
A Novel Monoclonal Antibody Targeting Cancer-Specific Plectin Has Potent Antitumor Activity in Ovarian Cancer

 , ,
, ,
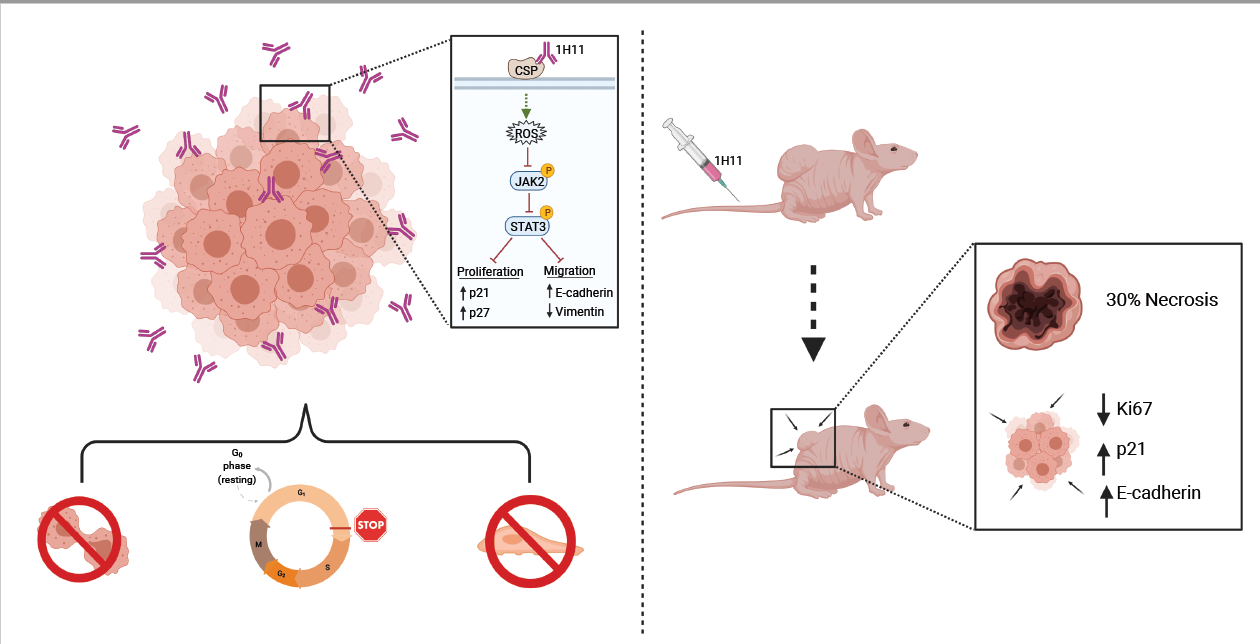
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patient Survival Analysis
2.2. Cell Culture and Reagents
2.3. Generation and Characterization of 1H11
2.4. Immunofluorescence Staining for Endosome Markers
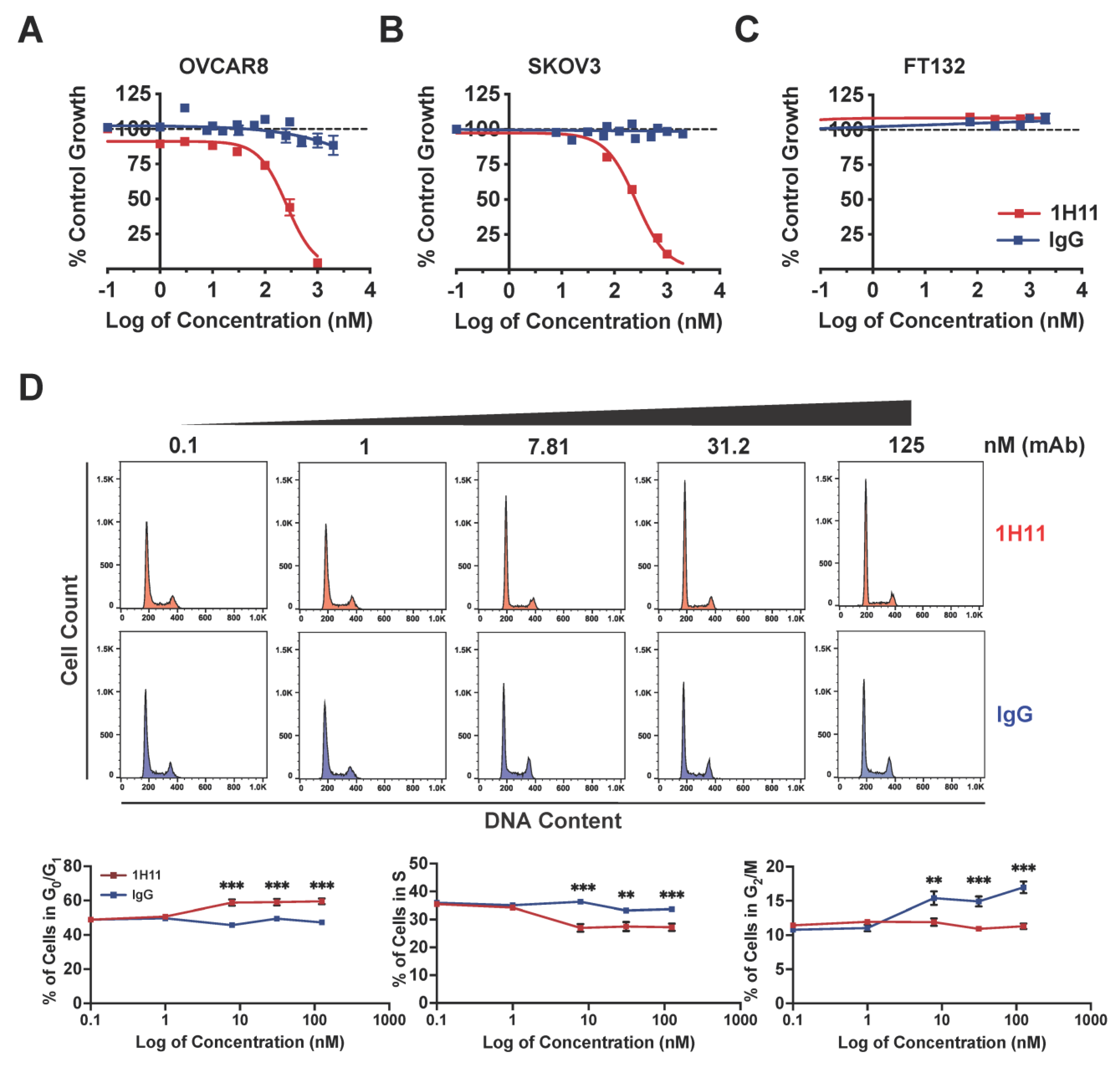
2.5. Cell Viability
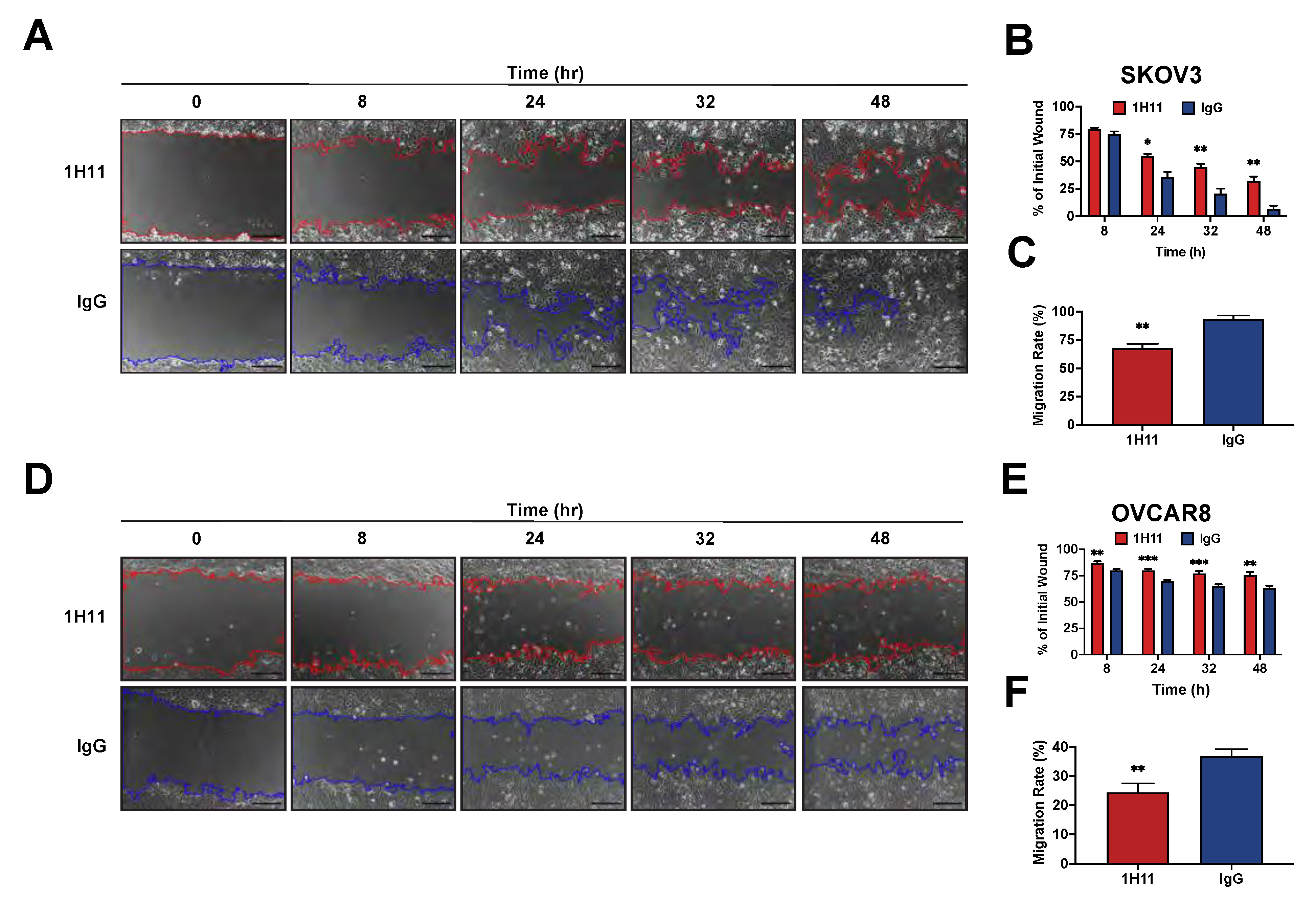
2.6. Cell Migration Assay
2.7. Cell Cycle Phase Distribution
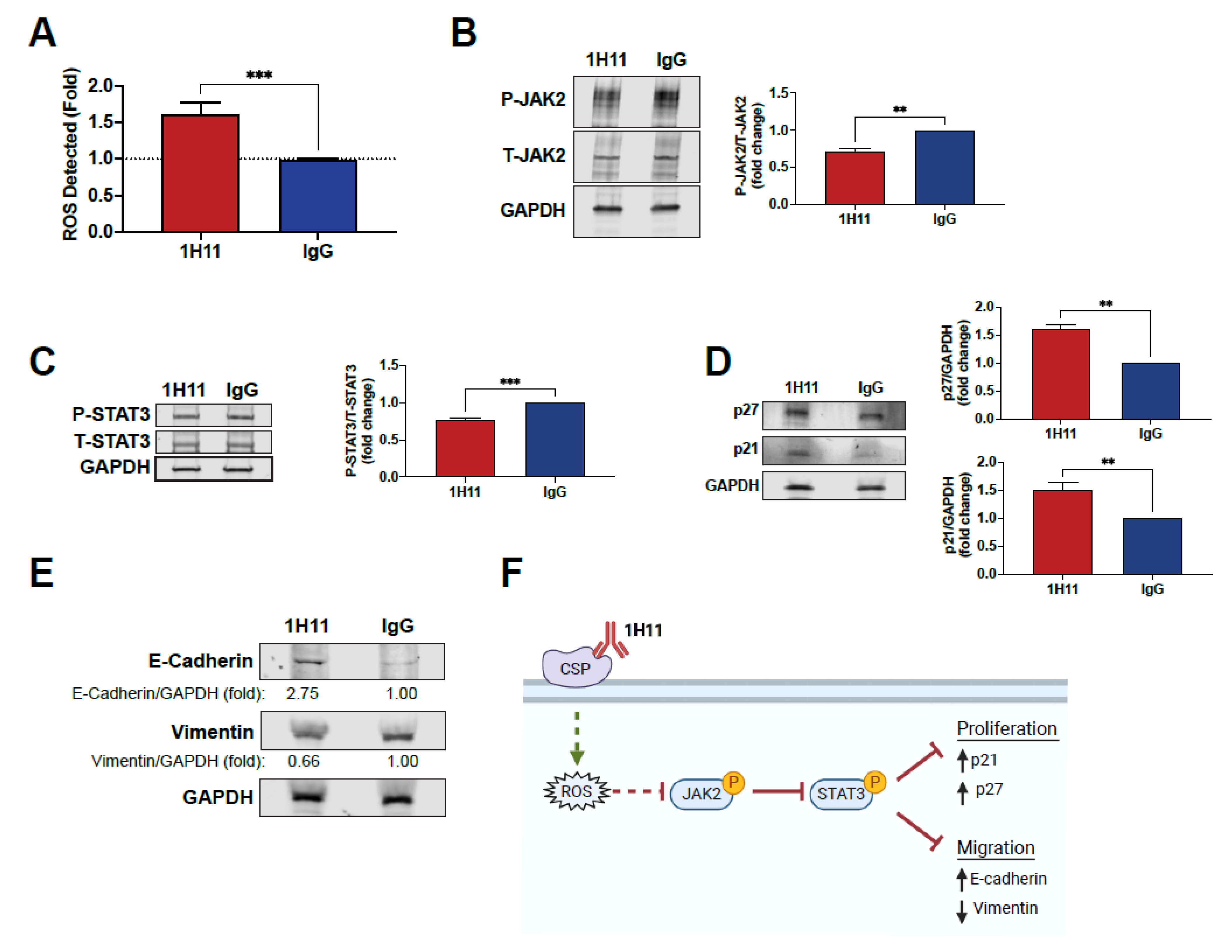
2.8. Reactive Oxygen Species (ROS) Detection
2.9. Western Blot Analysis
2.10. Therapeutic Efficacy of Anti-CSP mAb Treatment In Vivo
2.11. H&E and IHC Analysis
2.12. Statistical Analysis
3. Results
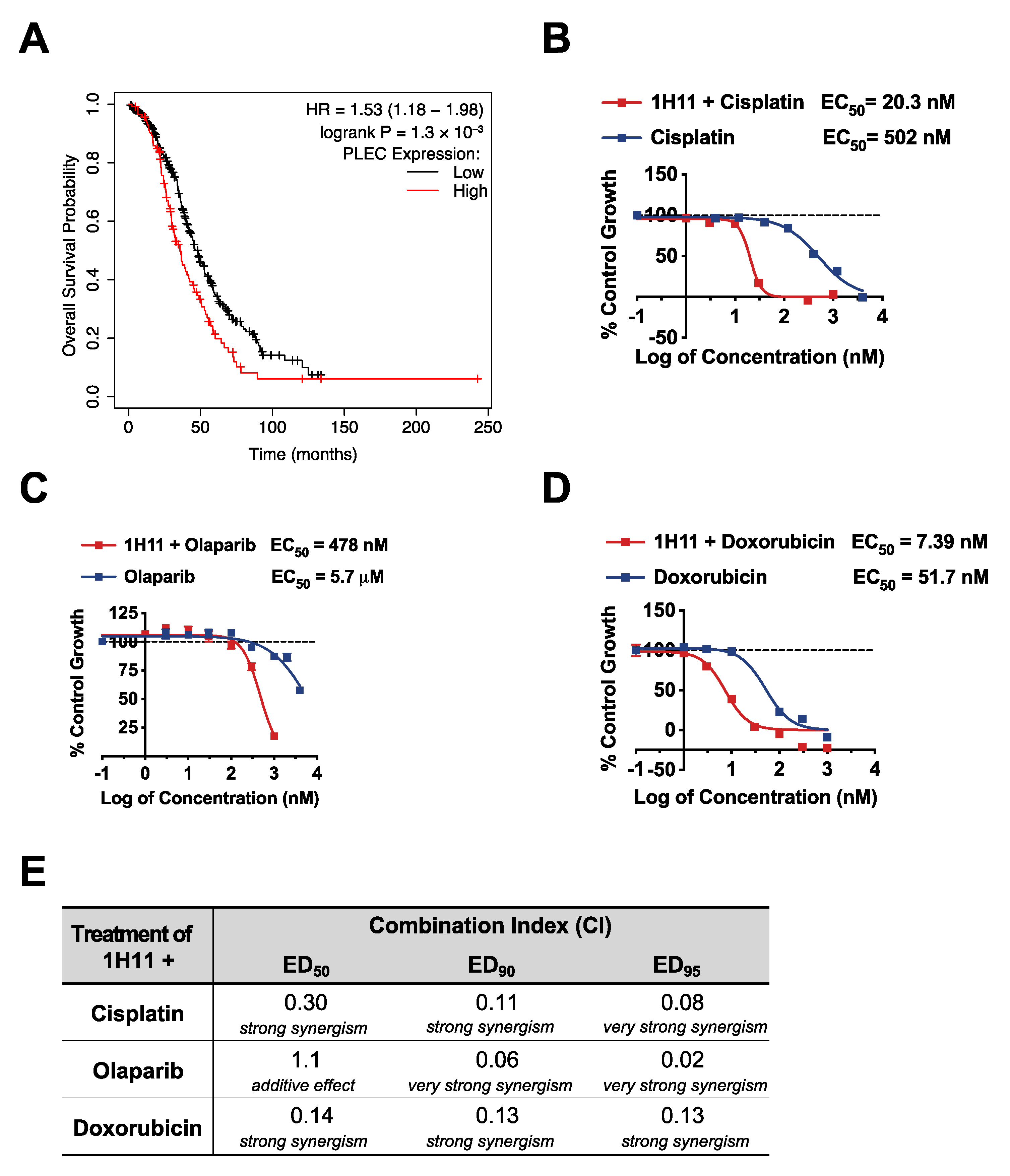
3.1. Plectin as a Prognostic Marker in OC
3.2. Generation and Characterization of 1H11
3.3. Targeting CSP Inhibits OC Cell Viability and Migration and Induces G0/G1 Arrest
3.4. H11 Induces ROS Accumulation and Suppression of JAK2-STAT3 Signaling
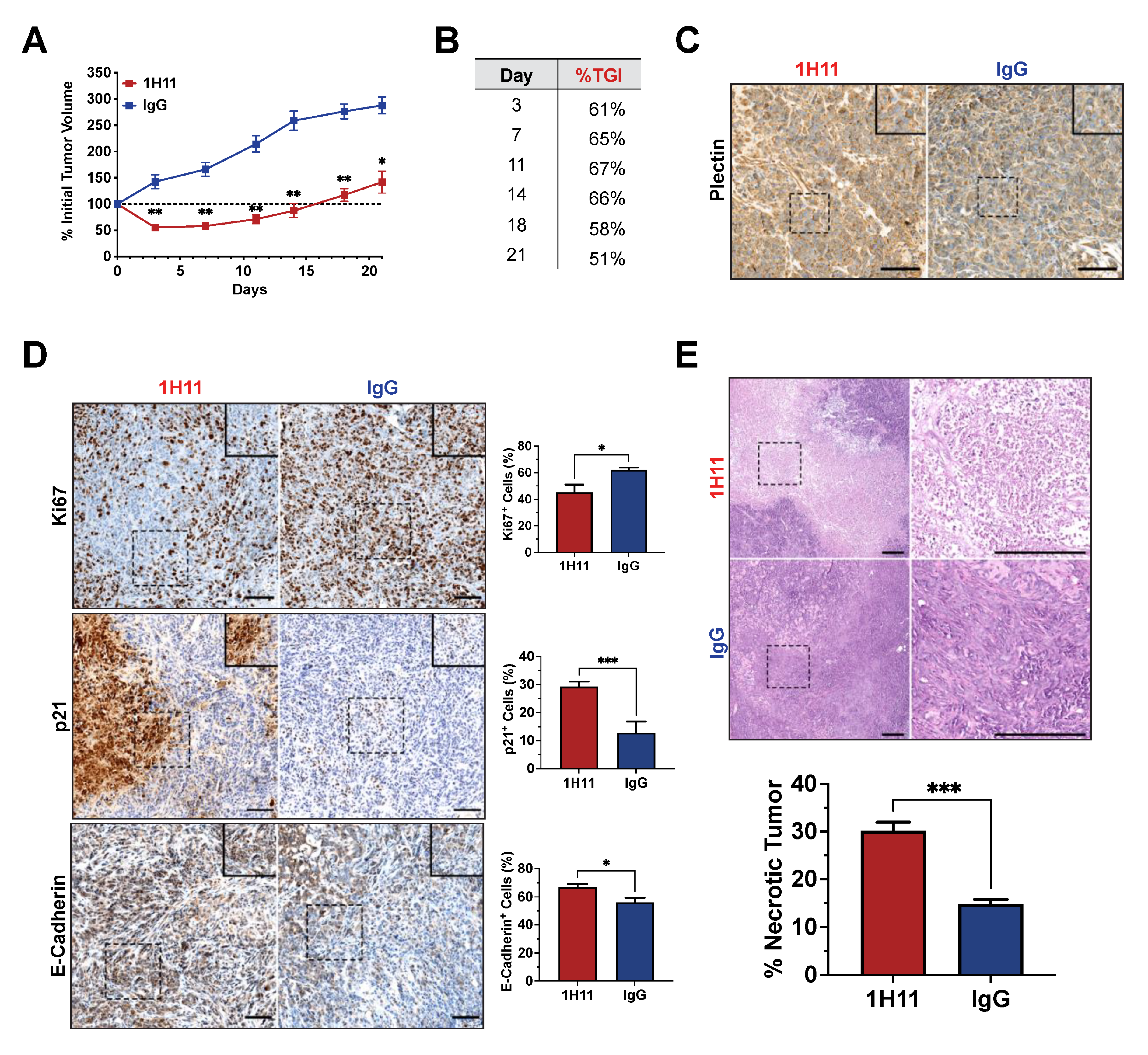
3.5. H11 Treatment Inhibits OC Tumor Growth In Vivo
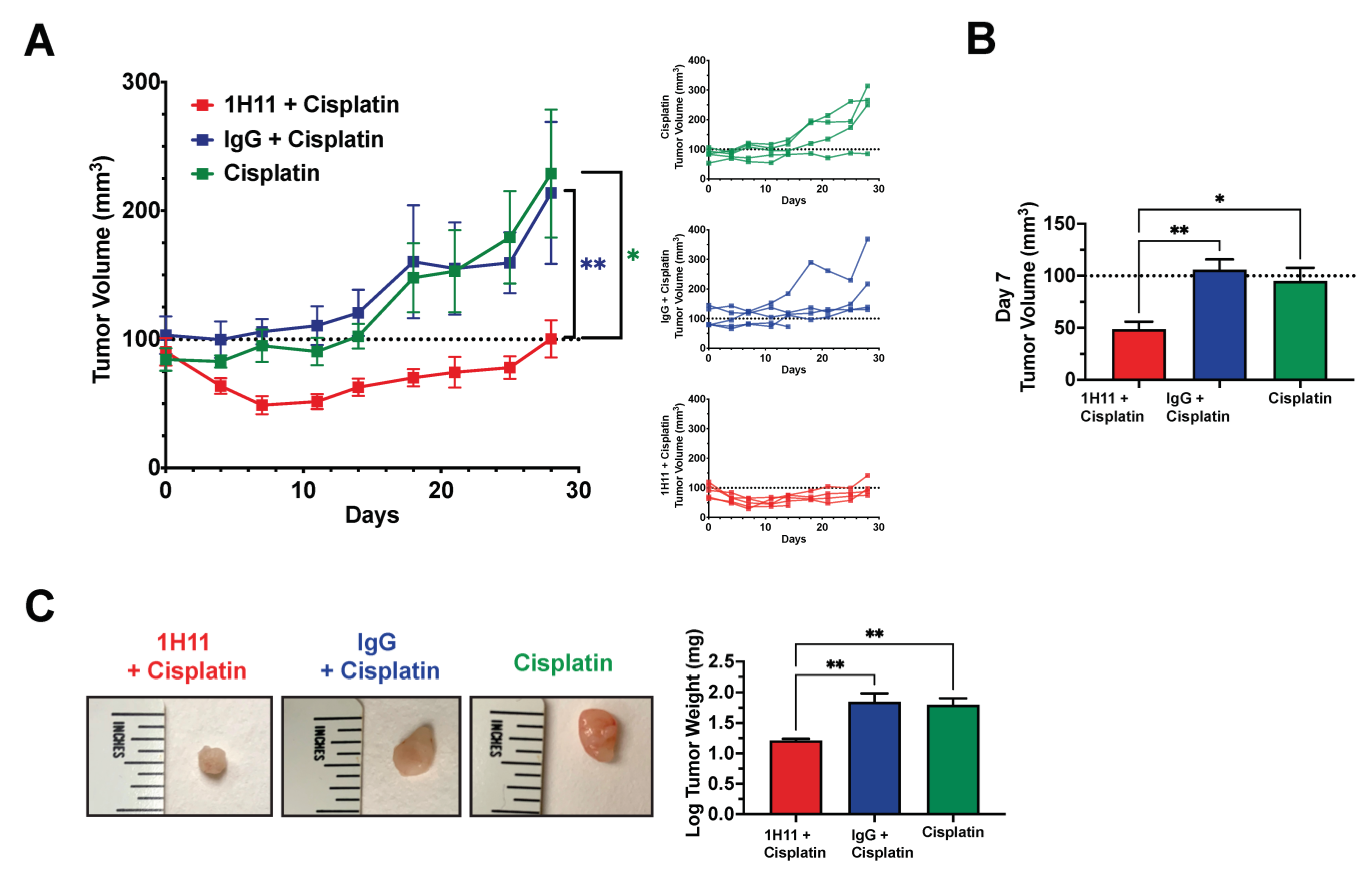
3.6. H11 Increases Chemosensitivity in OC In Vitro and In Vivo
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kelly, K.A.; Bardeesy, N.; Anbazhagan, R.; Gurumurthy, S.; Berger, J.; Alencar, H.; DePinho, R.A.; Mahmood, U.; Weissleder, R. Targeted Nanoparticles for Imaging Incipient Pancreatic Ductal Adenocarcinoma. PLoS Med. 2008, 5, e85. [Google Scholar] [CrossRef]
- Bausch, D.; Thomas, S.; Mino-Kenudson, M.; Fernández-del, C.C.; Bauer, T.W.; Williams, M.; Warshaw, A.L.; Thayer, S.P.; Kelly, K.A. Plectin-1 as a Novel Biomarker for Pancreatic Cancer. Clin. Cancer Res. 2011, 17, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Chan, K.C.; Waters, A.M.; Bess, M.; Harned, A.; Wei, B.-R.; Loncarek, J.; Luke, B.T.; Orsburn, B.C.; Hollinger, B.D.; et al. Comparative Proteomics of a Model MCF10A-KRasG12V Cell Line Reveals a Distinct Molecular Signature of the KRasG12V Cell Surface. Oncotarget 2016, 7, 86948–86971. [Google Scholar] [CrossRef] [Green Version]
