
The Interactions of Insulin and Vitamin A Signaling Systems for the Regulation of Hepatic Glucose and Lipid Metabolism


Abstract
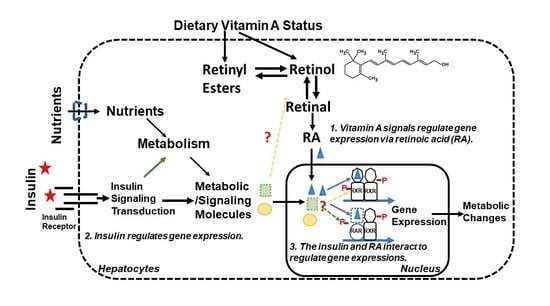
:
1. Introduction
2. Insulin and VA
2.1. The History of Diabetes and Insulin
2.2. VA and Its Signaling
2.3. The Impacts of VA Status on Body Weight and Metabolism
2.4. The Effects of RA on Metabolism
3. The Regulation and Activation of RAR and RXR on Metabolism
3.1. The Changes of RARs and RXRs in Metabolic Disease Models
3.2. Targeting RARs and RXRs to Control Metabolism
3.3. RAR Phosphorylation
3.4. RXR Phosphorylation
4. Insulin and RA Interactions for Regulating Gene Expression in Hepatocytes
5. Conclusions and Future Perspectives
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Federation, I.D. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussel, Belgium, 2019. [Google Scholar]
- Kotwas, A.; Karakiewicz, B.; Zabielska, P.; Wieder-Huszla, S.; Jurczak, A. Epidemiological factors for type 2 diabetes mellitus: Evidence from the Global Burden of Disease. Arch. Public Health 2021, 79, 1–7. [Google Scholar] [CrossRef]
- McGarry, J.D. Glucose-fatty acid interactions in health and disease. Am. J. Clin. Nutr. 1998, 67, 500S–504S. [Google Scholar] [CrossRef] [Green Version]
- Needham, J.; Hartley, H.B. Sir Frederick Gowland Hopkins, OM, FRS (1861-1947) Centenary Lecture held on 20 November 1961 in the University of Cambridge. Notes Rec. R. Soc. Lond. 1962, 17, 117–162. [Google Scholar]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef]
- Ross, A.C. Retinoid Production and Catabolism: Role of Diet in Regulating Retinol Esterification and Retinoic Acid Oxidation. J. Nutr. 2003, 133, 291S–296S. [Google Scholar] [CrossRef] [Green Version]
- Napoli, J.L. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2012, 1821, 152–167. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.C.; Harrison, E. Vitamin A and Carotenoids. In Handbook of Vitamins, 4th ed.; McCormick, D., Rucker, R., Suttie, J., Zempleni, J., Eds.; Dekker/CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Chen, W.; Chen, G. The Roles of Vitamin A in the Regulation of Carbohydrate, Lipid, and Protein Metabolism. J. Clin. Med. 2014, 3, 453–479. [Google Scholar] [CrossRef] [Green Version]
- Blaner, W.S. Vitamin A signaling and homeostasis in obesity, diabetes, and metabolic disorders. Pharmacol. Ther. 2019, 197, 153–178. [Google Scholar] [CrossRef]
- Bliss, M. The Discovery of Insulin. Anniversary Edition; University of Chicago Press: Chicago, IL, USA, 1982. [Google Scholar]
- Banting, F.G.; Best, C.H.; Collip, J.B.; Campbell, W.R.; Fletcher, A. Pancreatic Extracts in the Treatment of Diabetes Mellitus: Preliminary Report. Can. Med. Assoc. J. 1962, 87, 1062–1067. [Google Scholar] [PubMed]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.M.; Granner, D.K. Regulation of gene expression by insulin. Physiol. Rev. 1996, 76, 1109–1161. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Selective versus Total Insulin Resistance: A Pathogenic Paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Guan, H.P.; Chen, G. Chapter Six - Factors Affecting Insulin-Regulated Hepatic Gene Expression. In Progress in Molecular Biology and Translational Science: Glucose Homeostatis and the Pathogenesis of Diabetes Mellitus; Ya-Xiong, T., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 121, pp. 165–215. [Google Scholar]
- Ahmed, A.M. History of Diabetes Mellitus. Saudi Med. J. 2002, 23, 373. [Google Scholar]
- Covington, M.B. Traditional Chinese Medicine in the Treatment of Diabetes. Diabetes Spectr. 2001, 14, 154–159. [Google Scholar] [CrossRef] [Green Version]
- Himsworth, H. Diabetes Mellitus: Its Differentiation into Insulin-Sensitive and Insulin-Insensitive Types. Lancet 1936, 227, 127–130. [Google Scholar] [CrossRef]
- Walker, J. Frederick Sanger (1918–2013). Nature 2014, 505, 27. [Google Scholar] [CrossRef] [Green Version]
- Kahn, C.R.; Roth, J. Rosalyn Sussman Yalow (1921–2011). Proc. Natl. Acad. Sci. USA 2012, 109, 669–670. [Google Scholar] [CrossRef] [Green Version]
- Johnson, I. Human insulin from recombinant DNA technology. Science 1983, 219, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Shulman, G. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- McKern, N.M.; Lawrence, M.; Streltsov, V.A.; Lou, M.-Z.; Adams, T.; Lovrecz, G.O.; Elleman, T.C.; Richards, K.M.; Bentley, J.D.; Pilling, P.A.; et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 2006, 443, 218–221. [Google Scholar] [CrossRef]
