
Induced Pluripotent Stem Cells to Model Juvenile Myelomonocytic Leukemia: New Perspectives for Preclinical Research


Abstract
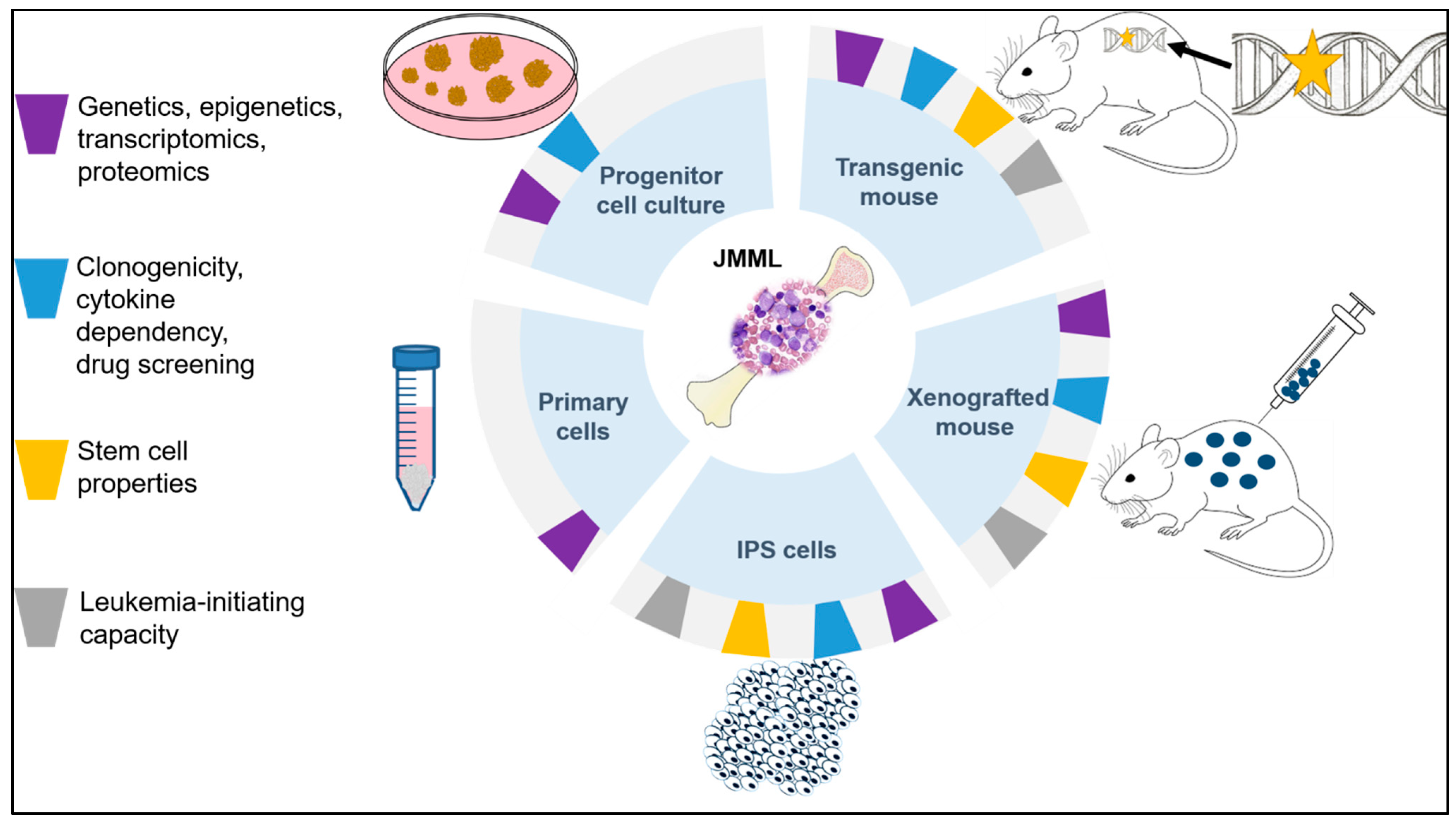
:1. Introduction
2. Previous Strategies of Modeling JMML
2.1. In Vitro Approaches
2.2. In Vivo Approaches
3. IPSCs as an Emerging Approach to Model Human Disease
3.1. Methods Used for Reprogramming
3.2. Types of Reprogrammed Cells
4. Differentiation of IPSCs to Hematopoietic Cells
5. Role of Preexistent Genetics and Epigenetics on Leukemia Formation
6. Applications of JMML-Derived IPSCs
7. Outlook: The Future of IPSCs in JMML Research
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Niemeyer, C.M.; Flotho, C. Juvenile myelomonocytic leukemia: Who’s the driver at the wheel? Blood 2019, 133, 1060–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krombholz, C.F.; Aumann, K.; Kollek, M.; Bertele, D.; Fluhr, S.; Kunze, M.; Niemeyer, C.M.; Flotho, C.; Erlacher, M. Long-term serial xenotransplantation of juvenile myelomonocytic leukemia recapitulates human disease in Rag2−/−γc−/− mice. Haematologica 2016, 101, 597–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieglitz, E.; Mazor, T.; Olshen, A.B.; Geng, H.; Gelston, L.C.; Akutagawa, J.; Lipka, D.B.; Plass, C.; Flotho, C.; Chehab, F.F.; et al. Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat. Commun. 2017, 8, 2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, N.; Okuno, Y.; Yoshida, K.; Shiraishi, Y.; Nagae, G.; Suzuki, K.; Narita, A.; Sakaguchi, H.; Kawashima, N.; Wang, X.; et al. Integrated molecular profiling of juvenile myelomonocytic leukemia. Blood 2018, 131, 1576–1586. [Google Scholar] [CrossRef] [PubMed]
- Lipka, D.B.; Witte, T.; Toth, R.; Yang, J.; Wiesenfarth, M.; Nöllke, P.; Fischer, A.; Brocks, D.; Gu, Z.; Park, J.; et al. RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia. Nat. Commun. 2017, 8, 2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locatelli, F.; Niemeyer, C.M. How I treat juvenile myelomonocytic leukemia. Blood 2015, 125, 1083–1090. [Google Scholar] [CrossRef]
- Chang, T.Y.; Dvorak, C.C.; Loh, M.L. Bedside to bench in juvenile myelomonocytic leukemia: Insights into leukemogenesis from a rare pediatric leukemia. Blood 2014, 124, 2487–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stieglitz, E.; Taylor-Weiner, A.N.; Chang, T.Y.; Gelston, L.C.; Wang, Y.-D.; Mazor, T.; Esquivel, E.; Yu, A.; Seepo, S.; Olsen, S.; et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat. Genet. 2015, 47, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.M. JMML genomics and decisions. Hematol. Am. Soc. Hematol. Educ. Program 2018, 2018, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Caye, A.; Strullu, M.; Guidez, F.; Cassinat, B.; Gazal, S.; Fenneteau, O.; Lainey, E.; Nouri, K.; Nakhaei-Rad, S.; Dvorsky, R.; et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat. Genet. 2015, 47, 1334–1340. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Okuno, Y.; Muramatsu, H.; Yoshida, K.; Shiraishi, Y.; Takahashi, M.; Kon, A.; Sanada, M.; Chiba, K.; Tanaka, H.; et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat. Genet. 2013, 45, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Furlan, I.; Batz, C.; Flotho, C.; Mohr, B.; Lübbert, M.; Suttorp, M.; Niemeyer, C.M. Intriguing response to azacitidine in a patient with juvenile myelomonocytic leukemia and monosomy 7. Blood 2009, 113, 2867–2868. [Google Scholar] [CrossRef]
- Krombholz, C.F.; Gallego-Villar, L.; Sahoo, S.S.; Panda, P.K.; Wlodarski, M.W.; Aumann, K.; Hartmann, M.; Lipka, D.B.; Daskalakis, M.; Plass, C.; et al. Azacitidine is effective for targeting leukemia-initiating cells in juvenile myelomonocytic leukemia. Leukemia 2019, 33, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Cseh, A.; Niemeyer, C.M.; Yoshimi, A.; Dworzak, M.; Hasle, H.; van den Heuvel-Eibrink, M.M.; Locatelli, F.; Masetti, R.; Schmugge, M.; Groß-Wieltsch, U.; et al. Bridging to transplant with azacitidine in juvenile myelomonocytic leukemia: A retrospective analysis of the EWOG-MDS study group. Blood 2015, 125, 2311–2313. [Google Scholar] [CrossRef] [Green Version]
- Orphanet. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=86834 (accessed on 30 July 2021).
