
Membrane Interference Against HIV-1 by Intrinsic Antiviral Factors: The Case of IFITMs


Abstract
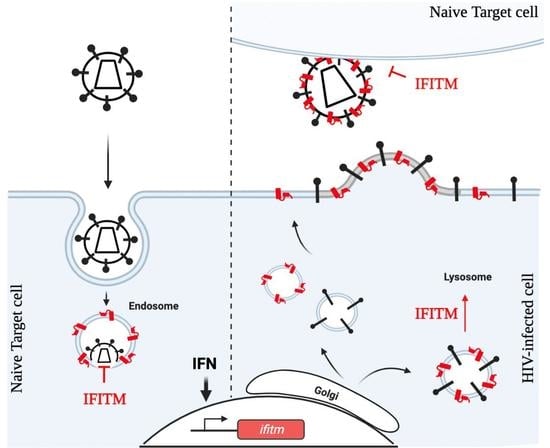
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. An Historical Overview of IFITMs
3. IFITMs Inhibition of HIV-1
3.1. Target Cell Protection
3.2. Negative Imprinting of Virions Particles Infectivity: The Production of Virion Particles of Decreased Infectivity
3.3. Translation Inhibition of HIV-1
4. Molecular Basis of IFITMs Inhibition
4.1. Direct Membrane Rigidification
4.2. Indirect Biophysical Changes Due to Lipid Alteration
4.3. Env-Mediated Trafficking/Processing Defects
5. HIV-1 Resistance towards IFITMs
6. Final Considerations and Perspectives
Beyond Viral Inhibition: IFITMs as Double-Edge Swords?
Funding
Acknowledgments
Conflicts of Interest
References
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef]
- Ramirez, P.W.; Sharma, S.; Singh, R.; Stoneham, C.A.; Vollbrecht, T.; Guatelli, J. Plasma Membrane-Associated Restriction Factors and Their Counteraction by HIV-1 Accessory Proteins. Cells 2019, 8, 1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffinet, C. Cellular Antiviral Factors that Target Particle Infectivity of HIV-1. Curr. HIV Res. 2016, 14, 211–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almén, S.M.; Bringeland, N.; Fredriksson, R.; Schiöth, H.B. The Dispanins: A Novel Gene Family of Ancient Origin that Contains 14 Human Members. PLoS ONE 2012, 7, e31961. [Google Scholar] [CrossRef]
- Friedman, R.L.; Manly, S.P.; McMahon, M.; Kerr, I.M.; Stark, G.R. Transcriptional and Posttranscriptional Regulation of Interferon-Induced Gene Expression in Human Cells. Cell 1984, 38, 745–755. [Google Scholar] [CrossRef]
- Siegrist, F.; Ebeling, M.; Certa, U. The Small Interferon-Induced Transmembrane Genes and Proteins. J. Interferon Cytokine Res. 2011, 31, 183–197. [Google Scholar] [CrossRef]
- Reid, L.E.; Brasnett, A.H.; Gilbert, C.S.; Porter, A.C.; Gewert, D.R.; Stark, G.R.; Kerr, I.M. A Single DNA Response Element Can Confer Inducibility by Both Alpha- and Gamma-Interferons. Proc. Natl. Acad. Sci. USA 1989, 86, 840–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillén-Navarro, E.; Ballesta-Martínez, M.J.; Valencia, M.; Bueno, A.M.; Martinez-Glez, V.; López-González, V.; Burnyte, B.; Utkus, A.; Lapunzina, P.; Ruiz-Perez, V.L. Two Mutations in IFITM5 Causing Distinct Forms of Osteogenesis Imperfecta. Am. J. Med. Genet. A 2014, 164, 1136–1142. [Google Scholar] [CrossRef]
- Coomer, C.A.; Rahman, K.; Compton, A.A. CD225 Proteins: A Family Portrait of Fusion Regulators. Trends Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.-N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. IFITM Proteins Are Incorporated onto HIV-1 Virion Particles and Negatively Imprint Their Infectivity. Retrovirology 2014, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.C.; Zhong, G.; Huang, I.-C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.; Zhang, C.; Wang, W.; Cai, X.; Yu, L.; Wu, F.; Zhang, L.; Tian, C. Combined Approaches of EPR and NMR Illustrate Only One Transmembrane Helix in the Human IFITM3. Sci. Rep. 2016, 6, 24029. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.S.; Yamaguchi, Y.L.; Tsoi, B.; Lickert, H.; Tam, P.P.L. IFITM/Mil/Fragilis Family Proteins IFITM1 and IFITM3 Play Distinct Roles in Mouse Primordial Germ Cell Homing and Repulsion. Dev. Cell 2005, 9, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Lange, U.C.; Adams, D.J.; Lee, C.; Barton, S.; Schneider, R.; Bradley, A.; Surani, M.A. Normal Germ Line Establishment in Mice Carrying a Deletion of the Ifitm/Fragilis Gene Family Cluster. Mol. Cell. Biol. 2008, 28, 4688–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Peng, Z.; Zhou, C.; Qiu, G.; Tang, H.; Sun, Y.; Wang, X.; Li, Q.; Le, X.; Xie, K. Gene-Expression Profiling in Chinese Patients with Colon Cancer by Coupling Experimental and Bioinformatic Genomewide Gene-Expression Analyses: Identification and Validation of IFITM3 as a Biomarker of Early Colon Carcinogenesis. Cancer 2008, 113, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, C.; Miyamoto, N.; Yamamoto, H.; Imai, K.; Shinomura, Y. Detection of Fecal Interferon-Induced Transmembrane Protein Messenger RNA for Colorectal Cancer Screening. Oncol. Lett. 2011, 2, 95–100. [Google Scholar] [CrossRef]
- Li, H.; Yang, L.-L.; Wu, C.-C.; Xiao, Y.; Mao, L.; Chen, L.; Zhang, W.-F.; Sun, Z.-J. Expression and Prognostic Value of IFIT1 and IFITM3 in Head and Neck Squamous Cell Carcinoma. Am. J. Clin. Pathol. 2020, 153, 618–629. [Google Scholar] [CrossRef]
- Hu, J.; Wang, S.; Zhao, Y.; Guo, Q.; Zhang, D.; Chen, J.; Li, J.; Fei, Q.; Sun, Y. Mechanism and Biological Significance of the Overexpression of IFITM3 in Gastric Cancer. Oncol. Rep. 2014, 32, 2648–2656. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, R.; Cui, W.; Pang, Y.; Liu, C.; Cui, L.; Qian, T.; Quan, L.; Dai, Y.; Jiao, Y.; et al. High IFITM3 Expression Predicts Adverse Prognosis in Acute Myeloid Leukemia. Cancer Gene Ther. 2020, 27, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tang, F.; Bian, E.; Zhang, Y.; Ji, X.; Yang, Z.; Zhao, B. IFITM3/STAT3 Axis Promotes Glioma Cells Invasion and Is Modulated by TGF-β. Mol. Biol. Rep. 2020, 47, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Feng, Q.; Liao, W.; Liang, Y.; Gong, C.; Li, E.; He, W.; Yuan, R.; Wu, L. IFITM3 Promotes Hepatocellular Carcinoma Invasion and Metastasis by Regulating MMP9 through P38/MAPK Signaling. FEBS Open Bio 2018, 8, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Xiao, Z.; Jiang, W.; Chen, G.; Wang, Z. Overexpression of IFITM3 Predicts Poor Prognosis in Stage IIA Esophageal Squamous Cell Carcinoma after Ivor Lewis Esophagectomy. Thorac. Cancer 2017, 8, 592–599. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Wang, H.; He, H.; Niu, H.; Li, Y. Interferon Induced Transmembrane Protein 3 Regulates the Growth and Invasion of Human Lung Adenocarcinoma. Thorac. Cancer 2017, 8, 337–343. [Google Scholar] [CrossRef]
- Andreu, P.; Colnot, S.; Godard, C.; Laurent-Puig, P.; Lamarque, D.; Kahn, A.; Perret, C.; Romagnolo, B. Identification of the IFITM Family as a New Molecular Marker in Human Colorectal Tumors. Cancer Res. 2006, 66, 1949–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajapaksa, U.S.; Jin, C.; Dong, T. Malignancy and IFITM3: Friend or Foe? Front. Oncol. 2020, 10, 593245. [Google Scholar] [CrossRef]