- Cohen, P. The twentieth century struggle to decipher insulin signalling. Nat. Rev. Mol. Cell Biol. 2006, 7, 867–873. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. & Metab. 2002, 283, E413–E422. [Google Scholar]
- Hanke, S.; Mann, M. The phosphotyrosine interactome of the insulin receptor family and its substrates IRS-1 and IRS-2. Mol. & Cell. Proteom. 2009, 8, 519–534. [Google Scholar]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avruch, J. MAP kinase pathways: The first twenty years. Biochim. Biophys. Acta Mol. Cell. Res. 2007, 1773, 1150–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Cantley, L. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Osborne, J.K.; Zaganjor, E.; Cobb, M.H. Signal control through Raf: In sickness and in health. Cell Res. 2011, 22, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Iynedjian, P.B. Mammalian glucokinase and its gene. Biochem. J. 1993, 293, 1–13. [Google Scholar] [CrossRef]
- Magnuson, M.; Andreone, T.L.; Printz, R.L.; Koch, S.; Granner, D.K. Rat glucokinase gene: Structure and regulation by insulin. Proc. Natl. Acad. Sci. USA 1989, 86, 4838–4842. [Google Scholar] [CrossRef] [Green Version]
- Schmitt-Ney, M. The FOXO’s Advantages of Being a Family: Considerations on Function and Evolution. Cells 2020, 9, 787. [Google Scholar] [CrossRef] [Green Version]
- Guo, S. Insulin signaling, resistance, and metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2013, 220, T1–T23. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Yan, F.; Feng, Z.; Lazarovici, P.; Zheng, W. Signaling Network of Forkhead Family of Transcription Factors (FOXO) in Dietary Restriction. Cells 2019, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Zečić, A.; Braeckman, B.P. DAF-16/FoxO in Caenorhabditis elegans and Its Role in Metabolic Remodeling. Cells 2020, 9, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, I.; Bashmakov, Y.; Ikemoto, S.; Horton, J.D.; Brown, M.S.; Goldstein, J.L. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 13656–13661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, J.; Goldstein, J.; Brown, M. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liang, G.; Ou, J.; Goldstein, J.L.; Brown, M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 11245–11250. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, I.; Matsuda, M.; Hammer, R.E.; Bashmakov, Y.; Brown, M.S.; Goldstein, J.L. Decreased IRS-2 and Increased SREBP-1c Lead to Mixed Insulin Resistance and Sensitivity in Livers of Lipodystrophic and ob/ob Mice. Mol. Cell 2000, 6, 77–86. [Google Scholar] [CrossRef]
- Ross, A.C.; Caballero, B.; Cousins, R.J.; Tucker, K.L.; Ziegler, T.R. Modern Nutrition in Health and Disease; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012. [Google Scholar]
- Lakshman, M.R. Alpha and omega of carotenoid cleavage. J. Nutr. 2004, 134, 241S–245S. [Google Scholar] [CrossRef]
- Wyss, A. Carotene Oxygenases: A New Family of Double Bond Cleavage Enzymes. J. Nutr. 2004, 134, 246S–250S. [Google Scholar] [CrossRef] [Green Version]
- Moise, A.R.; Noy, N.; Palczewski, K.; Blaner, W.S. Delivery of Retinoid-Based Therapies To Target Tissues. Biochemistry 2007, 46, 4449–4458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Bain, D.L.; Heneghan, A.F.; Connaghan-Jones, K.D.; Miura, M.T. Nuclear Receptor Structure: Implications for Function. Annu. Rev. Physiol. 2007, 69, 201–220. [Google Scholar] [CrossRef] [PubMed]
- Benbrook, D.M.; Chambon, P.; Rochette-Egly, C.; Asson-Batres, M.A. History of Retinoic Acid Receptors. Biochem. Retin. Acid Recept. I Struct. Act. Funct. Mol. Level 2014, 70, 1–20. [Google Scholar] [CrossRef]
- Wolf, G. Tissue-specific increases in endogenous all-trans retinoic acid: Possible contributing factor in ethanol toxicity. Nutr. Rev. 2010, 68, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.; Evans, R. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Makita, T.; Hernandez-Hoyos, G.; Chen, T.H.-P.; Wu, H.; Rothenberg, E.V.; Sucov, H.M. A developmental transition in definitive erythropoiesis: Erythropoietin expression is sequentially regulated by retinoic acid receptors and HNF4. Genes Dev. 2001, 15, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, S.W.; Suino-Powell, K.; Zhou, X.E.; E. Kretschman, J.; Reynolds, R.; Vonrhein, C.; Xu, Y.; Wang, L.; Tsai, S.Y.; Tsai, M.-J.; et al. Identification of COUP-TFII Orphan Nuclear Receptor as a Retinoic Acid–Activated Receptor. PLoS Biol. 2008, 6, e227. [Google Scholar] [CrossRef]