- Chan, R.J.; Cooper, T.; Kratz, C.P.; Weiss, B.; Loh, M.L. Juvenile myelomonocytic leukemia: A report from the 2nd International JMML Symposium. Leuk. Res. 2009, 33, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakashita, K.; Kato, I.; Daifu, T.; Saida, S.; Hiramatsu, H.; Nishinaka, Y.; Ebihara, Y.; Ma, F.; Matsuda, K.; Saito, S.; et al. In vitro expansion of CD34(+)CD38(−) cells under stimulation with hematopoietic growth factors on AGM-S3 cells in juvenile myelomonocytic leukemia. Leukemia 2015, 29, 606–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandre-Babbe, S.; Paluru, P.; Aribeana, C.; Chou, S.T.; Bresolin, S.; Lu, L.; Sullivan, S.K.; Tasian, S.K.; Weng, J.; Favre, H.; et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood 2013, 121, 4925–4929. [Google Scholar] [CrossRef]
- Mulero-Navarro, S.; Sevilla, A.; Roman, A.C.; Lee, D.-F.; D’Souza, S.L.; Pardo, S.; Riess, I.; Su, J.; Cohen, N.; Schaniel, C.; et al. Myeloid dysregulation in a human induced pluripotent stem cell model of PTPN11-associated juvenile myelomonocytic leukemia. Cell Rep. 2015, 13, 504–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasian, S.K.; Casas, J.A.; Posocco, D.; Gandre-Babbe, S.; Gagne, A.L.; Liang, G.; Loh, M.L.; Weiss, M.J.; French, D.L.; Chou, S.T. Mutation-specific signaling profiles and kinase inhibitor sensitivities of juvenile myelomonocytic leukemia revealed by induced pluripotent stem cells. Leukemia 2019, 33, 181–190. [Google Scholar] [CrossRef]
- Shigemura, T.; Matsuda, K.; Kurata, T.; Sakashita, K.; Okuno, Y.; Muramatsu, H.; Yue, F.; Ebihara, Y.; Tsuji, K.; Sasaki, K.; et al. Essential role of PTPN11 mutation in enhanced haematopoietic differentiation potential of induced pluripotent stem cells of juvenile myelomonocytic leukaemia. Br. J. Haematol. 2019, 187, 163–173. [Google Scholar] [CrossRef]
- Pearson, S.; Guo, B.; Pierce, A.; Azadbakht, N.; Brazzatti, J.A.; Patassini, S.; Mulero-Navarro, S.; Meyer, S.; Flotho, C.; Gelb, B.D.; et al. Proteomic analysis of an induced pluripotent stem cell model reveals strategies to treat juvenile myelomonocytic leukemia. J. Proteome Res. 2020, 19, 194–203. [Google Scholar] [CrossRef]
- Altman, A.J.; Palmer, C.G.; Baehner, R.L. Juvenile “chronic granulocytic” leukemia: A panmyelopathy with prominent monocytic involvement and circulating monocyte colony-forming cells. Blood 1974, 43, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Levin, S.; Vogel, R.; Cohen, I.J.; Wallach, B.; Nir, E.; Zaizov, R. Juvenile and adult types of chronic granulocytic leukemia of childhood: Growth patterns and characteristics of granulocyte-macrophage colony forming cells. Am. J. Hematol. 1981, 10, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Miura, Y.; Mizoguchi, H.; Ijima, H.; Eguchi, M.; Kaku, H.; Ide, C. Characterization of hemopoietic precursor cells in juvenile-type chronic myelocytic leukemia. Leuk. Res. 1982, 6, 43–53. [Google Scholar] [CrossRef]
- Estrov, Z.; Grunberger, T.; Chan, H.S.; Freedman, M.H. Juvenile chronic myelogenous leukemia: Characterization of the disease using cell cultures. Blood 1986, 67, 1382–1387. [Google Scholar] [CrossRef] [Green Version]
- Gualtieri, R.J.; Castleberry, R.P.; Gibbons, J.; Miller, D.M.; Berkow, R.L.; Parmley, R.T.; Banks, J. Cell culture studies and oncogene expression in juvenile chronic myelogenous leukemia. Exp. Hematol. 1988, 16, 613–619. [Google Scholar] [PubMed]
- Emanuel, P.D.; Bates, L.J.; Castleberry, R.P.; Gualtieri, R.J.; Zuckerman, K.S. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood 1991, 77, 925–929. [Google Scholar] [CrossRef] [Green Version]
- Gualtieri, R.J.; Emanuel, P.D.; Zuckerman, K.S.; Martin, G.; Clark, S.C.; Shadduck, R.K.; Dracker, R.A.; Akabutu, J.; Nitschke, R.; Hetherington, M.L. Granulocyte-macrophage colony-stimulating factor is an endogenous regulator of cell proliferation in juvenile chronic myelogenous leukemia. Blood 1989, 74, 2360–2367. [Google Scholar] [CrossRef] [Green Version]
- Freedman, M.H.; Cohen, A.; Grunberger, T.; Bunin, N.; Luddy, R.E.; Saunders, E.F.; Shahidi, N.; Lau, A.; Estrov, Z. Central role of tumour necrosis factor, GM-CSF, and interleukin 1 in the pathogenesis of juvenile chronic myelogenous leukaemia. Br. J. Haematol. 1992, 80, 40–48. [Google Scholar] [CrossRef]
- Bagby, G.C.; Dinarello, C.A.; Neerhout, R.C.; Ridgway, D.; McCall, E. Interleukin 1-dependent paracrine granulopoiesis in chronic granulocytic leukemia of the juvenile type. J. Clin. Invest. 1988, 82, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Castleberry, R.P.; Emanuel, P.D.; Zuckerman, K.S.; Cohn, S.; Strauss, L.; Byrd, R.L.; Homans, A.; Chaffee, S.; Nitschke, R.; Gualtieri, R.J. A pilot study of isotretinoin in the treatment of juvenile chronic myelogenous leukemia. N. Engl. J. Med. 1994, 331, 1680–1684. [Google Scholar] [CrossRef]