- De Carvalho, V.P.; Grassi, M.L.; de Souza Palma, C.; Carrara, H.H.A.; Faça, V.M.; Dos Reis, F.J.C.; Poersch, A. The Contribution and Perspectives of Proteomics to Uncover Ovarian Cancer Tumor Markers. Transl. Res. 2019, 206, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.K.; Wilson, G.M.; Kirkemo, L.L.; Riley, N.M.; Coon, J.J.; Wells, J.A. Broad and Thematic Remodeling of the Surfaceome and Glycoproteome on Isogenic Cells Transformed with Driving Proliferative Oncogenes. Proc. Natl. Acad. Sci. USA 2020, 117, 7764–7775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinko, A.J.; Truillet, C.; Julien, O.; Diaz, J.E.; Horlbeck, M.A.; Whiteley, G.; Blonder, J.; Weissman, J.S.; Bandyopadhyay, S.; Evans, M.J.; et al. Targeting RAS-Driven Human Cancer Cells with Antibodies to Upregulated and Essential Cell-Surface Proteins. eLife 2018, 7, e31098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.J.; Smith, J.A.; Rezniczek, G.A.; Pan, S.; Chen, R.; Brentnall, T.A.; Wiche, G.; Kelly, K.A. Unexpected Gain of Function for the Scaffolding Protein Plectin Due to Mislocalization in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 19414–19419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, F.; Panneer, N.; Tutino, C.M.; Wu, M.; Skrabal, W.R.; Moskaluk, C.; Kelly, K.A. A Functional Proteomic Method for Biomarker Discovery. PLoS ONE 2011, 6, e22471. [Google Scholar] [CrossRef] [Green Version]
- Dasa, S.S.K.; Diakova, G.; Suzuki, R.; Mills, A.M.; Gutknecht, M.F.; Klibanov, A.L.; Slack-Davis, J.K.; Kelly, K.A. Plectin-Targeted Liposomes Enhance the Therapeutic Efficacy of a PARP Inhibitor in the Treatment of Ovarian Cancer. Theranostics 2018, 8, 2782–2798. [Google Scholar] [CrossRef]
- Raymond, A.C.; Gao, B.; Girard, L.; Minna, J.D.; Gomika Udugamasooriya, D. Unbiased Peptoid Combinatorial Cell Screen Identifies Plectin Protein as a Potential Biomarker for Lung Cancer Stem Cells. Sci. Rep. 2019, 9, 14954. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Bausch, D.; Mino-Kenudson, M.; Fernández-del Castillo, C.; Warshaw, A.L.; Kelly, K.A.; Thayer, S.P. Plectin-1 Is a Biomarker of Malignant Pancreatic Intraductal Papillary Mucinous Neoplasms. J. Gastrointest. Surg. 2009, 13, 1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moris, M.; Dawson, D.W.; Jiang, J.; Lewis, J.; Nassar, A.; Takeuchi, K.K.; Lay, A.R.; Zhai, Q.; Donahue, T.R.; Kelly, K.A.; et al. Plectin-1 