- Shaw, N.; Elholm, M.; Noy, N. Retinoic Acid Is a High Affinity Selective Ligand for the Peroxisome Proliferator-activated Receptor β/δ. J. Biol. Chem. 2003, 278, 41589–41592. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zhang, R.; Li, Y.; Zhu, B.; Chen, W.; Zhang, Y.; Chen, G. A RARE of hepatic Gck promoter interacts with RARa, HNF4a and COUP-TFII that affect retinoic acid- and insulin-induced Gck expression. J. Nutr. Biochem. 2014, 25, 964–976. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, R.; Li, Y.; Chen, W.; Zhang, Y.; Chen, G. Roles of vitamin A status and retinoids in glucose and fatty acid metabolism. Biochem. Cell Biol. 2012, 90, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Moore, T. Vitamin A and carotene: The vitamin A reserve of the adult human being in health and disease. Biochem. J. 1937, 31, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, G.; Lane, M.D.; Johnson, B.C. Studies on the function of vitamin A in metabolism. J. Biol. Chem. 1957, 225, 995–1008. [Google Scholar] [CrossRef]
- Singh, M.; Singh, V.N.; Venkitasubramanian, T. Early effects of feeding excess vitamin A: Hepatic glycogen, blood lactic acid, plasma nefa and glucose tolerance in rats. Life Sci. 1968, 7, 239–247. [Google Scholar] [CrossRef]
- Brown, E.F.; Morgan, A.F. The Effect of Vitamin A Deficiency upon the Nitrogen Metabolism of the Rat: Two Figures. J. Nutr. 1948, 35, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Howell, M.L.; Li, Y.; Li, R.; Chen, G. Vitamin A and Feeding Statuses Modulate the Insulin-Regulated Gene Expression in Zucker Lean and Fatty Primary Rat Hepatocytes. PLoS ONE 2014, 9, e100868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chen, G.; Liu, Y. Vitamin A status affects the plasma parameters and regulation of hepatic genes in streptozotocin-induced diabetic rats. Biochimie 2017, 137, 1–11. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, R.; Li, Y.; Chen, W.; Zhao, S.; Chen, G. Vitamin A status affects obesity development and hepatic expression of key genes for fuel metabolism in Zucker fatty rats. Biochem. Cell Biol. 2012, 90, 548–557. [Google Scholar] [CrossRef]
- Li, Y.; Li, R.; Chen, W.; Chen, G.; Information, P.E.K.F.C. Vitamin A status and its metabolism contribute to the regulation of hepatic genes during the cycle of fasting and refeeding in rats. J. Nutr. Biochem. 2015, 30, 33–43. [Google Scholar] [CrossRef]
- McCollum, E.; Davis, M. The necessity of certain lipins in the diet during growth. J. Biol. Chem. 1913, 15, 167–175. [Google Scholar] [CrossRef]
- Kang, H.W.; Bhimidi, G.R.; Odom, D.P.; Brun, P.-J.; Fernandez, M.-L.; McGrane, M.M. Altered lipid catabolism in the vitamin A deficient liver. Mol. Cell. Endocrinol. 2007, 271, 18–27. [Google Scholar] [CrossRef]
- Ziouzenkova, O.; Orasanu, G.; Sharlach, M.; E. Akiyama, T.; Berger, J.P.; Viereck, J.; Hamilton, J.; Tang, G.; Dolnikowski, G.; Vogel, S.; et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat. Med. 2007, 13, 695–702. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, Y.; Lu, D.; Li, N.-Q.; Ross, A.C. Retinoids synergize with insulin to induce hepatic Gck expression. Biochem. J. 2009, 419, 645–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klyuyeva, A.V.; Belyaeva, O.V.; Goggans, K.R.; Krezel, W.; Popov, K.M.; Kedishvili, N.Y. Changes in retinoid metabolism and signaling associated with metabolic remodeling during fasting and in type I diabetes. J. Biol. Chem. 2021, 296, 100323. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Li, R.; Chen, W.; Howell, M.; Zhang, R.; Chen, G. The Hepatic Raldh1 Expression Is elevated in Zucker Fatty Rats and Its Over-Expression Introduced the Retinal-Induced Srebp-1c Expression in INS-1 Cells. PLoS ONE 2012, 7, e45210. [Google Scholar] [CrossRef] [Green Version]
