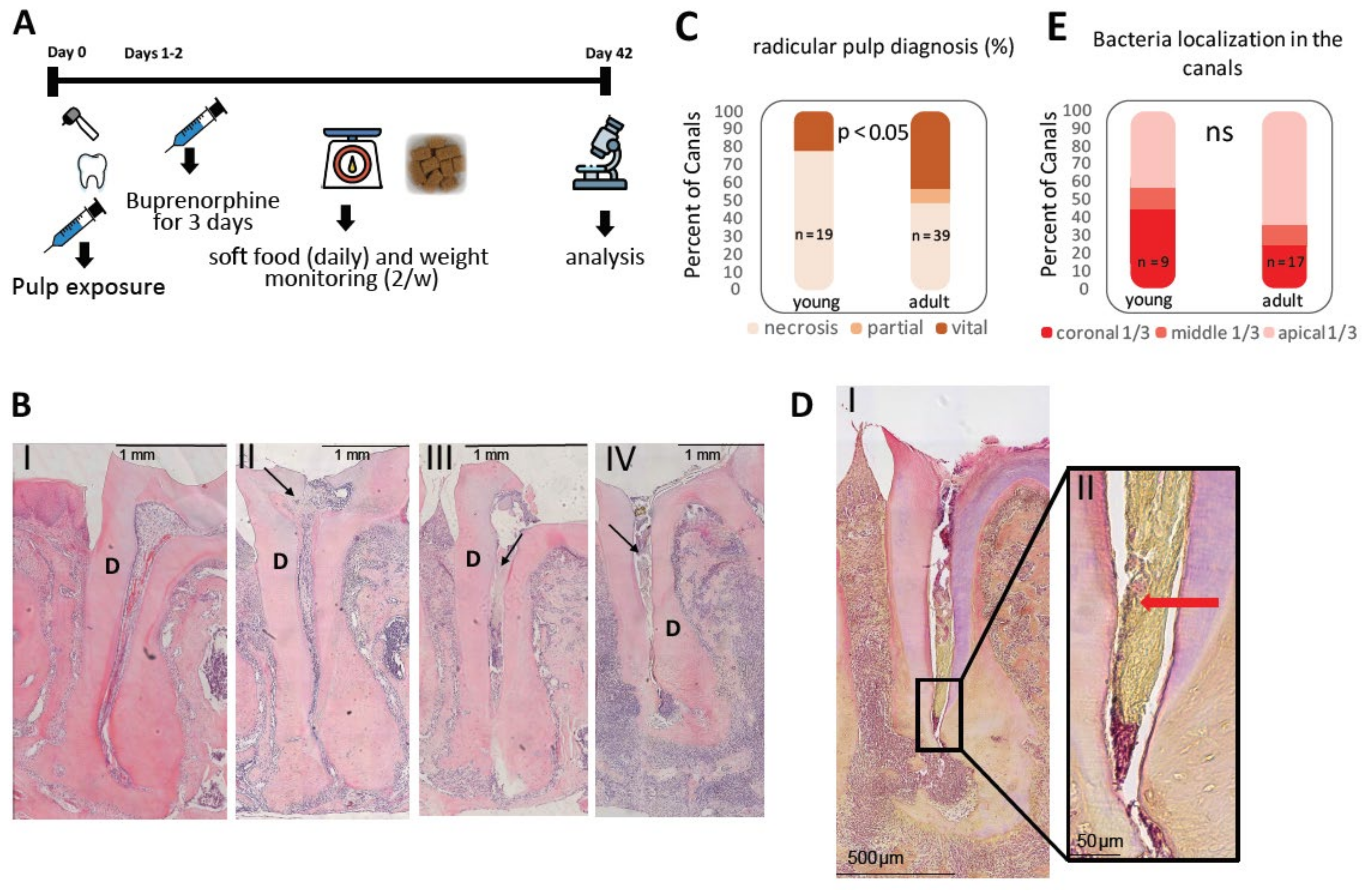
All animal experiments were approved by the animal ethics committee of the Hebrew University of Jerusalem (Institute review board (IRB) approval MD-19-15962-4). Female C57bl mice were used in all the experiments. The young group were mice aged 6–8 weeks, and the adult group were aged 8–10 months on the first day of the experiment. Apical periodontitis was induced as previously described [
6]. Briefly, mice were anesthetized with ketamine–medetomidime (concentration of 70 mg/kg ketamine hydrochloride (Vetoquinol, CAS 1867-669) and 0.9 mg/kg medetomidine hydrochloride ((Cepetor, CP-pharma GmbH, CAS 86347-15-1) diluted in PBS), and the pulp of the first mandibular molar (M1) on the right side was exposed in the mesial and distal pulp horns. M1 on the left side was used as a control (the pulp of the right side M2 was also exposed for flow cytometry experiments). The animals received an antisedant ((atipamezole hydrochloride (Eurovet Animal Health, CAS 104075-48-1), concentration of 4 mg/kg, diluted in PBS), at the end of the procedure. During the first three days of the experiment the animals received a daily dosage (0.05 mg/kg on the day of the procedure, and 0.1 mg/kg for another 2 days) of buprenorphine (vetmarket 163451) to relieve pain. During the course of the experiment the animals’ weight was monitored and the animals received soft food daily in order to reduce pain during chewing and to encourage biofilm formation. The teeth were left to become contaminated by the oral flora for 42 days as previously described [
6,
9]. After this time period, the animals were anesthetized with ketamine (162.5 mg/kg)-xylazine (75 mg/kg, Eurovent Animal Health, CAS 7361-61-7) and euthanized by cervical dislocation.