as a Biomarker of Malignant Progression in Intraductal Papillary Mucinous Neoplasms: A Multicenter Study. Pancreas 2016, 45, 1353–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawar, H.; Kashyap, M.K.; Sahasrabuddhe, N.A.; Renuse, S.; Harsha, H.C.; Kumar, P.; Sharma, J.; Kandasamy, K.; Marimuthu, A.; Nair, B.; et al. Quantitative Tissue Proteomics of Esophageal Squamous Cell Carcinoma for Novel Biomarker Discovery. Cancer Biol. Ther. 2011, 12, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Kannarkat, G.J.; Harris, T.; Kelly, K.; Fracasso, P.; Moskaluk, C. Evaluation of Plectin-1 Immunohistochemical Expression in Human Colon Cancer Tumor Progression. J. Clin. Oncol. 2009, 27, e22132. [Google Scholar] [CrossRef]
- Konkalmatt, P.R.; Deng, D.; Thomas, S.; Wu, M.T.; Logsdon, C.D.; French, B.A.; Kelly, K.A. Plectin-1 Targeted AAV Vector for the Molecular Imaging of Pancreatic Cancer. Front. Oncol. 2013, 3, 84. [Google Scholar] [CrossRef] [Green Version]
- Pal, K.; Al-suraih, F.; Gonzalez-Rodriguez, R.; Kumar Dutta, S.; Wang, E.; Shaun Kwak, H.; Caulfield, T.R.; Coffer, J.L.; Bhattacharya, S. Multifaceted Peptide Assisted One-Pot Synthesis of Gold Nanoparticles for Plectin-1 Targeted Gemcitabine Delivery in Pancreatic Cancer. Nanoscale 2017, 9, 15622–15634. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, Y.; Zhi, X.; Ma, T.; Liu, H.; Chen, B.W.; Zheng, X.; Xie, S.; Zhao, B.; Feng, X.; et al. Delivery of MiR-212 by Chimeric Peptide-Condensed Supramolecular Nanoparticles Enhances the Sensitivity of Pancreatic Ductal Adenocarcinoma to Doxorubicin. Biomaterials 2019, 192, 590–600. [Google Scholar] [CrossRef]
- Katada, K.; Tomonaga, T.; Satoh, M.; Matsushita, K.; Tonoike, Y.; Kodera, Y.; Hanazawa, T.; Nomura, F.; Okamoto, Y. Plectin Promotes Migration and Invasion of Cancer Cells and Is a Novel Prognostic Marker for Head and Neck Squamous Cell Carcinoma. J. Proteom. 2012, 75, 1803–1815. [Google Scholar] [CrossRef]
- Buckup, M.; Rice, M.A.; Hsu, E.-C.; Garcia-Marques, F.; Liu, S.; Aslan, M.; Bermudez, A.; Huang, J.; Pitteri, S.J.; Stoyanova, T. Plectin Is a Regulator of Prostate Cancer Growth and Metastasis. Oncogene 2021, 40, 663–676. [Google Scholar] [CrossRef]