- Alber, D.; Staeheli, P. Partial Inhibition of Vesicular Stomatitis Virus by the Interferon-Induced Human 9-27 Protein. J. Interferon Cytokine Res. 1996, 16, 375–380. [Google Scholar] [CrossRef]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [Green Version]
- Shapira, S.D.; Gat-Viks, I.; Shum, B.O.V.; Dricot, A.; de Grace, M.M.; Wu, L.; Gupta, P.B.; Hao, T.; Silver, S.J.; Root, D.E.; et al. A Physical and Regulatory Map of Host-Influenza Interactions Reveals Pathways in H1N1 Infection. Cell 2009, 139, 1255–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zani, A.; Yount, J.S. Antiviral Protection by IFITM3 In Vivo. Curr. Clin. Microbiol. Rep. 2018, 5, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. IFITMs Restrict the Replication of Multiple Pathogenic Viruses. J. Mol. Biol. 2013, 425, 4937–4955. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Schwartz, O.; Compton, A.A. More than Meets the I: The Diverse Antiviral and Cellular Functions of Interferon-Induced Transmembrane Proteins. Retrovirology 2017, 14, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.-L.; Liang, C. The IFITM Proteins Inhibit HIV-1 Infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM Proteins Incorporated into HIV-1 Virions Impair Viral Fusion and Spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef] [Green Version]
- Tartour, K.; Nguyen, X.-N.; Appourchaux, R.; Assil, S.; Barateau, V.; Bloyet, L.-M.; Burlaud Gaillard, J.; Confort, M.-P.; Escudero-Perez, B.; Gruffat, H.; et al. Interference with the Production of Infectious Viral Particles and Bimodal Inhibition of Replication Are Broadly Conserved Antiviral Properties of IFITMs. PLoS Pathog. 2017, 13, e1006610. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-Y.J.; Fu, R.M.; Liang, C.; Sloan, R.D. IFITM Proteins Inhibit HIV-1 Protein Synthesis. Sci. Rep. 2018, 8, 14551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Markosyan, R.M.; Zheng, Y.-M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM Proteins Restrict Viral Membrane Hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 Restricts Influenza A Virus Entry by Blocking the Formation of Fusion PoRes. Following Virus-Endosome Hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suddala, K.C.; Lee, C.C.; Meraner, P.; Marin, M.; Markosyan, R.M.; Desai, T.M.; Cohen, F.S.; Brass, A.L.; Melikyan, G.B. Interferon-Induced Transmembrane Protein 3 Blocks Fusion of Sensitive but Not Resistant Viruses by Partitioning into Virus-Carrying Endosomes. PLoS Pathog. 2019, 15, e1007532. [Google Scholar] [CrossRef] [Green Version]
- Spence, J.S.; He, R.; Hoffmann, H.-H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 Directly Engages and Shuttles Incoming Virus Particles to Lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef]
- Bright, N.A.; Gratian, M.J.; Luzio, J.P. Endocytic Delivery to Lysosomes Mediated by Concurrent Fusion and Kissing Events in Living Cells. Curr. Biol. 2005, 15, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, Y.; Helenius, A. Virus Entry at a Glance. J. Cell. Sci. 2013, 126, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Pan, Q.; Ding, S.; Rong, L.; Liu, S.-L.; Geng, Y.; Qiao, W.; Liang, C. The N-Terminal Region of IFITM3 Modulates Its Antiviral Activity by Regulating IFITM3 Cellular Localization. J. Virol. 