- Cambier, N.; Menot, M.L.; Schlageter, M.H.; Balitrand, N.; Leblanc, T.; Bordigoni, P.; Rohrlich, P.; Lamagnère, J.P.; Donadieu, J.; Herbelin, C.; et al. All trans retinoic acid abrogates spontaneous monocytic growth in juvenile chronic myelomonocytic leukaemia. Hematol. J. 2001, 2, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Estrov, Z.; Lau, A.S.; Williams, B.R.; Freedman, M.H. Recombinant human interferon alpha-2 and juvenile chronic myelogenous leukemia: Cell receptor binding, enzymatic induction, and growth suppression in vitro. Exp. Hematol. 1987, 15, 127–132. [Google Scholar] [PubMed]
- Emanuel, P.D.; Snyder, R.C.; Wiley, T.; Gopurala, B.; Castleberry, R.P. Inhibition of juvenile myelomonocytic leukemia cell growth in vitro by farnesyltransferase inhibitors. Blood 2000, 95, 639–645. [Google Scholar] [CrossRef]
- Iversen, P.O.; Hart, P.H.; Bonder, C.S.; Lopez, A.F. Interleukin (IL)-10, but not IL-4 or IL-13, inhibits cytokine production and growth in juvenile myelomonocytic leukemia cells. Cancer Res. 1997, 57, 476–480. [Google Scholar]
- Zhu, J.Y.; Abate, M.; Rice, P.W.; Cole, C.N. The ability of simian virus 40 large T antigen to immortalize primary mouse embryo fibroblasts cosegregates with its ability to bind to p53. J. Virol. 1991, 65, 6872–6880. [Google Scholar] [CrossRef] [Green Version]
- Garbe, J.; Wong, M.; Wigington, D.; Yaswen, P.; Stampfer, M.R. Viral oncogenes accelerate conversion to immortality of cultured conditionally immortal human mammary epithelial cells. Oncogene 1999, 18, 2169–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexler, H.G.; Quentmeier, H. The LL-100 cell lines panel: Tool for molecular leukemia-lymphoma research. Int. J. Mol. Sci. 2020, 21, 5800. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mi, Y.; Mueller, T.; Kreibich, S.; Williams, E.G.; Van Drogen, A.; Borel, C.; Frank, M.; Germain, P.-L.; Bludau, I.; et al. Multi-omic measurements of heterogeneity in HeLa cells across laboratories. Nat. Biotechnol. 2019, 37, 314–322. [Google Scholar] [CrossRef]
- Yu, K.; Chen, B.; Aran, D.; Charalel, J.; Yau, C.; Wolf, D.M.; van’t Veer, L.J.; Butte, A.J.; Goldstein, T.; Sirota, M. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat. Commun. 2019, 10, 3574. [Google Scholar] [CrossRef] [Green Version]
- Ozgyin, L.; Horvath, A.; Hevessy, Z.; Balint, B.L. Extensive epigenetic and transcriptomic variability between genetically identical human B-lymphoblastoid cells with implications in pharmacogenomics research. Sci. Rep. 2019, 9, 4889. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Shih, T.S.; Schmitt, E.M.; Bronson, R.T.; Bernards, A.; Weinberg, R.A. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 1994, 7, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Vik, T.A.; Ryder, J.W.; Srour, E.F.; Jacks, T.; Shannon, K.; Clapp, D.W. Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J. Exp. Med. 1998, 187, 1893–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Wang, S.; Yu, W.-M.; Chan, G.; Araki, T.; Bunting, K.D.; Neel, B.G.; Qu, C.-K. A germline gain-of-function mutation in Ptpn11 (Shp-2) phosphatase induces myeloproliferative disease by aberrant activation of hematopoietic stem cells. Blood 2010, 116, 3611–3621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Liu, X.; Yu, W.-M.; Meyerson, H.J.; Guo, C.; Gerson, S.L.; Qu, C.-K. Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J. Exp. Med. 2011, 208, 1977–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, B.S.; Tuveson, D.A.; Kong, N.; Le, D.T.; Kogan, S.C.; Rozmus, J.; Le Beau, M.M.; Jacks, T.E.; Shannon, K.M. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc. Natl. Acad. Sci. USA 2004, 101, 597–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, I.T.; Kutok, J.L.; Williams, I.R.; Cohen, S.; Kelly, L.; Shigematsu, H.; Johnson, L.; Akashi, K.; Tuveson, D.A.; Jacks, T.; et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J. Clin. Invest. 2004, 113, 528–538. [Google Scholar] [CrossRef] [Green Version]
- Tarnawsky, S.P.; Kobayashi, M.; Chan, R.J.; Yoder, M.C. Mice expressing KrasG12D in hematopoietic multipotent progenitor cells develop neonatal myeloid leukemia. J. Clin. Invest. 2017, 127, 3652–3656. [Google Scholar] [CrossRef] [Green Version]
- Lapidot, T.; Grunberger, T.; Vormoor, J.; Estrov, Z.; Kollet, O.; Bunin, N.; Zaizov, R.; Williams, D.E.; Freedman, M.H. Identification of human juvenile chronic myelogenous leukemia stem cells capable of initiating the disease in primary and secondary SCID mice. Blood 1996, 88, 2655–2664. [Google Scholar] [CrossRef] [Green Version]