For histological sections, mandibular bone blocks including the molars were cut and the soft tissues were peeled off. The bone blocks were rinsed with PBS and left overnight in PFA (paraformaldehyde) 4%, then rinsed thoroughly with phosphate buffered saline (PBS). For histology and micro-CT n = 15 (young), 29 (adult). For flow cytometry, see relevant detailed section following.
2.2. Micro-CT
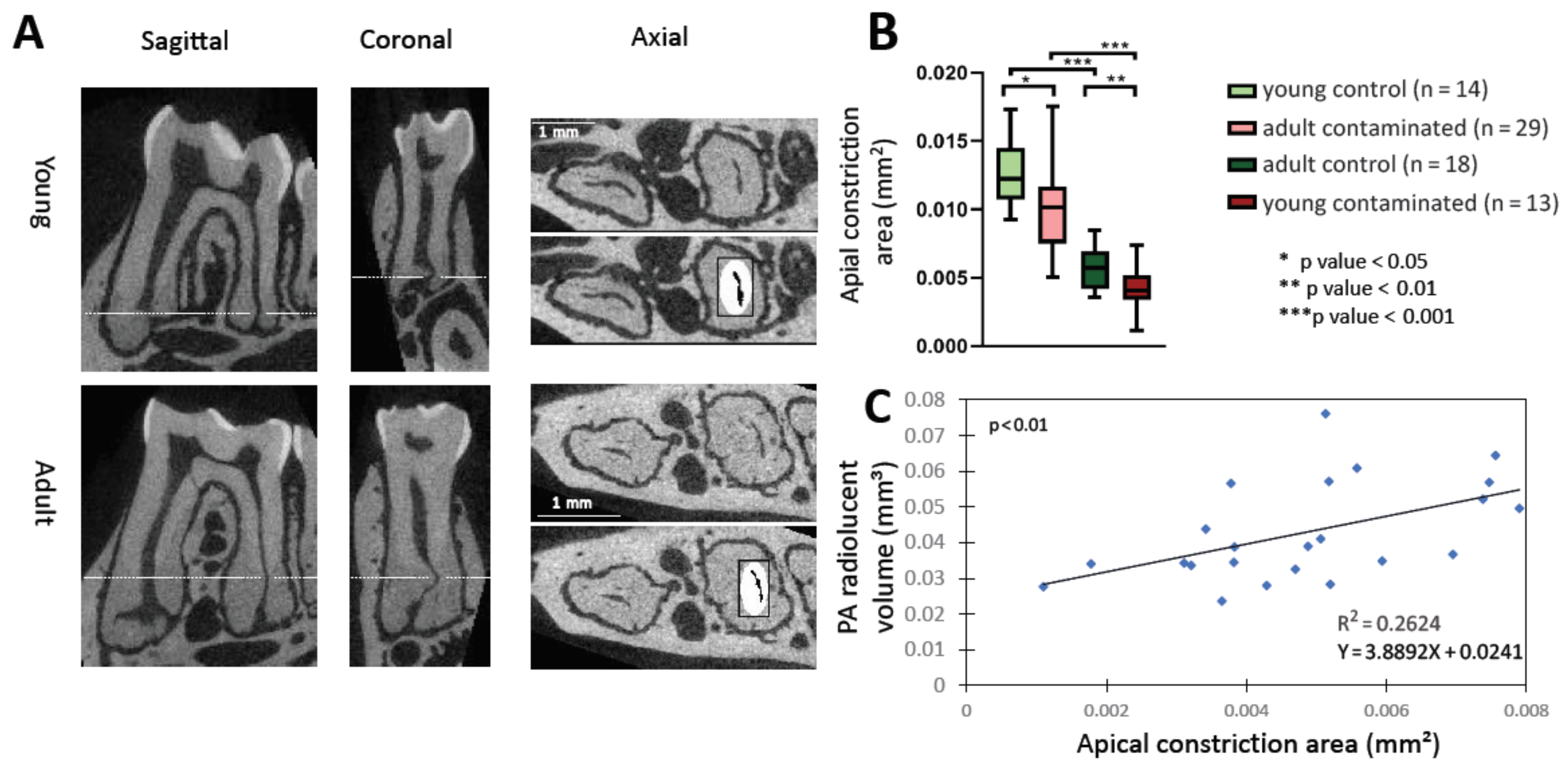
Samples were scanned for micro-computed tomography (micro-CT) (µCT 40, Scanco Medical AG, Bassersdorf, Switzerland) using the following parameters: energy of 70 kV, intensity of 114 μA, exposure time of 200 ms, and slice increments of 6 μm in each plane (voxel size of 6 μm3). The area of the distal apical constriction (AC) and volume of the distal canal and distal apical bone resorption were calculated using Scanco software. Each scanned tooth was aligned by 3 points, on 2 planes: (1) the mesial apical constriction, (2) the distal apical constriction, (3) the mesial coronal opening of the canal. This gave sagittal slices with a central slice including the pulp chamber, both mesial and distal canals, and the apical foramens which were used for PA lesion analysis. The sagittal aligned samples were flipped to obtain axial slices, which were used for canal volume and apical constriction analyses. For PA lesion volume calculation: the radiolucent area around the distal periapical region was manually delineated in 79 adjacent slices; 39 buccal and 39 lingual to the central slice of each tooth. The volume (3D analysis) of the normal periodontal ligament (PDL) (in control teeth) and the PA lesions (in contaminated teeth) was calculated. In addition, in order to evaluate the accurate area of the inflammatory lesion, we extracted the average volume of the normal PDL area from the PA volume of each contaminated tooth (according to age groups). For apical constriction calculation: the position of the apical constriction of the distal canal was assessed by the sagittal and coronal