2012, 86, 13697–13707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of Transmitted Founder HIV-1 to IFITM-Mediated Restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Appourchaux, R.; Delpeuch, M.; Zhong, L.; Burlaud-Gaillard, J.; Tartour, K.; Savidis, G.; Brass, A.; Etienne, L.; Roingeard, P.; Cimarelli, A. Functional Mapping of Regions Involved in the Negative Imprinting of Virion Particle Infectivity and in Target Cell Protection by Interferon-Induced Transmembrane Protein 3 against HIV-1. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV Enters Cells via Endocytosis and Dynamin-Dependent Fusion with Endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Khandkar, M. Endocytosis of Human Immunodeficiency Virus 1 (HIV-1) in Astrocytes: A Fiery Path to Its Destination. Microb. Pathog. 2015, 78, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fackler, O.T.; Peterlin, B.M. Endocytic Entry of HIV-1. Curr. Biol. 2000, 10, 1005–1008. [Google Scholar] [CrossRef] [Green Version]
- Sloan, R.D.; Kuhl, B.D.; Mesplède, T.; Münch, J.; Donahue, D.A.; Wainberg, M.A. Productive Entry of HIV-1 during Cell-to-Cell Transmission via Dynamin-Dependent Endocytosis. J. Virol. 2013, 87, 8110–8123. [Google Scholar] [CrossRef] [Green Version]
- Herold, N.; Anders-Ößwein, M.; Glass, B.; Eckhardt, M.; Müller, B.; Kräusslich, H.-G. HIV-1 Entry in SupT1-R5, CEM-Ss, and Primary CD4+ T Cells Occurs at the Plasma Membrane and Does Not Require Endocytosis. J. Virol. 2014, 88, 13956–13970. [Google Scholar] [CrossRef] [Green Version]
- Stein, B.S.; Gowda, S.D.; Lifson, J.D.; Penhallow, R.C.; Bensch, K.G.; Engleman, E.G. PH-Independent HIV Entry into CD4-Positive T Cells via Virus Envelope Fusion to the Plasma Membrane. Cell 1987, 49, 659–668. [Google Scholar] [CrossRef]
- McClure, M.O.; Marsh, M.; Weiss, R.A. Human Immunodeficiency Virus Infection of CD4-Bearing Cells Occurs by a PH-Independent Mechanism. EMBO J. 1988, 7, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Hulseberg, C.E.; Fénéant, L.; Szymańska, K.M.; White, J.M. Lamp1 Increases the Efficiency of Lassa Virus Infection by Promoting Fusion in Less Acidic Endosomal Compartments. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Rojek, J.M.; Sanchez, A.B.; Nguyen, N.T.; de la Torre, J.-C.; Kunz, S. Different Mechanisms of Cell Entry by Human-Pathogenic Old World and New World Arenaviruses. J. Virol. 2008, 82, 7677–7687. [Google Scholar] [CrossRef] [Green Version]
- Rahman, K.; Coomer, C.A.; Majdoul, S.; Ding, S.Y.; Padilla-Parra, S.; Compton, A.A. Homology-Guided Identification of a Conserved Motif Linking the Antiviral Functions of IFITM3 to Its Oligomeric State. Elife 2020, 9, e58537. [Google Scholar] [CrossRef]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.-M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. IFITM Proteins Restrict HIV-1 Infection by Antagonizing the Envelope Glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahi, Y.S.; Yimer, D.; Shi, G.; Majdoul, S.; Rahman, K.; Rein, A.; Compton, A.A. IFITM3 Reduces Retroviral Envelope Abundance and Function and Is Counteracted by GlycoGag. mBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Li, S.; Li, R.; Chai, Q.; Zhang, L.; Wang, B.; Yu, C.; Zheng, Y.-H. The Retroviral Accessory Proteins S2, Nef, and GlycoMA Use Similar Mechanisms for Antagonizing the Host Restriction Factor SERINC5. J. Biol. Chem. 2019, 294, 7013–7024. [Google Scholar] [CrossRef]