- Bershad, S.; Rubinstein, A.; Paterniti, J.R.; Le, N.A.; Poliak, S.C.; Heller, B.; Ginsberg, H.N.; Fleischmajer, R.; Brown, W.V. Changes in plasma lipids and lipoproteins during isotretinoin therapy for acne. N. Engl. J. Med 1985, 313, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Noy, N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor β/δ and retinoic acid receptor. Mol. Cell. Biol. 2009, 29, 3286–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.C.; DeSantis, D.; Soltanian, H.; Croniger, C.M.; Noy, N. Retinoic Acid Upregulates Preadipocyte Genes to Block Adipogenesis and Suppress Diet-Induced Obesity. Diabetes 2012, 61, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Mercader, J.; Ribot, J.; Murano, I.; Felipe, F.; Cinti, S.; Bonet, M.L.; Palou, A. Remodeling of White Adipose Tissue after Retinoic Acid Administration in Mice. Endocrinology 2006, 147, 5325–5332. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, H.; Ikeda, Y.; Ebata, Y.; Kojima, C.; Katsuma, R.; Tsuruyama, T.; Sakabe, T.; Shomori, K.; Komeda, N.; Oshiro, S.; et al. Retinoids ameliorate insulin resistance in a leptin-dependent manner in mice. Hepatology 2012, 56, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wong, K.; Walsh, K.; Gao, B.; Zang, M. Retinoic Acid Receptor β Stimulates Hepatic Induction of Fibroblast Growth Factor 21 to Promote Fatty Acid Oxidation and Control Whole-body Energy Homeostasis in Mice. J. Biol. Chem. 2013, 288, 10490–10504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codner, E.; Loviscach, M.; Ciaraldi, T.; Rehman, N.; Carter, L.; Mudaliar, S.; Henry, R. Retinoid X receptor expression in skeletal muscle of nondiabetic, obese and type 2 diabetic individuals. Metabolism 2001, 50, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chu, W.; Hemphill, C.; Hasstedt, S.J.; Elbein, S.C. Mutation screening and association of human retinoid X receptor γ variation with lipid levels in familial type 2 diabetes. Mol. Genet. Metab. 2002, 76, 14–22. [Google Scholar] [CrossRef]
- Rieusset, J.; Roques, M.; Bouzakri, K.; Chevillotte, E.; Vidal, H. Regulation of p85α phosphatidylinositol-3-kinase expression by peroxisome proliferator-activated receptors (PPARs) in human muscle cells. FEBS Lett. 2001, 502, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Snyder, R.; Thekkumkara, T. 13-cis-Retinoic acid specific down-regulation of angiotensin type 1 receptor in rat liver epithelial and aortic smooth muscle cells. J. Mol. Endocrinol. 2011, 48, 99–114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, T.; Hu, X.; Chen, G. Vitamin A and Diabetes. J. Med. Food. 2021, 24, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.-J.; Plutzky, J. Retinoid Metabolism and Diabetes Mellitus. Diabetes Metab. J. 2012, 36, 167–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Wang, H.; Zhou, J.; Qiu, S.; Cai, T.; Li, H.; Shen, Z.; Hu, Y.; Ding, B.; Luo, M.; et al. Vitamin A and Its Multi-Effects on Pancreas: Recent Advances and Prospects. Front. Endocrinol. 2021, 12, 620941. [Google Scholar] [CrossRef]
- Juang, J.-H.; Van, Y.-H.; Kuo, C.-H.; Lin, M.-Y.; Liu, Y.-H.; Chang, H.-Y. Prevention and Reversal of Diabetes by All-Trans Retinoid Acid and Exendin-4 in NOD Mice. Int. J. Endocrinol. 2014, 2014, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Moreno, A.; Quintanar-Escorza, M.A.; Garza, R.G.; Hady, K.; Valenzuela, A.M.; Marszalek, J.E.; Sharara-Núñez, I.; Delgadillo-Guzmán, D.; Keita, H. All-trans retinoic acid improves pancreatic cell proliferation on induced type 1 diabetic rats. Fundam. Clin. Pharmacol. 2019, 34, 345–351. [Google Scholar] [CrossRef]
- Ewa, O.-B.; Karolina, G.; Aleksandra, D.; Natalia, J. Considering The Role of Vitamin A in Glucose Metabolism. J. Endocrinol. Diabetes 2018, 5, 1–4. [Google Scholar] [CrossRef]
- Morgenstern, J.; Fleming, T.; Kliemank, E.; Brune, M.; Nawroth, P.; Fischer, A. Quantification of All-Trans Retinoic Acid by Liquid Chromatography–Tandem Mass Spectrometry and Association with Lipid Profile in Patients with Type 2 Diabetes. Metabolites 2021, 11, 60. [Google Scholar] [CrossRef]
- Park, S.; Lee, Y.J.; Ko, E.H.; Kim, J.-W. Regulation of retinoid X receptor gamma expression by fed state in mouse liver. Biochem. Biophys. Res. Commun. 2015, 458, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Waki, H.; Kamon, J.; Murakami, K.; Motojima, K.; Komeda, K.; Miki, H.; Kubota, N.; Terauchi, Y.; Tsuchida, A.; et al. Inhibition of RXR and PPARγ ameliorates diet-induced obesity and type 2 diabetes. J. Clin. Investig. 2001, 108, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Kamei, Y.; Akaike, F.; Suganami, T.; Kanai, S.; Hattori, M.; Manabe, Y.; Fujii, N.; Takai-Igarashi, T.; Tadaishi, M.; et al. Increased Systemic Glucose Tolerance with Increased Muscle Glucose Uptake in Transgenic Mice Overexpressing RXRγ in Skeletal Muscle. PLoS ONE 2011, 6, e20467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.-J.Y.; Han, G.; Cai, Y.; Dai, T.; Konishi, T.; Leng, A.-S. Hepatocyte Retinoid X Receptor-α-Deficient Mice Have Reduced Food Intake, Increased Body Weight, and Improved Glucose Tolerance. Endocrinology 2003, 144, 605–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhou, S.; Gustafsson, J. Nuclear receptors: Recent drug discovery for cancer therapies. Endocr. Rev. 2019, 40, 1207–1249. [Google Scholar] [CrossRef]