sections, and the axial slice with the smallest area of the canal in that position was chosen for two-dimensional (2D) analysis. The area of the canal in the chosen section was calculated by the volume of interest (VOI) minus the bone volume (BV). Similarly, the distal canal volume was calculated (3D) by subtracting the BV from the VOI in all the sequential axial slices from the coronal section of the canal (determined by the sagittal slices) until the slice of the apical constriction. To statistically compare the results, a Student’s t-test was used.
2.3. Histological Slide Preperation
After the CT scan, bone blocks, rinsed with PBS, were placed in EDTA (0.5M, pH 8) for 10 days. EDTA was replaced every 3–4 days. For Paraffin sections (hematoxylin eosin (H&E), Brown and Brenn (B&B), tartrate-resistant acid phosphatase (TRAP) staining) tissues were dehydrated with increasing concentrations of ethanol, then placed in xylene, and in paraffin blocks. Paraffin blocks were sliced to a thickness of 6 microns. For cryosections (immunofluorescent staining): after ethylenediaminetetraacetic acid (EDTA) treatment, tissues were placed in a 30% sucrose solution overnight in 4 °C, and then in Optimal cutting temperature compound (OCT compound) in −80 °C. Cryo-blocks were sliced to a thickness of 10 microns and kept in −20 °C until staining.
2.3.1. H&E Staining
Hematoxylin eosin (H&E) staining was done as previously described [
10]. Slides were deparaffinized (Xylene 10 min twice) and rehydrated with descending concentrations of ethanol to double distilled water (DDW) (100%—5 min, 90 and 70%—2 min each). Thereafter, they were stained with hematoxylin (Sigma-Aldrich, Jerusalem, Israel, MHS 16) for 1 min, rinsed with tap water, stained with eosin (Sigma-Aldrich, Jerusalem, Israel, HT110116) for 30 s and rinsed with tap water. Then, the slides were placed in DDW and dehydrated with increasing concentrations of ethanol (70, 90, and 100%—2 min each), followed by xylene, and covered with a coverslip with entelane.