- Iversen, P.O.; Lewis, I.D.; Turczynowicz, S.; Hasle, H.; Niemeyer, C.; Schmiegelow, K.; Bastiras, S.; Biondi, A.; Hughes, T.P.; Lopez, A.F. Inhibition of granulocyte-macrophage colony-stimulating factor prevents dissemination and induces remission of juvenile myelomonocytic leukemia in engrafted immunodeficient mice. Blood 1997, 90, 4910–4917. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, Y.; Ito, M.; Yamamoto, T.; Yan, X.Y.; Yagasaki, H.; Kamachi, Y.; Kudo, K.; Kojima, S. Engraftment of NOD/SCID/gammac(null) mice with multilineage neoplastic cells from patients with juvenile myelomonocytic leukaemia. Br. J. Haematol. 2005, 130, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Yoshimi, A.; Balasis, M.E.; Vedder, A.; Feldman, K.; Ma, Y.; Zhang, H.; Lee, S.C.-W.; Letson, C.; Niyongere, S.; Lu, S.X.; et al. Robust patient-derived xenografts of MDS/MPN overlap syndromes capture the unique characteristics of CMML and JMML. Blood 2017, 130, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Lowry, W.E.; Richter, L.; Yachechko, R.; Pyle, A.D.; Tchieu, J.; Sridharan, R.; Clark, A.T.; Plath, K. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc. Natl. Acad. Sci. USA 2008, 105, 2883–2888. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 2011, 471, 230–234. [Google Scholar] [CrossRef]
- Stillitano, F.; Hansen, J.; Kong, C.-W.; Karakikes, I.; Funck-Brentano, C.; Geng, L.; Scott, S.; Reynier, S.; Wu, M.; Valogne, Y.; et al. Modeling susceptibility to drug-induced long QT with a panel of subject-specific induced pluripotent stem cells. eLife 2017, 6, e19406. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.-G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Stadtfeld, M.; Nagaya, M.; Utikal, J.; Weir, G.; Hochedlinger, K. Induced pluripotent stem cells generated without viral integration. Science 2008, 322, 945–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [Green Version]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, C.-H.; Moon, J.-I.; Chung, Y.-G.; Chang, M.-Y.; Han, B.-S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.-H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [Green Version]
- Subramanyam, D.; Lamouille, S.; Judson, R.L.; Liu, J.Y.; Bucay, N.; Derynck, R.; Blelloch, R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 443–448. [Google Scholar] [CrossRef]
- Mali, P.; Chou, B.-K.; Yen, J.; Ye, Z.; Zou, J.; Dowey, S.; Brodsky, R.A.; Ohm, J.E.; Yu, W.; Baylin, S.B.; et al. Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells 2010, 28, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narsinh, K.H.; Jia, F.; Robbins, R.C.; Kay, M.A.; Longaker, M.T.; Wu, J.C. Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat. Protoc. 2011, 6, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Hansen, N.F.; Zhao, L.; Du, Y.; Zou, C.; Donovan, F.X.; Chou, B.-K.; Zhou, G.; Li, S.; Dowey, S.N.; et al. Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell 2012, 10, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-P.; Han, D.W.; Kim, J.; Schöler, H.R. Biological importance of OCT transcription factors in reprogramming and development. Exp. Mol. Med. 2021, 53, 1018–1028. [Google Scholar] [CrossRef]
- Velychko, S.; Adachi, K.; Kim, K.-P.; Hou, Y.; MacCarthy, C.M.; Wu, G.; Schöler, H.R. Excluding Oct4 from Yamanaka cocktail unleashes the developmental potential of iPSCs. Cell Stem Cell 2019, 25, 737–753.e4. [Google Scholar] [CrossRef] [Green Version]
- Malik, N.; Rao, M.S. A review of the methods for human iPSC derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, N.; Gros, E.; Li, H.-R.; Kumar, S.; Deacon, D.C.; Maron, C.; Muotri, A.R.; Chi, N.C.; Fu, X.-D.; Yu, B.D.; et al. Efficient generation of human iPSCs by a synthetic self-replicative RNA. Cell Stem Cell 2013, 13, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Kumar, N.; Kalsan, M.; Saini, A.; Chandra, R. Mechanism of induction: Induced pluripotent stem cells (iPSCs). J. Stem Cells 2015, 10, 43–62. [Google Scholar]
- Felfly, H.; Haddad, G.G. Hematopoietic stem cells: Potential new applications for translational medicine. J. Stem Cells 2014, 9, 163–197. [Google Scholar]