- Mirza, A.Z.; AlThagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef]
- Ren, G.; Kim, T.; Kim, H.-S.; Young, M.E.; Muccio, D.D.; Atigadda, V.R.; Blum, S.; Tse, H.M.; Habegger, K.M.; Bhatnagar, S.; et al. A Small Molecule, UAB126, Reverses Diet-Induced Obesity and its Associated Metabolic Disorders. Diabetes 2020, 69, 2003–2016. [Google Scholar] [CrossRef]
- Guleria, R.S.; Singh, A.B.; Nizamutdinova, I.T.; Souslova, T.; Mohammad, A.A.; Kendall, J.A.; Baker, K.M.; Pan, J. Activation of retinoid receptor-mediated signaling ameliorates diabetes-induced cardiac dysfunction in Zucker diabetic rats. J. Mol. Cell. Cardiol. 2013, 57, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-H.; Zheng, B.; Han, M.; Miao, S.-B.; Wen, J. Synthetic retinoid Am80 inhibits interaction of KLF5 with RARα through inducing KLF5 dephosphorylation mediated by the PI3K/Akt signaling in vascular smooth muscle cells. FEBS Lett. 2009, 583, 1231–1236. [Google Scholar] [CrossRef] [Green Version]
- Robciuc, M.R.; Skrobuk, P.; Anisimov, A.; Olkkonen, V.M.; Alitalo, K.; Eckel, R.H.; Koistinen, H.A.; Jauhiainen, M.; Ehnholm, C. Angiopoietin-Like 4 Mediates PPAR Delta Effect on Lipoprotein Lipase-Dependent Fatty Acid Uptake but Not on Beta-Oxidation in Myotubes. PLoS ONE 2012, 7, e46212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, R.; Davies, P.J.A.; Crombie, D.L.; Bischoff, E.D.; Cesario, R.M.; Jow, L.; Hamann, L.G.; Boehm, M.F.; Mondon, C.E.; Nadzan, A.M.; et al. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature 1997, 386, 407–410. [Google Scholar] [CrossRef]
- Lenhard, J.M.; Lancaster, M.E.; Paulik, M.A.; Weiel, J.E.; Binz, J.G.; Sundseth, S.S.; Gaskill, B.A.; Lightfoot, R.M.; Brown, H.R. The RXR agonist LG100268 causes hepatomegaly, improves glycaemic control and decreases cardiovascular risk and cachexia in diabetic mice suffering from pancreatic beta-cell dysfunction. Diabetologia 1999, 42, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Cline, G.W.; Shulman, G.; Leibowitz, M.D.; Davies, P.J.A. Effects of Rexinoids on Glucose Transport and Insulin-mediated Signaling in Skeletal Muscles of Diabetic (db/db) Mice. J. Biol. Chem. 2004, 279, 19721–19731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-L.; Sennitt, M.; Hislop, D.; Crombie, D.; Heyman, R.; Cawthorne, M. Retinoid X receptor agonists have anti-obesity effects and improve insulin sensitivity in Zucker fa/fa rats. Int. J. Obes. 2000, 24, 997–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogilvie, K.M.; Saladin, R.; Nagy, T.; Urcan, M.S.; Heyman, R.A.; Leibowitz, M.D. Activation of the Retinoid X Receptor Suppresses Appetite in the Rat. Endocrinology 2004, 145, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Cesario, R.M.; Klausing, K.; Razzaghi, H.; Crombie, D.; Rungta, D.; Heyman, R.A.; Lala, D.S. The Rexinoid LG100754 Is a Novel RXR:PPARγ Agonist and Decreases Glucose Levelsin Vivo. Mol. Endocrinol. 2001, 15, 1360–1369. [Google Scholar] [CrossRef] [Green Version]
- Leibowitz, M.D.; Ardecky, R.J.; Boehm, M.F.; Broderick, C.L.; Carfagna, M.A.; Crombie, D.L.; D’Arrigo, J.; Etgen, G.J.; Faul, M.M.; Grese, T.A.; et al. Biological Characterization of a Heterodimer-Selective Retinoid X Receptor Modulator: Potential Benefits for the Treatment of Type 2 Diabetes. Endocrinology 2006, 147, 1044–1053. [Google Scholar] [CrossRef]
- Samarut, E.; Amal, I.; Markov, G.V.; Stote, R.; Dejaegere, A.; Laudet, V.; Rochette-Egly, C. Evolution of Nuclear Retinoic Acid Receptor Alpha (RAR) Phosphorylation Sites. Serine Gain Provides Fine-Tuned Regulation. Mol. Biol. Evol. 2011, 28, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Rochette-Egly, C.; Adam, S.; Rossignol, M.; Egly, J.-M.; Chambon, P. Stimulation of RARα Activation Function AF-1 through Binding to the General Transcription Factor TFIIH and Phosphorylation by CDK7. Cell 1997, 90, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Bour, G.; Lalevee, S.; Rochette-Egly, C. Protein kinases and the proteasome join in the combinatorial control of transcription by nuclear retinoic acid receptors. Trends Cell Biol. 2007, 17, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Bastien, J.; Adam-Stitah, S.; Riedl, T.; Egly, J.