2.3.2. B&B Staining
Brown and Brenn (B&B) staining was done as previously described [
10]. Slides were deparaffinized (xylene (Gadot, CAS 1330-20-7) 10 min twice) and rehydrated with descending concentrations of ethanol (100%—5 min, 90 and 70%—2 min each) to tap water (for 10 min). Thereafter, they were stained with a sequence of the following solutions; Harris haematoxylin (Sigma HHS32)—5 min, acid alcohol (1% HCl in 70% ethanol)—5 s, saturated lithium carbonate (Sigma 62470) in DDW—5 s, Hucker’s solution (0.2% crystal violet (sigma 61135)), 2% ethanol, 0.8% ammonium oxalate ((sigma 221716) in DDW)—2 min, iodine solution (0.33% iodine (BDH CAS 7553-56-2)), 0.66% potassium iodide ((J.T. Baker CAS 7681-11-0) in DDW)—15 s, ether acetone (50% Ether (Daejung CAS 60-29-7)), and 50% acetone (Gadot, CAS 67-64-1)—10 s, basic fuchsin (0.007% basic fuchsin (Sigma 857343, 7.31% ethanol in DDW))—3 min, acetone—10 s, picric acetone (0.1% picric acid in acetone EMS 26105-06)—20 s, acetone–xylene I (33.3% acetone, and 66.6% xylene)—5 s, acetone–xylene II (25% acetone, 75% xylene)—5 s, and xylene—1 min. The slides were mounted with entelane (mercury 1.07961) and covered with a coverslip. For analysis, each canal was divided into three parts, and evaluation was done by manually determining the most apical localization of bacteria in each canal. Statistics were performed using Fisher’s exact test.
2.4. H&E Periapical Computerized Analysis
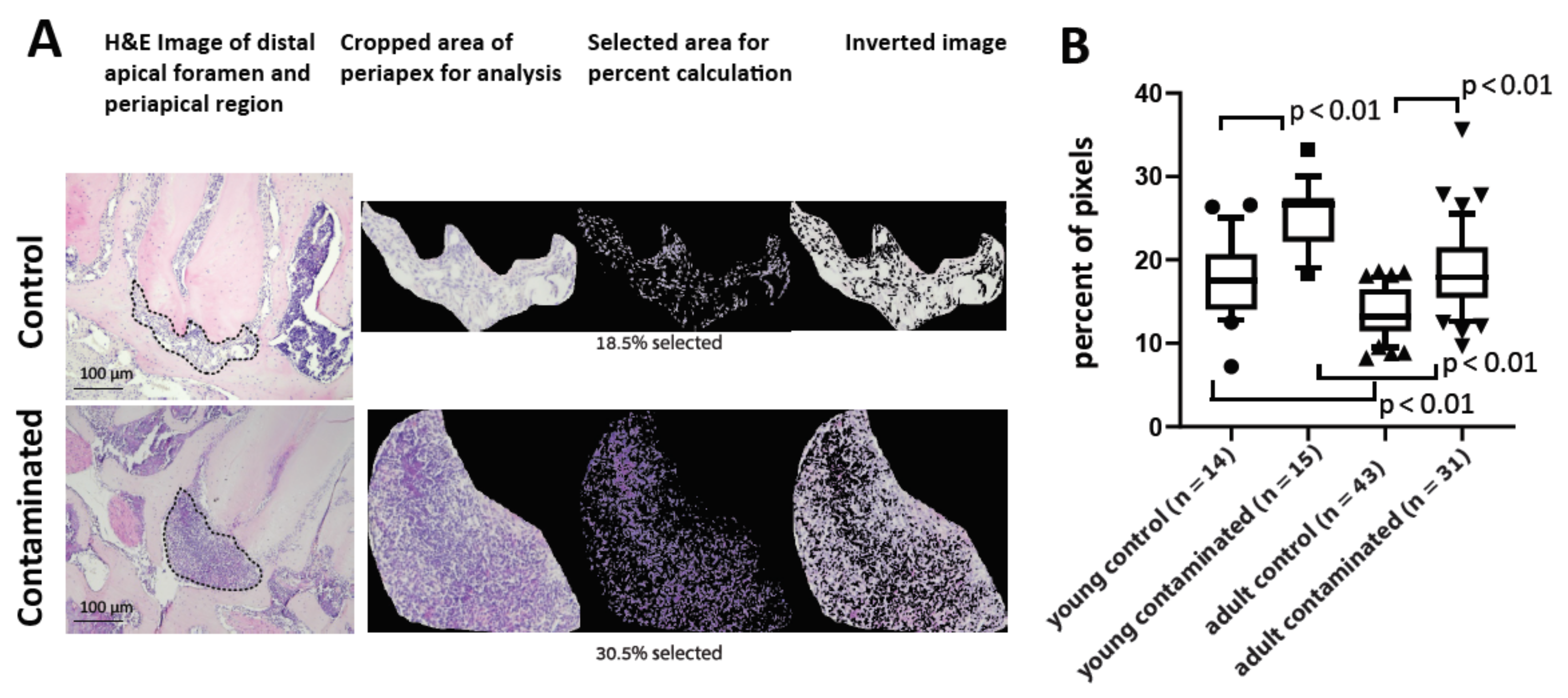
A computer program was developed for colorimetric evaluation of the cell density of the periapical region on the H&E slides. A python computer program was developed for colorimetric evaluation of the cell density of the periapical region on the H&E slides (the code is available on github via the following link:
https://github.com/elishevag16/cell-density-analysis, (accessed on 3 February 2021). The program enables one, for each slide, to calibrate the parameters of HSV (hue, saturation, value) range. The parameters were calibrated for each slide to receive an optimal range in which the nuclei of the cells were maximally included, and the background colors were minimized. Then, the periapical region was cropped, and the computer calculated cell density as a percentage of the included pixels from all pixels (in the cropped area). The output was used for comparison between the different experimental groups as a proxy for cell density in area of interest. The technique was validated by showing a significant difference between treated and control teeth, and by showing a correlation with the micro-CT results.