- Guo, X.; Steinkühler, J.; Marin, M.; Li, X.; Lu, W.; Dimova, R.; Melikyan, G.B. Interferon-Induced Transmembrane Protein 3 Blocks Fusion of Diverse Enveloped Viruses by Altering Mechanical Properties of Cell Membranes. ACS Nano 2021. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Frangos, J.A.; Chachisvilis, M. Laurdan Fluorescence Senses Mechanical Strain in the Lipid Bilayer Membrane. Biochem. Biophys. Res. Commun. 2006, 347, 838–841. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-Y.; Chin, C.R.; Everitt, A.R.; Clare, S.; Perreira, J.M.; Savidis, G.; Aker, A.M.; John, S.P.; Sarlah, D.; Carreira, E.M.; et al. Amphotericin B Increases Influenza A Virus Infection by Preventing IFITM3-Mediated Restriction. Cell Rep. 2013, 5, 895–908. [Google Scholar] [CrossRef] [Green Version]
- Heaton, N.S.; Randall, G. Multifaceted Roles for Lipids in Viral Infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef]
- Chesarino, N.M.; Compton, A.A.; McMichael, T.M.; Kenney, A.D.; Zhang, L.; Soewarna, V.; Davis, M.; Schwartz, O.; Yount, J.S. IFITM 3 RequiRes. An Amphipathic Helix for Antiviral Activity. EMBO Rep. 2017, 18, 1740–1751. [Google Scholar] [CrossRef] [PubMed]
- Amini-Bavil-Olyaee, S.; Choi, Y.J.; Lee, J.H.; Shi, M.; Huang, I.-C.; Farzan, M.; Jung, J.U. The Antiviral Effector IFITM3 Disrupts Intracellular Cholesterol Homeostasis to Block Viral Entry. Cell Host Microbe 2013, 13, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Wrensch, F.; Winkler, M.; Pöhlmann, S. IFITM Proteins Inhibit Entry Driven by the MERS-Coronavirus Spike Protein: Evidence for Cholesterol-Independent Mechanisms. Viruses 2014, 6, 3683–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.E.; Levine, T.P. VAP, a Versatile Access Point for the Endoplasmic Reticulum: Review and Analysis of FFAT-like Motifs in the VAPome. Biochim. Biophys. Acta 2016, 1861, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Prosser, D.C.; Tran, D.; Gougeon, P.-Y.; Verly, C.; Ngsee, J.K. FFAT Rescues VAPA-Mediated Inhibition of ER-to-Golgi Transport and VAPB-Mediated ER Aggregation. J. Cell. Sci. 2008, 121, 3052–3061. [Google Scholar] [CrossRef] [Green Version]
- Di Mattia, T.; Martinet, A.; Ikhlef, S.; McEwen, A.G.; Nominé, Y.; Wendling, C.; Poussin-Courmontagne, P.; Voilquin, L.; Eberling, P.; Ruffenach, F.; et al. FFAT Motif Phosphorylation Controls Formation and Lipid Transfer Function of Inter-Organelle Contacts. EMBO J. 2020, 39, e104369. [Google Scholar] [CrossRef] [PubMed]
- Weber-Boyvat, M.; Trimbuch, T.; Shah, S.; Jäntti, J.; Olkkonen, V.M.; Rosenmund, C. ORP/Osh Mediate Cross-Talk between ER-Plasma Membrane Contact Site Components and Plasma Membrane SNAREs. Cell Mol. Life Sci. 2021, 78, 1689–1708. [Google Scholar] [CrossRef]
- Quon, E.; Sere, Y.Y.; Chauhan, N.; Johansen, J.; Sullivan, D.P.; Dittman, J.S.; Rice, W.J.; Chan, R.B.; Di Paolo, G.; Beh, C.T.; et al. Endoplasmic Reticulum-Plasma Membrane Contact Sites Integrate Sterol and Phospholipid Regulation. PLoS Biol. 2018, 16, e2003864. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Nguyen, H.T.; Ding, H.; Wang, J.; Zou, S.; Liu, L.; Guha, D.; Gabuzda, D.; Ho, D.D.; Kappes, J.C.; et al. Dual Pathways of Human Immunodeficiency Virus Type 1 Envelope Glycoprotein Trafficking Modulate the Selective Exclusion of Uncleaved Oligomers from Virions. J. Virol. 2021, 95. [Google Scholar] [CrossRef]