- McInroy, L.; Määttä, A. Plectin Regulates Invasiveness of SW480 Colon Carcinoma Cells and Is Targeted to Podosome-like Adhesions in an Isoform-Specific Manner. Exp. Cell Res. 2011, 317, 2468–2478. [Google Scholar] [CrossRef] [PubMed]
- Sutoh Yoneyama, M.; Hatakeyama, S.; Habuchi, T.; Inoue, T.; Nakamura, T.; Funyu, T.; Wiche, G.; Ohyama, C.; Tsuboi, S. Vimentin Intermediate Filament and Plectin Provide a Scaffold for Invadopodia, Facilitating Cancer Cell Invasion and Extravasation for Metastasis. Eur. J. Cell Biol. 2014, 93, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Burch, T.C.; Watson, M.T.; Nyalwidhe, J.O. Variable Metastatic Potentials Correlate with Differential Plectin and Vimentin Expression in Syngeneic Androgen Independent Prostate Cancer Cells. PLoS ONE 2013, 8, e65005. [Google Scholar] [CrossRef]
- Musrap, N.; Karagiannis, G.S.; Saraon, P.; Batruch, I.; Smith, C.; Diamandis, E.P. Proteomic Analysis of Cancer and Mesothelial Cells Reveals an Increase in Mucin 5AC during Ovarian Cancer and Peritoneal Interaction. J. Proteom. 2014, 103, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Wesley, T.; Berzins, S.; Kannourakis, G.; Ahmed, N. The Attributes of Plakins in Cancer and Disease: Perspectives on Ovarian Cancer Progression, Chemoresistance and Recurrence. Cell Commun. Signal. 2021, 19, 55. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA. Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Heintz, A.; Odicino, F.; Maisonneuve, P.; Quinn, M.; Benedet, J.; Creasman, W.; Ngan, H.; Pecorelli, S.; Beller, U. Carcinoma of the Ovary. Int. J. Gynecol. Obstet. 2006, 95, S161–S192. [Google Scholar] [CrossRef]
- Romero, I.; Bast, R.C. Minireview: Human Ovarian Cancer: Biology, Current Management, and Paths to Personalizing Therapy. Endocrinology 2012, 153, 1593–1602. [Google Scholar] [CrossRef] [Green Version]
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in Ovarian Cancer Therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Győrffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online Survival Analysis Software to Assess the Prognostic Value of Biomarkers Using Transcriptomic Data in Non-Small-Cell Lung Cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [Green Version]