2.5. Tartrate-Resistant Acid Phosphatase (TRAP)
Paraffin slides were stained for osteoclasts with TRAP staining, as previously described [
11]. Slides were deparaffinized (xylene 2 min × 2 (and rehydrated with descending concentrations of ethanol (100% × 3, 90%, 70%—2 min each) to tap water. Tissues were marked with a pap pen, and covered with TRAP incubation solution (50 mL TRAP buffer: sodium acetate (J.T.Baker CAS 127-09-3) 16.4 g/L and sodium tartarate (Sigma CAS 6106-24-7) 11.6 g/L in DDW, titrated to pH 5 with 2.5 mL AS-BI substrate mix: 6 g/L naphthol AS-BI phosphate sodium salt (Sigma N2250), 10% N’N-dimethylformamide (Sigma 227056), 70% methanol (Gadot CAS 67-56-1) and 20% DDW, mixed with 0.025 g Fast red TR salt (Sigma CAS 89453-69-0). The slides were incubated for 30 min at 37 degrees Celsius until red staining was detected under a microscope. The slides were washed with DDW, counterstained with hematoxylin for 30 s, washed with tap water and covered with a coverslip with hydromount 17,966 (EMS).
For analysis, the periapical bone border length was measured using ImageJ software. The number of TRAP-positive multinucleated (≥3 nuclei) cells was counted manually around the necrotic canals, and the number of osteoclasts per mm on the bone border was calculated as previously described [
9,
12]
2.6. Immunofluorescent Staining
Cryosections were stained with lymphocyte antigen 6 complex locus G6D (Ly6G) primary antibody, and a conjugate fluorescent secondary antibody, as previously described [
13]. Cryosections were defrosted at room temperature for 1 h, followed by 10 min at 37 degrees Celsius. The remaining OCT was rinsed with PBS. Tissues were marked with P pap pen, and incubated at room temperature in a wet chamber with blocking solution (5% FCS (biological industries, Beit Haemek)) and 1% Triton ((Sigma-Aldrich, Jerusalem, Israel, CAS 9002-93-1) in pbs) for 1 h. Thereafter, tissues were incubated overnight at 4 °C in a wet chamber with the primary antibody vs. Ly6G (BD biosciences, 551459) diluted at a ratio of 1:100 in blocking solution. PBS rinses were performed, and the tissues were incubated for 2 h at room temperature in a wet chamber with the secondary antibody (Invitrogen A11077) diluted at a ratio of 1:200 in blocking solution. Slides were rinsed, stained with 5 µM DAPI (abcam AB-ab228549) at room temperature for 10 min, rinsed, and covered with a coverslip with mounting medium (VECTASHIELD
® Mounting Medium (H-1000, Zotal)). Images were obtained with NIKON-Ti X40 magnification (N.A. 0.75 dry), using Photometrics Prime 95B camera.