- Park, B.; Yoo, K.H.; Kim, C. Hematopoietic stem cell expansion and generation: The ways to make a breakthrough. Blood Res. 2015, 50, 194–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in pluripotent stem cells: History, mechanisms, technologies, and applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.-H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Su, R.-J.; Baylink, D.J.; Neises, A.; Kiroyan, J.B.; Meng, X.; Payne, K.J.; Tschudy-Seney, B.; Duan, Y.; Appleby, N.; Kearns-Jonker, M.; et al. Efficient generation of integration-free ips cells from human adult peripheral blood using BCL-XL together with Yamanaka factors. PLoS ONE 2013, 8, e64496. [Google Scholar] [CrossRef] [Green Version]
- Su, R.J.; Neises, A.; Zhang, X.-B. Generation of iPS cells from human peripheral blood mononuclear cells using episomal vectors. Methods Mol. Biol. 2016, 1357, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Greber, B.; Araúzo-Bravo, M.J.; Meyer, J.; Park, K.I.; Zaehres, H.; Schöler, H.R. Direct reprogramming of human neural stem cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef]
- Hanna, J.; Markoulaki, S.; Schorderet, P.; Carey, B.W.; Beard, C.; Wernig, M.; Creyghton, M.P.; Steine, E.J.; Cassady, J.P.; Foreman, R.; et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 2008, 133, 250–264. [Google Scholar] [CrossRef] [Green Version]
- Loh, Y.-H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T Cells from human peripheral blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ye, Z.; Kim, Y.; Sharkis, S.; Jang, Y.-Y. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology 2010, 51, 1810–1819. [Google Scholar] [CrossRef]
- Chao, M.P.; Gentles, A.J.; Chatterjee, S.; Lan, F.; Reinisch, A.; Corces, M.R.; Xavy, S.; Shen, J.; Haag, D.; Chanda, S.; et al. Human AML-iPSCs reacquire leukemic properties after differentiation and model clonal variation of disease. Cell Stem Cell 2017, 20, 329–344.e7. [Google Scholar] [CrossRef] [Green Version]
- Kumano, K.; Arai, S.; Hosoi, M.; Taoka, K.; Takayama, N.; Otsu, M.; Nagae, G.; Ueda, K.; Nakazaki, K.; Kamikubo, Y.; et al. Generation of induced pluripotent stem cells from primary chronic myelogenous leukemia patient samples. Blood 2012, 119, 6234–6242. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Cheng, H.; Wang, Y.; Zheng, Y.; Liu, Y.; Liu, K.; Xu, J.; Hao, S.; Yuan, W.; Zhao, T.; et al. Reprogramming of Notch1-induced acute lymphoblastic leukemia cells into pluripotent stem cells in mice. Blood Cancer J. 2016, 6, e444. [Google Scholar] [CrossRef] [Green Version]
- Amabile, G.; Welner, R.S.; Nombela-Arrieta, C.; D’Alise, A.M.; Di Ruscio, A.; Ebralidze, A.K.; Kraytsberg, Y.; Ye, M.; Kocher, O.; Neuberg, D.S.; et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood 2013, 121, 1255–1264. [Google Scholar] [CrossRef]
- Engle, S.J.; Blaha, L.; Kleiman, R.J. Best practices for translational disease modeling using human iPSC-derived neurons. Neuron 2018, 100, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Mohsen-Kanson, T.; Hafner, A.-L.; Wdziekonski, B.; Takashima, Y.; Villageois, P.; Carrière, A.; Svensson, M.; Bagnis, C.; Chignon-Sicard, B.; Svensson, P.-A.; et al. Differentiation of human induced pluripotent stem cells into brown and white adipocytes: Role of Pax3. Stem Cells 2014, 32, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Negoro, R.; Yamashita, T.; Kawai, K.; Ichikawa, M.; Mori, T.; Nakatsu, N.; Harada, K.; Ito, S.; Yamada, H.; et al. Generation of human iPSC-derived intestinal epithelial cell monolayers by CDX2 transduction. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 513–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, I.; Klich, K.; Arauzo-Bravo, M.J.; Radstaak, M.; Santourlidis, S.; Ghanjati, F.; Radke, T.F.; Psathaki, O.E.; Hargus, G.; Kramer, J.; et al. Erythroid differentiation of human induced pluripotent stem cells is independent of donor cell type of origin. Haematologica 2015, 100, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.S.; Hanson, E.T.; Lewis, R.L.; Auerbach, R.; Thomson, J.A. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2001, 98, 10716–10721. [Google Scholar] [CrossRef] [Green Version]
- Vodyanik, M.A.; Bork, J.A.; Thomson, J.A.; Slukvin, I.I. Human embryonic stem cell-derived CD34+ cells: Efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood 2005, 105, 617–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, P.; Hematti, P. Simultaneous generation of CD34+ primitive hematopoietic cells and CD73+ mesenchymal stem cells from human embryonic stem cells cocultured with murine OP9 stromal cells. Exp. Hematol. 2007, 35, 146–154. [Google Scholar] [CrossRef]
- De Smedt, M.; Hoebeke, I.; Plum, J. Human bone marrow CD34+ progenitor cells mature to T cells on OP9-DL1 stromal cell line without thymus microenvironment. Blood Cells Mol. Dis. 2004, 33, 227–232. [Google Scholar] [CrossRef]