-M.; Chambon, P.; Rochette-Egly, C. TFIIH Interacts with the Retinoic Acid Receptor γ and Phosphorylates Its AF-1-activating Domain through cdk7. J. Biol. Chem. 2000, 275, 21896–21904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannì, M.; Kopf, E.; Bastien, J.; Oulad-Abdelghani, M.; Garattini, E.; Chambon, P.; Rochette-Egly, C. Down-regulation of the Phosphatidylinositol 3-Kinase/Akt Pathway Is Involved in Retinoic Acid-induced Phosphorylation, Degradation, and Transcriptional Activity of Retinoic Acid Receptor γ2. J. Biol. Chem. 2002, 277, 24859–24862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Tanoury, Z.; Gaouar, S.; Piskunov, A.; Ye, T.; Urban, S.; Jost, B.; Keime, C.; Davidson, I.; Dierich, A.; Rochette-Egly, C. Phosphorylation of the retinoic acid receptor RARγ2 is crucial for the neuronal differentiation of mouse embryonic stem cells. J. Cell Sci. 2014, 127, 2095–2105. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, H.; Xia, D.; Moore, N.L.; Uray, I.P.; Kim, H.; Ma, L.; Weigel, N.L.; Brown, P.H.; Kurie, J.M. Akt phosphorylates and suppresses the transactivation of retinoic acid receptor α. Biochem. J. 2006, 395, 653–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivas, H.; Juroske, D.M.; Kalyankrishna, S.; Cody, D.D.; Price, R.E.; Xu, X.-C.; Narayanan, R.; Weigel, N.L.; Kurie, J.M. c-Jun N-Terminal Kinase Contributes to Aberrant Retinoid Signaling in Lung Cancer Cells by Phosphorylating and Inducing Proteasomal Degradation of Retinoic Acid Receptor α. Mol. Cell. Biol. 2005, 25, 1054–1069. [Google Scholar] [CrossRef] [Green Version]
- Chebaro, Y.; Amal, I.; Rochel, N.; Rochette-Egly, C.; Stote, R.H.; Dejaegere, A. Phosphorylation of the Retinoic Acid Receptor Alpha Induces a Mechanical Allosteric Regulation and Changes in Internal Dynamics. PLoS Comput. Biol. 2013, 9, e1003012. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Ichikawa-Tomikawa, N.; Kashiwagi, K.; Endo, C.; Tanaka, S.; Sawada, N.; Watabe, T.; Higashi, T.; Chiba, H. Cell adhesion signals regulate the nuclear receptor activity. Proc. Natl. Acad. Sci. USA 2019, 116, 24600–24609. [Google Scholar] [CrossRef]
- Gaillard, E.; Bruck, N.; Brelivet, Y.; Bour, G.; Lalevee, S.; Bauer, A.; Poch, O.; Moras, D.; Rochette-Egly, C. Phosphorylation by PKA potentiates retinoic acid receptor activity by means of increasing interaction with and phosphorylation by cyclin H/cdk7. Proc. Natl. Acad. Sci. USA 2006, 103, 9548–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruck, N.; Vitoux, D.; Ferry, C.; Duong, V.; Bauer, A.; De Thé, H.; Rochette-Egly, C. A coordinated phosphorylation cascade initiated by p38MAPK/MSK1 directs RARα to target promoters. EMBO J. 2008, 28, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabiee, A.; Krüger, M.; Ardenkjær-Larsen, J.; Kahn, C.R.; Emanuelli, B. Distinct signalling properties of insulin receptor substrate (IRS)-1 and IRS-2 in mediating insulin/IGF-1 action. Cell. Signal. 2018, 47, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mascrez, B.; Mark, M.; Krezel, W.; Dupe, V.; LeMeur, M.; Ghyselinck, N.; Chambon, P. Differential contributions of AF-1 and AF-2 activities to the developmental functions of RXR alpha. Development 2001, 128, 2049–2062. [Google Scholar] [CrossRef]
- Bastien, J.; Adam-Stitah, S.; Plassat, J.-L.; Chambon, P.; Rochette-Egly, C. The Phosphorylation Site Located in the A Region of Retinoic X Receptor α Is Required for the Antiproliferative Effect of Retinoic Acid (RA) and the Activation of RA Target Genes in F9 Cells. J. Biol. Chem. 2002, 277, 28683–28689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardenkjær-Larsen, J.; Rupar, K.; Sinkevičiūtė, G.; Petersen, P.S.S.; Villarroel, J.; Lundh, M.; Barres, R.; Rabiee, A.; Emanuelli, B. Insulin-induced serine 22 phosphorylation of retinoid X receptor alpha is dispensable for adipogenesis in brown adipocytes. Adipocyte 2020, 9, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Zhong, G.; Zhang, H.; Yu, B.; Wei, F.; Luo, L.; Kang, Y.; Wu, J.; Jiang, J.; Li, Y.; et al. LncRNA DANCR upregulates PI3K/AKT signaling through activating serine phosphorylation of RXRA. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Gao, W.; Tang, J.; Zhang, H.; Zhou, Y.; Liu, J.; Chen, K.; Liu, F.; Li, W.; To, S.K.Y.; et al. The Roles of GSK-3β in Regulation of Retinoid Signaling and Sorafenib Treatment Response in Hepatocellular Carcinoma. Theranostics 2020, 10, 1230–1244. [Google Scholar] [CrossRef] [PubMed]