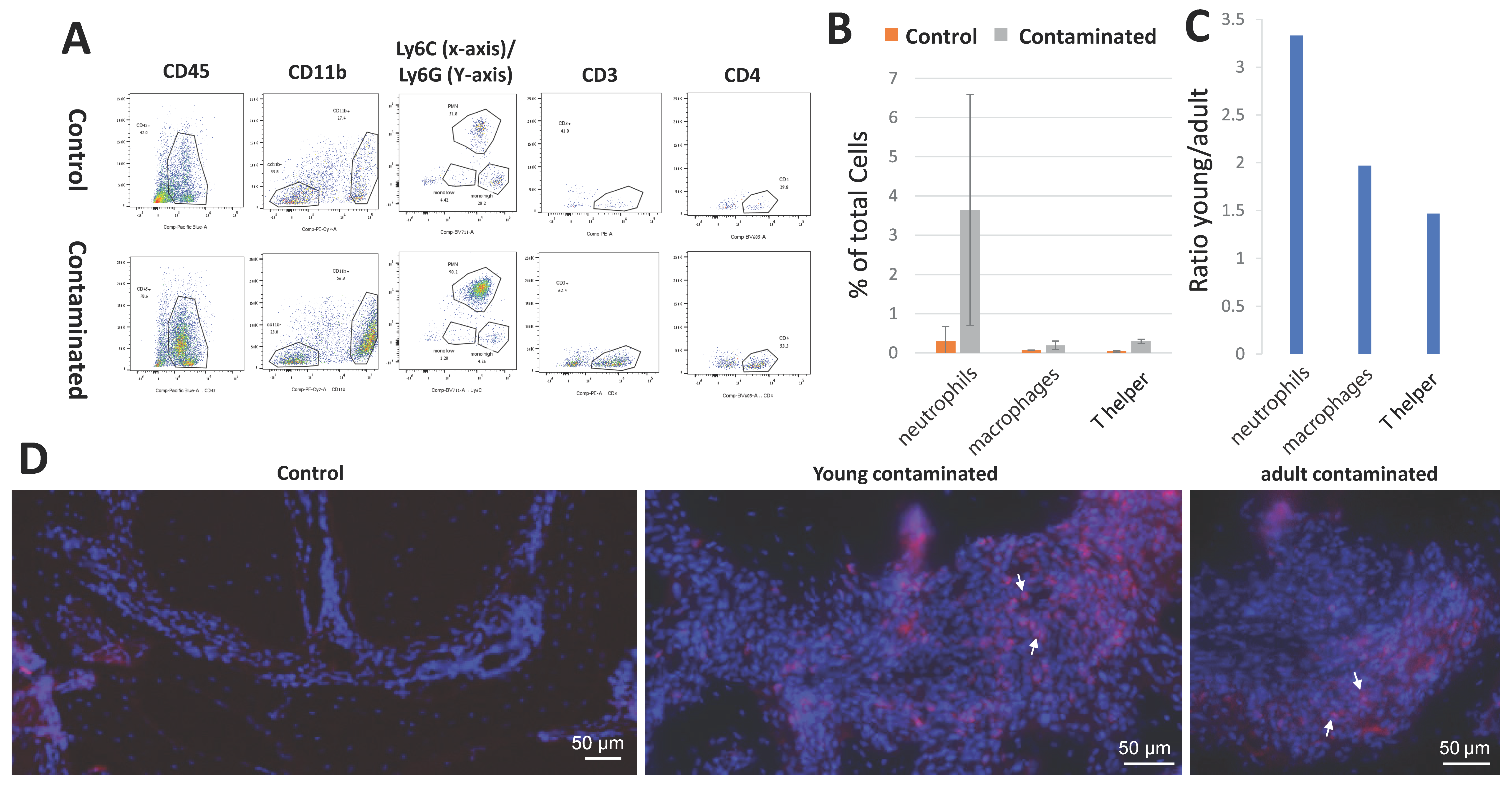
2.7. Flow Cytometry
The cells were isolated from the periapical region of the first mandibular molars as previously described [
14]. After the animals had been euthanized, jaws were extracted, and the soft tissues were peeled off. Under magnification, the first and second mandibular molars were extracted, and cleaned from remnants of bone and gingiva. The tissue was placed in working solution (2% FCS in PBS) with DNAse I (Sigma DN25) 1 mg/mL and Collagenase II (Worthington LS004177). Each tube consisted of a pool of 3–5 mice, and each group was run 3 times on sequential days (
n = 10 (young), 15 (Adult)). The solution with the teeth was incubated in a shaker at 37 °C for 25 min, and EDTA (0.5 mM, pH 8) was added (at a concentration of 1%) and incubated for another 10 min at the same conditions. The solution was filtered (with 70 micron filters), centrifuged (4 °C, 1000 rpm, 5 min), upper liquid was removed, and cells were stained with an antibody mix (Pacific Blue™ anti-mouse CD45.2 antibody (109820), PE/Cy7 anti-mouse/human CD11b antibody (101216), Brilliant Violet 711™ anti-mouse Ly-6C Antibody (128037), APC anti-mouse Ly-6G Antibody (127614), PE anti-mouse CD3 Antibody (100206), PE anti-mouse CD3 Antibody (100206), all from Biolegend, diluted 1:250 for each antibody in working solution) for 20 min on ice, rinsed, and fixated with cell preservative (Streck 213350) at a ratio of 1:6. The stained samples were run in the BD LSRFortessa™ cell analyzer (BD Biosciences, US) flow cytometer and analyzed using FlowJo software (Tree Star, BD Biosciences, US).