- Guo, N.-N.; Liu, L.-P.; Zheng, Y.-W.; Li, Y.-M. Inducing human induced pluripotent stem cell differentiation through embryoid bodies: A practical and stable approach. World J. Stem Cells 2020, 12, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, M.; Yan, Y.; Yang, S.-T. Neural differentiation from pluripotent stem cells: The role of natural and synthetic extracellular matrix. World J. Stem Cells 2014, 6, 11–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, A.J.; Zhong, J.; Lipke, E.A. Enhanced stem cell-derived cardiomyocyte differentiation in suspension culture by delivery of nitric oxide using S-nitrosocysteine. Biotechnol. Bioeng. 2016, 113, 882–894. [Google Scholar] [CrossRef]
- Nakahara, M.; Matsuyama, S.; Saeki, K.; Nakamura, N.; Saeki, K.; Yogiashi, Y.; Yoneda, A.; Koyanagi, M.; Kondo, Y.; Yuo, A. A feeder-free hematopoietic differentiation system with generation of functional neutrophils from feeder- and cytokine-free primate embryonic stem cells. Clon. Stem Cells 2008, 10, 341–354. [Google Scholar] [CrossRef]
- French, A.; Yang, C.-T.; Taylor, S.; Watt, S.M.; Carpenter, L. Human induced pluripotent stem cell-derived B lymphocytes express sIgM and can be generated via a hemogenic endothelium intermediate. Stem Cells Dev. 2015, 24, 1082–1095. [Google Scholar] [CrossRef]
- Murdoch, B.; Gallacher, L.; Chadwick, K.; Fellows, F.; Bhatia, M. Human embryonic-derived hematopoietic repopulating cells require distinct factors to sustain in vivo repopulating function. Exp. Hematol. 2002, 30, 598–605. [Google Scholar] [CrossRef]
- Bhatia, M.; Bonnet, D.; Kapp, U.; Wang, J.C.; Murdoch, B.; Dick, J.E. Quantitative analysis reveals expansion of human hematopoietic repopulating cells after short-term ex vivo culture. J. Exp. Med. 1997, 186, 619–624. [Google Scholar] [CrossRef]
- Bhardwaj, G.; Murdoch, B.; Wu, D.; Baker, D.P.; Williams, K.P.; Chadwick, K.; Ling, L.E.; Karanu, F.N.; Bhatia, M. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat. Immunol. 2001, 2, 172–180. [Google Scholar] [CrossRef]
- Bhatia, M.; Bonnet, D.; Wu, D.; Murdoch, B.; Wrana, J.; Gallacher, L.; Dick, J.E. Bone morphogenetic proteins regulate the developmental program of human hematopoietic stem cells. J. Exp. Med. 1999, 189, 1139–1148. [Google Scholar] [CrossRef] [Green Version]
- Chadwick, K.; Wang, L.; Li, L.; Menendez, P.; Murdoch, B.; Rouleau, A.; Bhatia, M. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood 2003, 102, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.-P.; Malik, A.K.; Solar, G.P.; Sherman, D.; Liang, X.H.; Meng, G.; Hong, K.; Marsters, J.C.; Ferrara, N. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 2002, 417, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Broudy, V.C.; Lin, N.L.; Kaushansky, K. Thrombopoietin (c-mpl ligand) acts synergistically with erythropoietin, stem cell factor, and interleukin-11 to enhance murine megakaryocyte colony growth and increases megakaryocyte ploidy in vitro. Blood 1995, 85, 1719–1726. [Google Scholar] [CrossRef] [Green Version]
- Briddell, R.A.; Bruno, E.; Cooper, R.J.; Brandt, J.E.; Hoffman, R. Effect of c-kit ligand on in vitro human megakaryocytopoiesis. Blood 1991, 78, 2854–2859. [Google Scholar] [CrossRef] [PubMed]
- Debili, N.; Massé, J.M.; Katz, A.; Guichard, J.; Breton-Gorius, J.; Vainchenker, W. Effects of the recombinant hematopoietic growth factors interleukin-3, interleukin-6, stem cell factor, and leukemia inhibitory factor on the megakaryocytic differentiation of CD34+ cells. Blood 1993, 82, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wodnar-Filipowicz, A.; Lyman, S.D.; Gratwohl, A.; Tichelli, A.; Speck, B.; Nissen, C. Flt3 ligand level reflects hematopoietic progenitor cell function in aplastic anemia and chemotherapy-induced bone marrow aplasia. Blood 1996, 88, 4493–4499. [Google Scholar] [CrossRef] [Green Version]
- Allouche, M. Basic fibroblast growth factor and hematopoiesis. Leukemia 1995, 9, 937–942. [Google Scholar]
- Quesniaux, V.F.; Clark, S.C.; Turner, K.; Fagg, B. Interleukin-11 stimulates multiple phases of erythropoiesis in vitro. Blood 1992, 80, 1218–1223. [Google Scholar] [CrossRef] [Green Version]