- Adam-Stitah, S.; Penna, L.; Chambon, P.; Rochette-Egly, C. Hyperphosphorylation of the Retinoid X Receptor α by Activated c-Jun NH2-terminal Kinases. J. Biol. Chem. 1999, 274, 18932–18941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sueyoshi, T.; Sakuma, T.; Shindo, S.; Fashe, M.; Kanayama, T.; Ray, M.; Moore, R.; Negishi, M. A phosphorylation-deficient mutant of retinoid X receptor α at Thr 167 alters fasting response and energy metabolism in mice. Lab. Investig. 2019, 99, 1470–1483. [Google Scholar] [CrossRef] [PubMed]
- Jusu, S.; Presley, J.F.; Kremer, R. Phosphorylation of Human Retinoid X Receptor α at Serine 260 Impairs Its Subcellular Localization, Receptor Interaction, Nuclear Mobility, and 1α,25-Dihydroxyvitamin D3-dependent DNA Binding in Ras-transformed Keratinocytes. J. Biol. Chem. 2017, 292, 1490–1509. [Google Scholar] [CrossRef] [Green Version]
- Solomon, C.; White, J.H.; Kremer, R. Mitogen-activated protein kinase inhibits 1,25-dihydroxyvitamin D3–dependent signal transduction by phosphorylating human retinoid X receptor α. J. Clin. Investig. 1999, 103, 1729–1735. [Google Scholar] [CrossRef] [Green Version]
- Matsushima-Nishiwaki, R.; Okuno, M.; Adachi, S.; Sano, T.; Akita, K.; Moriwaki, H.; Friedman, S.L.; Kojima, S. Phosphorylation of retinoid X receptor α at serine 260 impairs its metabolism and function in human hepatocellular carcinoma. Cancer Res. 2001, 61, 7675–7682. [Google Scholar] [PubMed]
- Yoshimura, K.; Muto, Y.; Shimizu, M.; Matsushima-Nishiwaki, R.; Okuno, M.; Takano, Y.; Tsurumi, H.; Kojima, S.; Okano, Y.; Moriwaki, H. Phosphorylated retinoid X receptor α loses its heterodimeric activity with retinoic acid receptor β. Cancer Sci. 2007, 98, 1868–1874. [Google Scholar] [CrossRef]
- Himsworth, H.P. Dietetic factors influencing the glucose tolerance and the activity of insulin. J. Physiol. 1934, 81, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Mingrone, G.; Panunzi, S.; De Gaetano, A.; Guidone, C.; Iaconelli, A.; Leccesi, L.; Nanni, G.; Pomp, A.; Castagneto, M.; Ghirlanda, G.; et al. Bariatric Surgery versus Conventional Medical Therapy for Type 2 Diabetes. N. Engl. J. Med. 2012, 366, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Schauer, P.R.; Kashyap, S.R.; Wolski, K.; Brethauer, S.A.; Kirwan, J.P.; Pothier, C.E.; Thomas, S.; Abood, B.; Nissen, S.E.; Bhatt, D.L. Bariatric Surgery versus Intensive Medical Therapy in Obese Patients with Diabetes. N. Engl. J. Med. 2012, 366, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Mingrone, G.; Panunzi, S.; De Gaetano, A.; Guidone, C.; Iaconelli, A.; Nanni, G.; Castagneto, M.; Bornstein, S.; Rubino, F. Bariatric–metabolic surgery versus conventional medical treatment in obese patients with type 2 diabetes: 5 year follow-up of an open-label, single-centre, randomised controlled trial. Lancet 2015, 386, 964–973. [Google Scholar] [CrossRef]
- Cummings, D.E.; Arterburn, D.E.; Westbrook, E.O.; Kuzma, J.N.; Stewart, S.D.; Chan, C.P.; Bock, S.N.; Landers, J.T.; Kratz, M.; Foster-Schubert, K.E.; et al. Gastric bypass surgery vs intensive lifestyle and medical intervention for type 2 diabetes: The CROSSROADS randomised controlled trial. Diabetologia 2016, 59, 945–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halperin, F.; Ding, S.-A.; Simonson, D.C.; Panosian, J.; Goebel-Fabbri, A.; Wewalka, M.; Hamdy, O.; Abrahamson, M.; Clancy, K.; Foster, K.; et al. Roux-en-Y Gastric Bypass Surgery or Lifestyle With Intensive Medical Management in Patients With Type 2 Diabetes. JAMA Surg. 2014, 149, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Ikramuddin, S.; Korner, J.; Lee, W.-J.; Connett, J.E.; Inabnet, W.B.; Billington, C.J.; Thomas, A.J.; Leslie, D.B.; Chong, K.; Jeffery, R.W.; et al. Roux-en-Y Gastric Bypass vs Intensive Medical Management for the Control of Type 2 Diabetes, Hypertension, and Hyperlipidemia. JAMA 2013, 309, 2240–2249. [Google Scholar] [CrossRef]
- Courcoulas, A.P.; Goodpaster, B.H.; Eagleton, J.K.; Belle, S.H.; Kalarchian, M.; Lang, W.; Toledo, F.; Jakicic, J.M. Surgical vs Medical Treatments for Type 2 Diabetes Mellitus. JAMA Surg. 2014, 149, 707–715. [Google Scholar] [CrossRef]
- Courcoulas, A.P.; Belle, S.H.; Neiberg, R.H.; Pierson, S.; Eagleton, J.K.; Kalarchian, M.A.; Delany, J.P.; Lang, W.; Jakicic, J.M. Three-Year Outcomes of Bariatric Surgery vs Lifestyle Intervention for Type 2 Diabetes Mellitus Treatment. JAMA Surg. 2015, 150, 931–940. [Google Scholar] [CrossRef]