2.8. Statistical Analysis
4. Discussion
The aim of our work was to study the effect of age on AP development. The experimental design used an established mouse model for induced AP [
7,
8] and compared two age groups termed young and adult. Surprisingly, only ~50% of adult mice canals, and ~80% of young mice canals presented necrotic pulp (
Figure 1C)—a prerequisite condition for AP. In most of the vital canals, pulp obliteration was observed (
Supplementary Figure S1), as previously reported [
18,
19], possibly decelerating the bacterial attack. The reason why adults presented fewer necrotic canals (compared to the young mice) was not tested in this work. One possible explanation could be age-related changes in the pulp resulting in a denser connective tissue [
15] that serves as a physical barrier for bacteria. Another biological explanation could be an immunological phenotype related to immune changes that occurs with age [
3], which correlates with the other results reported in this field of research. Regardless of the mechanism for this phenomenon, we discarded the vital canals in the following analyses which were aimed to study AP because they are not relevant for this examination.
Identification of bacteria in the tissue via B&B staining revealed no significant difference in the localization of bacteria in the canal between the age groups (
Figure 1E). These results show that the following AP age-related differences occur due to processes that are mediated by the host, rather than environmental (bacteria stimuli) processes. In this context, it is important to emphasize that B&B staining enabled us to identify the localization but not the quantity and composition of the canal bacteria, which can be evaluated by 16S. A differential age dependent microbiome [
23,
24,
25] could further contribute to the difference in AP progression between the age groups and should be further investigated.
The common cause of AP is bacteria or bacterial by products stimuli of the PA tissues [
2], which requires them to go through root canals and the AC. We, therefore, measured the size of the canals and AC in the different age and experimental groups (
Supplementary Figure S2 and
Figure 2). The adult group presented a significantly smaller canal volume and AC area compared to the young group (
Supplementary Figure S2A and
Figure 2B), most likely due to canal calcification occurring with age, as previously described [
16,
17]. There was a significant positive correlation between AC area and canal volume with the size of lesion, up to a canal volume of ~0.01 mm
3 and AC size of ~0.009 mm
2, and thereafter, the graph reached a plateau (
Supplementary Figures S2 and S3). Our interpretation of these results is that the canal volume and the AC area effects the influx of bacteria up to a certain point, when the size is no longer a limiting factor. Overall, according to our results, an age-related decrease in canal and AC size may be one of the causes of less severe AP that occurs with age. Although this observation may obscure the source of the attenuated inflammatory response in the adult mice (mechanical obstruction vs. immune system deterioration), it is important to point out that this mechanical effect also occurs in humans with age, and therefore, should be considered clinically. Moreover, it is possible that age-related canal calcification occurs as a result of an inflamm-aging process.
We developed a computer algorithm to quantify and compare the cell density in the PA region between the age groups (