- Grover, A.; Mancini, E.; Moore, S.; Mead, A.J.; Atkinson, D.; Rasmussen, K.D.; O’Carroll, D.; Jacobsen, S.E.W.; Nerlov, C. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J. Exp. Med. 2014, 211, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Cerdan, C.; McIntyre, B.A.S.; Mechael, R.; Levadoux-Martin, M.; Yang, J.; Lee, J.B.; Bhatia, M. Activin A promotes hematopoietic fated mesoderm development through upregulation of brachyury in human embryonic stem cells. Stem Cells Dev. 2012, 21, 2866–2877. [Google Scholar] [CrossRef] [Green Version]
- Zumkeller, W. The insulin-like growth factor system in hematopoietic cells. Leuk. Lymphoma 2002, 43, 487–491. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Kuczynski, W.I.; Onodera, K.; Moore, J.; Ratajczak, J.; Kregenow, D.A.; DeRiel, K.; Gewirtz, A.M. A reappraisal of the role of insulin-like growth factor I in the regulation of human hematopoiesis. J. Clin. Invest. 1994, 94, 320–327. [Google Scholar] [CrossRef]
- Metcalf, D. The molecular control of cell division, differentiation commitment and maturation in haemopoietic cells. Nature 1989, 339, 27–30. [Google Scholar] [CrossRef]
- Nianias, A.; Themeli, M. Induced pluripotent stem cell (iPSC)-derived lymphocytes for adoptive cell immunotherapy: Recent advances and challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat. Commun. 2021, 12, 430. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Varga, E.; Aarts, C.; Wust, T.; Kuijpers, T.; von Lindern, M.; van den Akker, E. Efficient production of erythroid, megakaryocytic and myeloid cells, using single cell-derived iPSC colony differentiation. Stem Cell Res. 2018, 29, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Féraud, O.; Valogne, Y.; Melkus, M.W.; Zhang, Y.; Oudrhiri, N.; Haddad, R.; Daury, A.; Rocher, C.; Larbi, A.; Duquesnoy, P.; et al. Donor dependent variations in hematopoietic differentiation among embryonic and induced pluripotent stem cell lines. PLoS ONE 2016, 11, e0149291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgomanoli, M.; Papapetrou, E.P. Modeling blood diseases with human induced pluripotent stem cells. Dis. Model. Mech. 2019, 12, dmm039321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, M.; Liebhaber, S.; Klusmann, J.-H.; Lachmann, N. Lost in translation: Pluripotent stem cell-derived hematopoiesis. EMBO Mol. Med. 2015, 7, 1388–1402. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Yamazaki, S.; Yamaguchi, T.; Okabe, M.; Masaki, H.; Takaki, S.; Otsu, M.; Nakauchi, H. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol. Ther. 2013, 21, 1424–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Zhang, G.; Gu, J.; Shao, X.; Dai, Y.; Li, J.; Pan, X.; Yao, S.; Xu, A.; Jin, Y.; et al. In vivo generated hematopoietic stem cells from genome edited induced pluripotent stem cells are functional in platelet-targeted gene therapy of murine hemophilia A. Haematologica 2020, 105, e175–e179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gładych, M.; Andrzejewska, A.; Oleksiewicz, U.; Estécio, M.R.H. Epigenetic mechanisms of induced pluripotency. Contemp. Oncol. 2015, 19, A30–A38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Hurk, M.; Kenis, G.; Bardy, C.; van den Hove, D.L.; Gage, F.H.; Steinbusch, H.W.; Rutten, B.P. Transcriptional and epigenetic mechanisms of cellular reprogramming to induced pluripotency. Epigenomics 2016, 8, 1131–1149. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Keller, A.; Dziedzicka, D.; Zambelli, F.; Markouli, C.; Sermon, K.; Spits, C.; Geens, M. Genetic and epigenetic factors which modulate differentiation propensity in human pluripotent stem cells. Hum. Reprod. Update 2018, 24, 162–175. [Google Scholar] [CrossRef]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Zhao, M.-T.; Jahanbani, F.; Shao, N.-Y.; Lee, W.H.; Chen, H.; Snyder, M.P.; Wu, J.C. Effects of cellular origin on differentiation of human induced pluripotent stem cell-derived endothelial cells. JCI Insight 2016, 1, e85558. [Google Scholar] [CrossRef]
- Melnick, A.M. Epigenetics in AML. Best Pract. Res. Clin. Haematol. 2010, 23, 463–468. [Google Scholar] [CrossRef]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Zhang, H.; Yang, H.; Koche, R.; Staber, P.B.; Cusan, M.; Levantini, E.; Welner, R.S.; Bach, C.S.; Zhang, J.; et al. Hematopoietic differentiation is required for initiation of acute myeloid leukemia. Cell Stem Cell 2015, 17, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amabile, G.; Di Ruscio, A.; Müller, F.; Welner, R.S.; Yang, H.; Ebralidze, A.K.; Zhang, H.; Levantini, E.; Qi, L.; Martinelli, G.; et al. Dissecting the role of aberrant DNA methylation in human leukaemia. Nat. Commun. 2015, 6, 7091. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, N.; Sakaguchi, H.; Yabe, M.; Hasegawa, D.; Hama, A.; Hasegawa, D.; Kato, M.; Noguchi, M.; Terui, K.; Takahashi, Y.; et al. Clinical outcomes after allogeneic hematopoietic stem cell transplantation in children with juvenile myelomonocytic leukemia: A report from the Japan Society for Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2020, 26, 902–910. [Google Scholar] [CrossRef]