- Győrffy, B.; Lánczky, A.; Szállási, Z. Implementing an Online Tool for Genome-Wide Validation of Survival-Associated Biomarkers in Ovarian-Cancer Using Microarray Data from 1287 Patients. Endocr. Relat. Cancer 2012, 19, 197–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vichai, V.; Kirtikara, K. Sulforhodamine B Colorimetric Assay for Cytotoxicity Screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Dai, W.; Liu, J.; Li, Y.-X.; Li, Y.-Y. DRAP: A Toolbox for Drug Response Analysis and Visualization Tailored for Preclinical Drug Testing on Patient-Derived Xenograft Models. J. Transl. Med. 2019, 17, 39. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallas-Potts, A.; Dawson, J.C.; Herrington, C.S. Ovarian Cancer Cell Lines Derived from Non-Serous Carcinomas Migrate and Invade More Aggressively than Those Derived from High-Grade Serous Carcinomas. Sci. Rep. 2019, 9, 5515. [Google Scholar] [CrossRef]
- Wernitznig, D.; Meier-Menches, S.M.; Cseh, K.; Theiner, S.; Wenisch, D.; Schweikert, A.; Jakupec, M.A.; Koellensperger, G.; Wernitznig, A.; Sommergruber, W.; et al. Plecstatin-1 Induces an Immunogenic Cell Death Signature in Colorectal Tumour Spheroids. Met. Integr. Biometal Sci. 2020, 12, 2121–2133. [Google Scholar] [CrossRef]
- Meier, S.M.; Kreutz, D.; Winter, L.; Klose, M.H.M.; Cseh, K.; Weiss, T.; Bileck, A.; Alte, B.; Mader, J.C.; Jana, S.; et al. An Organoruthenium Anticancer Agent Shows Unexpected Target Selectivity For Plectin. Angew. Chem. Int. Ed. 2017, 56, 8267–8271. [Google Scholar] [CrossRef]
- Meier-Menches, S.M.; Zappe, K.; Bileck, A.; Kreutz, D.; Tahir, A.; Cichna-Markl, M.; Gerner, C. Time-Dependent Shotgun Proteomics Revealed Distinct Effects of an Organoruthenium Prodrug and Its Activation Product on Colon Carcinoma Cells. Met. Integr. Biometal Sci. 2019, 11, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.-M.; Park, S.; Kim, J.-H. Leukotriene B4 Receptor-2 Promotes Invasiveness and Metastasis of Ovarian Cancer Cells through Signal Transducer and Activator of Transcription 3 (STAT3)-Dependent Up-Regulation of Matrix Metalloproteinase 2*. J. Biol. Chem. 2012, 287, 13840–13849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, V.; Camporeale, A. STAT3-Mediated Metabolic Reprograming in Cellular Transformation and Implications for Drug Resistance. Front. Oncol. 