- Drouin, A.; Migraine, J.; Durand, M.-A.; Moreau, A.; Burlaud-Gaillard, J.; Beretta, M.; Roingeard, P.; Bouvin-Pley, M.; Braibant, M. Escape of HIV-1 Envelope Glycoprotein from the Restriction of Infection by IFITM3. J. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Parrish, N.F.; Gao, F.; Li, H.; Giorgi, E.E.; Barbian, H.J.; Parrish, E.H.; Zajic, L.; Iyer, S.S.; Decker, J.M.; Kumar, A.; et al. Phenotypic Properties of Transmitted Founder HIV-1. Proc. Natl. Acad. Sci. USA 2013, 110, 6626–6633. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Pan, Q.; Ding, S.; Wang, Z.; Yu, J.; Finzi, A.; Liu, S.-L.; Liang, C. The V3 Loop of HIV-1 Env Determines Viral Susceptibility to IFITM3 Impairment of Viral Infectivity. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beitari, S.; Pan, Q.; Finzi, A.; Liang, C. Differential PressuRes. of SERINC5 and IFITM3 on HIV-1 Envelope Glycoprotein over the Course of HIV-1 Infection. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Magnus, C.; Regoes, R.R. Estimating the Stoichiometry of HIV Neutralization. PLoS Comput. Biol. 2010, 6, e1000713. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Kurteva, S.; Ren, X.; Lee, S.; Sodroski, J. Stoichiometry of Envelope Glycoprotein Trimers in the Entry of Human Immunodeficiency Virus Type 1. J. Virol. 2005, 79, 12132–12147. [Google Scholar] [CrossRef] [Green Version]
- Brandenberg, O.F.; Magnus, C.; Rusert, P.; Regoes, R.R.; Trkola, A. Different Infectivity of HIV-1 Strains Is Linked to Number of Envelope Trimers Required for Entry. PLoS Pathog. 2015, 11, e1004595. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Mothes, W. Structure and Dynamics of the Native HIV-1 Env Trimer. J. Virol. 2015, 89, 5752–5755. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Finzi, A.; Sodroski, J. The Conformational States of the HIV-1 Envelope Glycoproteins. Trends Microbiol. 2020, 28, 655–667. [Google Scholar] [CrossRef]
- Sakin, V.; Hanne, J.; Dunder, J.; Anders-Össwein, M.; Laketa, V.; Nikić, I.; Kräusslich, H.-G.; Lemke, E.A.; Müller, B. A Versatile Tool for Live-Cell Imaging and Super-Resolution Nanoscopy Studies of HIV-1 Env Distribution and Mobility. Cell Chem. Biol. 2017, 24, 635–645.e5. [Google Scholar] [CrossRef] [Green Version]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Müller, B.; Hell, S.W.; Kräusslich, H.-G. Maturation-Dependent HIV-1 Surface Protein Redistribution Revealed by Fluorescence Nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimi, H.; Johnson, J.; Flores, M.G.; Zhang, M.S.; O’Malley, Y.; Houtman, J.C.; Schlievert, P.M.; Haim, H. The Lipid Membrane of HIV-1 Stabilizes the Viral Envelope Glycoproteins and Modulates Their Sensitivity to Antibody Neutralization. J. Biol. Chem. 2020, 295, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, B.; Alsahafi, N.; Debbeche, O.; Prévost, J.; Ding, S.; Chapleau, J.-P.; Herschhorn, A.; Madani, N.; Princiotto, A.; Melillo, B.; et al. Residues in the Gp41 Ectodomain Regulate HIV-1 Envelope Glycoprotein Conformational Transitions Induced by Gp120-Directed Inhibitors. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Kovacs, J.M.; Peng, H.; Rits-Volloch, S.; Lu, J.; Park, D.; Zablowsky, E.; Seaman, M.S.; Chen, B. HIV-1 ENVELOPE. Effect of the Cytoplasmic Domain on Antigenic Characteristics of HIV-1 Envelope Glycoprotein. Science 2015, 349, 191–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfadhli, A.; Staubus, A.O.; Tedbury, P.R.; Novikova, M.; Freed, E.O.; Barklis, E. Analysis of HIV-1 Matrix-Envelope Cytoplasmic Tail Interactions. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Pezeshkian, N.; Groves, N.S.; van Engelenburg, S.B. Single-Molecule Imaging of HIV-1 Envelope Glycoprotein Dynamics and Gag Lattice Association Exposes Determinants Responsible for Virus Incorporation. Proc. Natl. Acad. Sci. USA 2019, 116, 25269–25277. [Google Scholar] [CrossRef] [Green Version]
- Zang, R.; Case, J.B.; Yutuc, E.; Ma, X.; Shen, S.; Gomez Castro, M.F.; Liu, Z.; Zeng, Q.; Zhao, H.; Son, J.; et al. Cholesterol 25-Hydroxylase Suppresses SARS-CoV-2 Replication by Blocking Membrane Fusion. Proc. Natl. Acad. Sci. USA 2020, 117, 32105–32113. [Google Scholar] [CrossRef]
- Henderson, S.; Fenton, T. APOBEC3 Genes: Retroviral Restriction Factors to Cancer Drivers. Trends Mol. Med. 2015, 21, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, R.; Lang, T.; Südhof, T.C. Membrane Fusion. Cell 2003, 112, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Wee, Y.S.; Weis, J.J.; Gahring, L.C.; Rogers, S.W.; Weis, J.H. Age-Related Onset of Obesity Corresponds with Metabolic Dysregulation and Altered Microglia Morphology in Mice Deficient for Ifitm Proteins. PLoS ONE 2015, 10, e0123218. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Robinson, M.E.; Ma, N.; Artadji, D.; Ahmed, M.A.; Xiao, G.; Sadras, T.; Deb, G.; Winchester, J.; Cosgun, K.N.; et al. IFITM3 Functions as a PIP3 Scaffold to Amplify PI3K Signalling in B Cells. Nature 2020, 588, 491–497. [Google Scholar] [CrossRef]
- Buchrieser, J.; Degrelle, S.A.; Couderc, T.; Nevers, Q.; Disson, O.; Manet, C.; Donahue, D.A.; Porrot, F.; Hillion, K.-H.; Perthame, E.; et al. IFITM Proteins Inhibit Placental Syncytiotrophoblast Formation and Promote Fetal Demise. Science 2019, 365, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Zani, A.; Zhang, L.; McMichael, T.M.; Kenney, A.D.; Chemudupati, M.; Kwiek, J.J.; Liu, S.-L.; Yount, J.S. Interferon-Induced Transmembrane Proteins Inhibit Cell Fusion Mediated by Trophoblast Syncytins. J. Biol. Chem. 2019, 294, 19844–19851. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marziali, F.; Cimarelli, A. Membrane Interference Against HIV-1 by Intrinsic Antiviral Factors: The Case of IFITMs. Cells 2021, 10, 1171. https://doi.org/10.3390/cells10051171
Marziali F, Cimarelli A. Membrane Interference Against HIV-1 by Intrinsic Antiviral Factors: The Case of IFITMs. Cells. 2021; 10(5):1171. https://doi.org/10.3390/cells10051171
Chicago/Turabian StyleMarziali, Federico, and Andrea Cimarelli. 2021. "Membrane Interference Against HIV-1 by Intrinsic Antiviral Factors: The Case of IFITMs" Cells 10, no. 5: 1171. https://doi.org/10.3390/cells10051171
APA StyleMarziali, F., & Cimarelli, A. (2021). Membrane Interference Against HIV-1 by Intrinsic Antiviral Factors: The Case of IFITMs. Cells, 10(5), 1171. https://doi.org/10.3390/cells10051171