- Chang, T.; Krisman, K.; Theobald, E.H.; Xu, J.; Akutagawa, J.; Lauchle, J.O.; Kogan, S.; Braun, B.S.; Shannon, K. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J. Clin. Investig. 2013, 123, 335–339. [Google Scholar] [CrossRef]
- Lyubynska, N.; Gorman, M.F.; Lauchle, J.O.; Hong, W.X.; Akutagawa, J.K.; Shannon, K.; Braun, B.S. A MEK inhibitor abrogates myeloproliferative disease in Kras mutant mice. Sci. Transl. Med. 2011, 3, 76ra27. [Google Scholar] [CrossRef] [Green Version]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.; Vu, B.T.; Qing, W.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scesa, G.; Adami, R.; Bottai, D. iPSC preparation and epigenetic memory: Does the tissue origin matter? Cells 2021, 10, 1470. [Google Scholar] [CrossRef]


{kind=link}
| Study | Reprogrammed Cells | Mutations | Reprogramming Technique | HSC Differentiation Protocol | Cytokine Combination | Remarks | Reference |
|---|---|---|---|---|---|---|---|
| Gandre–Babbe et al., 2013 | Ficoll-purified MNCs from BM or PB n = 2 | Somatic heterozygous missense mutations in PTPN11 | STEMCA lentivirus expressing doxycycline-regulated POU5F1, SOX2, MYC, and KLF4 | Embryoid bodies and adherent monolayer culture with supplementation of cytokines |
| First generation of IPSCs from JMML cells. In vitro differentiation of JMML-IPSCs produced myeloid cells with leukemic features including high proliferative capacity, activation of GM-CSF, and enhanced STAT5/ERK phosphorylation. The inhibition of MEK kinase in IPSC-derived JMML cells reduced their GM-CSF hypersensitivity | [18] |
| Mulero–Navarro et al., 2015 | Skin fibroblasts n = 2 | Germline mutations causing NS/MPD | Separate retroviruses expressing human POU5F1, SOX2, MYC, and KLF4 | Embryoid bodies and cytokine supplementation |
| NS/MPD-IPSC-derived myeloid cells carrying a PTPN11 mutation exhibited an upregulation of miR-223 and miR-15a, similar to BM mononuclear cells harboring PTPN11 mutations. Normal myelogenesis was reestablished via reducing miR-223’s function in NS/MPD IPSCs | [19] |
| Tasian et al., 2019 | Ficoll-purified MNCs from BM or PB n = 2 | Germline CBL, somatic PTPN11 | STEMCA lentivirus expressing doxycycline-regulated POU5F1, SOX2, MYC, and KLF4 | Embryoid bodies and cytokine supplementation |
| MEK, JAK, and PI3K/mTOR inhibitors resulted in different reactivity of IPSC-derived hematopoietic progenitors and signaling aberrations, depending on the driver mutation | [20] |
| Shigemura et al., 2019 | PB T cells n = 1 | PTPN11 | Sendai virus vector encoding the human transcription factors POU5F1, SOX2, MYC, and KLF4 | Co-culture system with stroma cells |
| Mutant IPSC colonies generated significantly more CD34+ and CD34+ CD45+ cells compared to non-mutant IPSC colonies. The PTPN11 mutation seems to govern hematopoietic differentiation in JMML | [21] |
| Pearson et al., 2020 | Skin fibroblasts n = 2 | NS/MPD | Separate retroviruses expressing human POU5F1, SOX2, MYC, and KLF4 | Embryoid bodies and cytokines supplementation |
| Establishment of proteomic screen in NS-derived IPSCs. Demonstration of additive effects of two drugs (JQ1 as differentiation enhancer and CBL0137 as apoptosis inducer) on NS/MPD cells. | [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wehbe, Z.; Ghanjati, F.; Flotho, C. Induced Pluripotent Stem Cells to Model Juvenile Myelomonocytic Leukemia: New Perspectives for Preclinical Research. Cells 2021, 10, 2335. https://doi.org/10.3390/cells10092335
Wehbe Z, Ghanjati F, Flotho C. Induced Pluripotent Stem Cells to Model Juvenile Myelomonocytic Leukemia: New Perspectives for Preclinical Research. Cells. 2021; 10(9):2335. https://doi.org/10.3390/cells10092335
Chicago/Turabian StyleWehbe, Zeinab, Foued Ghanjati, and Christian Flotho. 2021. "Induced Pluripotent Stem Cells to Model Juvenile Myelomonocytic Leukemia: New Perspectives for Preclinical Research" Cells 10, no. 9: 2335. https://doi.org/10.3390/cells10092335
APA StyleWehbe, Z., Ghanjati, F., & Flotho, C. (2021). Induced Pluripotent Stem Cells to Model Juvenile Myelomonocytic Leukemia: New Perspectives for Preclinical Research. Cells, 10(9), 2335. https://doi.org/10.3390/cells10092335