2015, 5, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Le, F.; Cheng, J.; Luo, C.; Zhang, X.; Wu, X.; Xu, F.; Zuo, Q.; Tan, B. Triptolide Inhibits JAK2/STAT3 Signaling and Induces Lethal Autophagy through ROS Generation in Cisplatin-resistant SKOV3/DDP Ovarian Cancer Cells. Oncol. Rep. 2021, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Liu, M.; Liu, Y.; Zhang, M.; Yang, G. Tetramethoxychalcone, a Chalcone Derivative, Suppresses Proliferation, Blocks Cell Cycle Progression, and Induces Apoptosis of Human Ovarian Cancer Cells. PLoS ONE 2014, 9, e106206. [Google Scholar] [CrossRef] [Green Version]
- Colomiere, M.; Ward, A.C.; Riley, C.; Trenerry, M.K.; Cameron-Smith, D.; Findlay, J.; Ackland, L.; Ahmed, N. Cross Talk of Signals between EGFR and IL-6R through JAK2/STAT3 Mediate Epithelial–Mesenchymal Transition in Ovarian Carcinomas. Br. J. Cancer 2009, 100, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Zhang, X.; Paladino, D.; Sengupta, B.; Ahmad, S.; Holloway, R.W.; Ingersoll, S.B.; Turkson, J. Hyperactive EGF Receptor, Jaks and Stat3 Signaling Promote Enhanced Colony-Forming Ability, Motility and Migration of Cisplatin-Resistant Ovarian Cancer Cells. Oncogene 2012, 31, 2309–2322. [Google Scholar] [CrossRef] [Green Version]
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial–Mesenchymal Transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, P.J.; Lazar, G.A. Next Generation Antibody Drugs: Pursuit of the “High-Hanging Fruit”. Nat. Rev. Drug Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef]
- Wu, C.-J.; Sundararajan, V.; Sheu, B.-C.; Huang, R.Y.-J.; Wei, L.-H. Activation of STAT3 and STAT5 Signaling in Epithelial Ovarian Cancer Progression: Mechanism and Therapeutic Opportunity. Cancers 2020, 12, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, R.; Chen, X.; Chen, L.; Wan, F.; Chen, K.; Sun, Y.; Zhu, X. STAT3 Signaling in Ovarian Cancer: A Potential Therapeutic Target. J. Cancer 2020, 11, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abubaker, K.; Luwor, R.B.; Zhu, H.; McNally, O.; Quinn, M.A.; Burns, C.J.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Inhibition of the JAK2/STAT3 Pathway in Ovarian Cancer Results in the Loss of Cancer Stem Cell-like Characteristics and a Reduced Tumor Burden. BMC Cancer 2014, 14, 317. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T.; Miyamoto, M.; Aoyama, T.; Soyama, H.; Goto, T.; Hirata, J.; Suzuki, A.; Nagaoka, I.; Tsuda, H.; Furuya, K.; et al. JAK2/STAT3 Pathway as a Therapeutic Target in Ovarian Cancers. Oncol. Lett. 2018, 15, 5772–5780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-T.; Ho, H.J.; Lin, J.-T.; Shieh, J.-J.; Wu, C.-Y. Simvastatin-Induced Cell Cycle Arrest through Inhibition of STAT3/SKP2 Axis and Activation of AMPK to Promote P27 and P21 Accumulation in Hepatocellular Carcinoma Cells. Cell Death Dis. 2017, 8, e2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marth, C.; Egle, D.; Auer, D.; Rössler, J.; Zeimet, A.G.; Vergote, I.; Daxenbichler, G. Modulation of CA-125 Tumor Marker Shedding in Ovarian Cancer Cells by Erlotinib or Cetuximab. Gynecol. Oncol. 2007, 105, 716–721. [Google Scholar] [CrossRef]
- Damiani, I.; Castiglioni, S.; Sochaj-Gregorczyk, A.; Bonacina, F.; Colombo, I.; Rusconi, V.; Otlewski, J.; Corsini, A.; Bellosta, S. Purification and In Vitro Evaluation of an Anti-HER2 Affibody-Monomethyl Auristatin E Conjugate in HER2-Positive Cancer Cells. Biology 2021, 10, 758. [Google Scholar] [CrossRef]
- McDermott, U.; Sharma, S.V.; Dowell, L.; Greninger, P.; Montagut, C.; Lamb, J.; Archibald, H.; Raudales, R.; Tam, A.; Lee, D.; et al. Identification of Genotype-Correlated Sensitivity to Selective Kinase Inhibitors by Using High-Throughput Tumor Cell Line Profiling. Proc. Natl. Acad. Sci. USA 2007, 104, 19936–19941. [Google Scholar] [CrossRef] [Green Version]
- Iijima, Y.; Bandow, K.; Sano, M.; Hino, S.; Kaneko, T.; Horie, N.; Sakagami, H. In Vitro Assessment of Antitumor Potential and Combination Effect of Classical and Molecular-Targeted Anticancer Drugs. Anticancer Res. 2019, 39, 6673–6684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, N.; Sessa, C.; du Bois, A.; Ledermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO–ESGO Consensus Conference Recommendations on Ovarian Cancer: Pathology and Molecular Biology, Early and Advanced Stages, Borderline Tumours and Recurrent Disease. Ann. Oncol. 2019, 30, 672–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crusz, S.M.; Miller, R.E. Targeted Therapies in Gynaecological Cancers. Histopathology 2020, 76, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Nero, C.; Ciccarone, F.; Pietragalla, A.; Duranti, S.; Daniele, G.; Salutari, V.; Carbone, M.V.; Scambia, G.; Lorusso, D. Ovarian Cancer Treatments Strategy: Focus on PARP Inhibitors and Immune Check Point Inhibitors. Cancers 2021, 13, 1298. [Google Scholar] [CrossRef] [PubMed]