- McGarry, J.D. Banting Lecture 2001: Dysregulation of Fatty Acid Metabolism in the Etiology of Type 2 Diabetes. Diabetes 2002, 51, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G. Liver lipid molecules induce PEPCK-C gene transcription and attenuate insulin action. Biochem. Biophys. Res. Commun. 2007, 361, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, R.; Chen, W.; Li, Y.; Chen, G. Retinoids induced Pck1 expression and attenuated insulin-mediated suppression of its expression via activation of retinoic acid receptor in primary rat hepatocytes. Mol. Cell. Biochem. 2011, 355, 1–8. [Google Scholar] [CrossRef]
- Li, R.; Chen, W.; Li, Y.; Zhang, Y.; Chen, G. Retinoids synergized with insulin to induce Srebp-1c expression and activated its promoter via the two liver X receptor binding sites that mediate insulin action. Biochem. Biophys. Res. Commun. 2011, 406, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Goff, M.; Chen, G. Long-term treatment with insulin and retinoic acid increased glucose utilization in L6 muscle cells via glycogenesis. Biochem. Cell Biol. 2020, 98, 683–697. [Google Scholar] [CrossRef]
- Boyd, A.S. An overview of the retinoids. Am. J. Med. 1989, 86, 568–574. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Proteins | Site | Materials | Methods | Doman | Kinase | Citations |
|---|---|---|---|---|---|---|
| Human RARα | Ser 77 | In vitro labeling | Activity assay | AF1 | Cyclin-dependent kinase 7 | [107] |
| Human RARα | Ser 96 | FLAG-tagger RARα, and GST-RARα | Immunoprecipitation and in vitro kinase assay | DBD reduced activity | Akt | [112] |
| Human RARα | Ser 369 | Synthetic phosphopeptides (not specified sequence) | Mouse monoclonal | LBD | PKA, and MSK1 | [116,117] |
| Human RARα | Thr181, Ser445, and Ser461 | Recombinant GST-RARα | In vitro kinase assay | Multiple increases in degradation | JNK | [113] |
| Human RARγ1 | Ser 77 and Ser 79 | Whole cell labeling | Immunoprecipitated | AF1 modulates AF1 activity depending on constructs | CDK7 | [109] |
| Zebrafish RARα | Ser 72 (Human Ser 77) | EEMVPSSPS(p) PPPPPRVYKPC | Mouse monoclonal | N-terminal proline-rich domain | CDK 7 | [106] |
| Mouse RARγ | Ser 66 and Ser 68 | Synthetic phosphopeptides | Rabbit polyclonal | AF-1 Caused degradation | P38 MAPK | [109,110,111] |
| Mouse RARγ2 | Ser 379 (human 371) | Mutant constructs | Immunoprecipitation | LBD Stimulates activity | Akt | [115] |
| Proteins | Site | Materials | Methods | Domain | Upstream Kinase | References |
|---|---|---|---|---|---|---|
| Mouse RXRα | Ser 22 | Mutant constructs and cell lines; SSLNS(p) PTGRGS phosphopeptide | Western blots; anti-phosphor antibody | AF1 required for RA-induced activity | Proline-dependent kinase | [119,120,121] |
| Human RXRα | Ser 49 and Ser 78 | synthesized peptide | Phospho-RXRA (Ser260) ( AB_2663160) | AF1 decreases its inhibition to PI3KC promoter | GSK3β | [122] |
| Human RXRα | Ser 78 and Thr 82 | Anti-phosphor antibodies, GST-RXRα | Immunoprecipitated | AF1 decrease interaction with RARα | GSK3β | [123] |
| Mouse RXRα | Ser 61, Ser 75, Thr 87 in AF1, and Ser 265 | Mutant constructs | Immunoprecipitation and in vitro kinase assay | AF1 and LBD | JNKs | [124] |
| Human RXRα | Thr167 | CKGFFKR-pT- VRKDLTY, human RXRα knock-in mice (RxrαT167A) | Phosphor specific antibody | DBD effects depending on the promoter contexts | PKC | [125] |
| Human RXRα | Ser 260 | Anti-phosphor antibodies Mutant constructs | Immunoprecipitation Immunofluorescent | LBD decreases interaction with VDR or RARβ | Ras-Raf-MAP kinase | [126,127,128,129] |
| Human RXRα | Thr 82 and Ser 260 | Mutant constructs | Immunoprecipitation, Western blot | AF1 and LBD decreases interaction with RARβ | MAPK | [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, G. The Interactions of Insulin and Vitamin A Signaling Systems for the Regulation of Hepatic Glucose and Lipid Metabolism. Cells 2021, 10, 2160. https://doi.org/10.3390/cells10082160
Chen G. The Interactions of Insulin and Vitamin A Signaling Systems for the Regulation of Hepatic Glucose and Lipid Metabolism. Cells. 2021; 10(8):2160. https://doi.org/10.3390/cells10082160
Chicago/Turabian StyleChen, Guoxun. 2021. "The Interactions of Insulin and Vitamin A Signaling Systems for the Regulation of Hepatic Glucose and Lipid Metabolism" Cells 10, no. 8: 2160. https://doi.org/10.3390/cells10082160
APA StyleChen, G. (2021). The Interactions of Insulin and Vitamin A Signaling Systems for the Regulation of Hepatic Glucose and Lipid Metabolism. Cells, 10(8), 2160. https://doi.org/10.3390/cells10082160