Figure 3). The intact control PDL presented a higher cell density in the young mice compared to the adults, as previously established [
26]. The average cell density in the AP area of the contaminated teeth was significantly higher in the young compared to the adult group, meaning that the young mice had more cells in the PA region to initiate inflammatory bone resorption. Another interesting observation in this experiment, as mentioned, is that the adults presented significantly higher levels of variance, a well-known phenomenon related to age, explained by cumulative environmental influences such as bacterial exposure [
27,
28].
In the pathophysiology of AP, osteoclasts are recruited and differentiated in a cascade caused by inflammatory cells and cytokines such as TNF-α [
29,
30]. During the maturation process of the osteoclast, it becomes a syncytium, a process which improves its bone resorption capability [
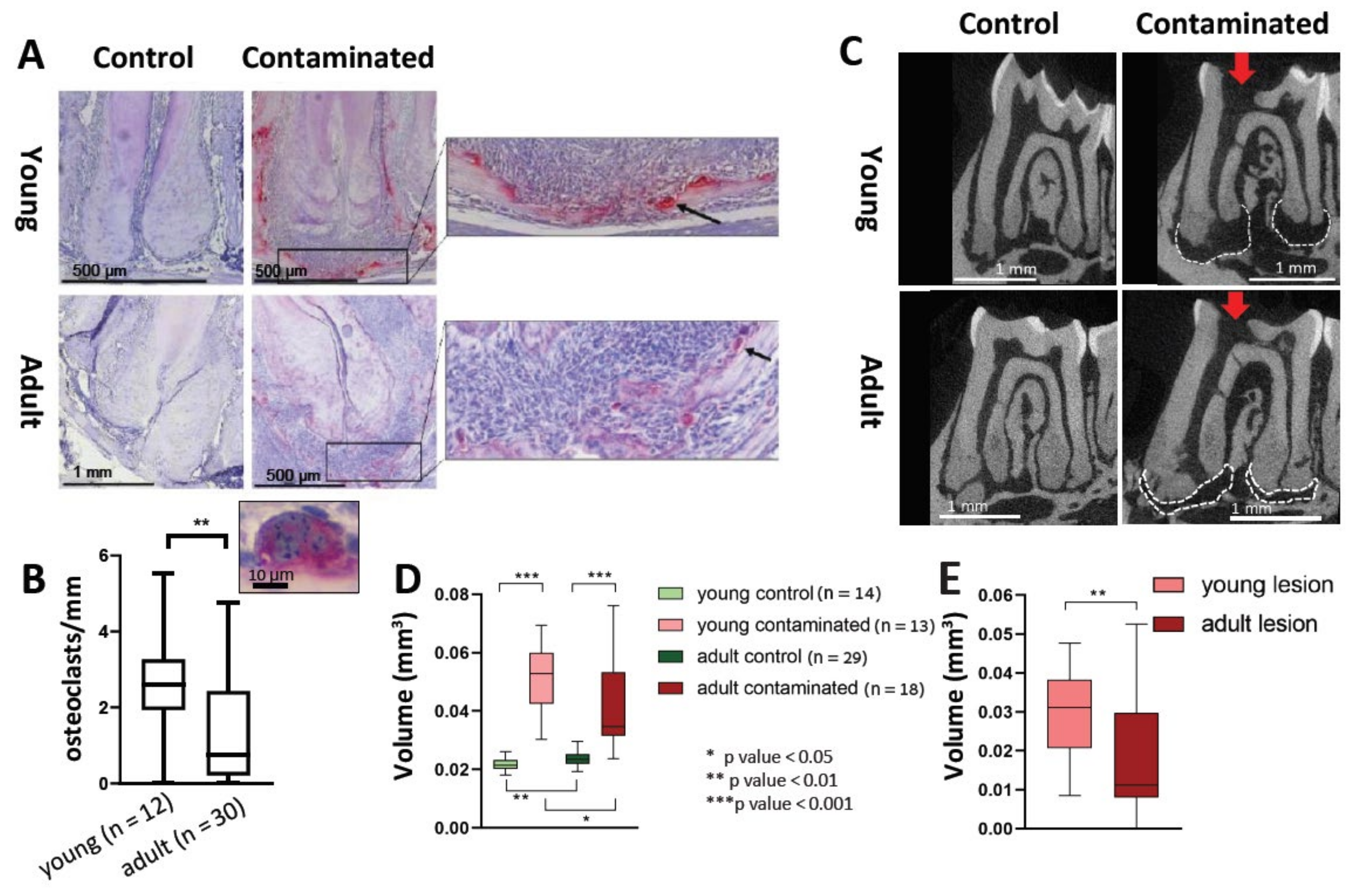
21]. We did not find a difference in the total number of TRAP-positive cells in the PA lesions between the young and adult mice. Nevertheless, our results revealed an increase in the number of multinucleated TRAP-positive cells (differentiated cells, as opposed to the mono-nucleated pre-osteoclasts) in the young mice compared to the adults (
Figure 5A,B). This result conveys an effect of age on the process of osteoclast differentiation, resulting in a reduced efficiency of bone resorption occurring with age. This phenomenon is in line with other studies showing an age-related deterioration in the differentiation capacity of stem cells, rendering them less prone to differentiation [
31]. Indeed, our micro-CT results (
Figure 5C–E) indicate reduced bone resorption in the lesion for the adult mice compared to the young.
Overall, our results—using several different methods—conclude that, on average, adult mice present lower levels of inflammatory parameters; necrosis, cell density of the periapical infiltrate, and alveolar bone resorption compared to young mice. We suggest that part of the reason for this phenomenon is related to reduced immune functioning that occurs with age [
32]. In a seemingly contradictory manner, the adult mice with the attenuated immune response reacted to the canal infection in a way that presented less tissue damage. Our interpretation of these results is that AP presents a unique phenomenon, as bacteria stimulate the immune system from a remote position (inside the canal or even in the dental tubuli) and are not in direct contact with the immune cells [
2]. Therefore, in contrast to other infectious diseases—where a strong immune system can eradicate the pathogen and overcome the infection—in AP, the disease is not self-healing, and eventually the immune system itself will cause damage.
To conclude, this study presents a model for studying the effect of age on AP in mice. We showed that age is positively correlated with a reduced inflammatory reaction and bone resorption, which may be due to both an age-related mechanical barrier and reduced immunity. Therefore, we suggest that patients’ age should perhaps be considered clinically for follow-up time after treatment, or for decision making about secondary treatment in asymptomatic teeth with a radiographic lesion. Moreover, we showed that neutrophils are a major player in AP in mice and are most significantly reduced with age. Clinical studies should be designed to investigate whether or not there is a similar age effect on AP progression in humans.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