- Randall, L.M.; Monk, B.J. Bevacizumab Toxicities and Their Management in Ovarian Cancer. Gynecol. Oncol. 2010, 117, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous Recombination Deficiency and Ovarian Cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Nie, J.; Huang, J.-P.; Zheng, G.-J.; Feng, B. Targeting STAT3 Inhibition to Reverse Cisplatin Resistance. Biomed. Pharmacother. 2019, 117, 109135. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lu, Y.; Pang, Y.; Li, M.; Cheng, X.; Chen, J. Propofol Enhances the Cisplatin-Induced Apoptosis on Cervical Cancer Cells via EGFR/JAK2/STAT3 Pathway. Biomed. Pharmacother. 2017, 86, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Capone, E.; Lattanzio, R.; Gasparri, F.; Orsini, P.; Rossi, C.; Iacobelli, V.; De Laurenzi, V.; Natali, P.G.; Valsasina, B.; Iacobelli, S.; et al. EV20/NMS-P945, a Novel Thienoindole Based Antibody-Drug Conjugate Targeting HER-3 for Solid Tumors. Pharmaceutics 2021, 13, 483. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, S.M.; Dimastromatteo, J.; Landen, C.N., Jr.; Kelly, K.A. A Novel Monoclonal Antibody Targeting Cancer-Specific Plectin Has Potent Antitumor Activity in Ovarian Cancer. Cells 2021, 10, 2218. https://doi.org/10.3390/cells10092218
Perez SM, Dimastromatteo J, Landen CN Jr., Kelly KA. A Novel Monoclonal Antibody Targeting Cancer-Specific Plectin Has Potent Antitumor Activity in Ovarian Cancer. Cells. 2021; 10(9):2218. https://doi.org/10.3390/cells10092218
Chicago/Turabian StylePerez, Samantha M., Julien Dimastromatteo, Charles N. Landen, Jr., and Kimberly A. Kelly. 2021. "A Novel Monoclonal Antibody Targeting Cancer-Specific Plectin Has Potent Antitumor Activity in Ovarian Cancer" Cells 10, no. 9: 2218. https://doi.org/10.3390/cells10092218
APA StylePerez, S. M., Dimastromatteo, J., Landen, C. N., Jr., & Kelly, K. A. (2021). A Novel Monoclonal Antibody Targeting Cancer-Specific Plectin Has Potent Antitumor Activity in Ovarian Cancer. Cells, 10(9), 2218. https://doi.org/10.3390/cells10092218