
Telomere Maintenance and the cGAS-STING Pathway in Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
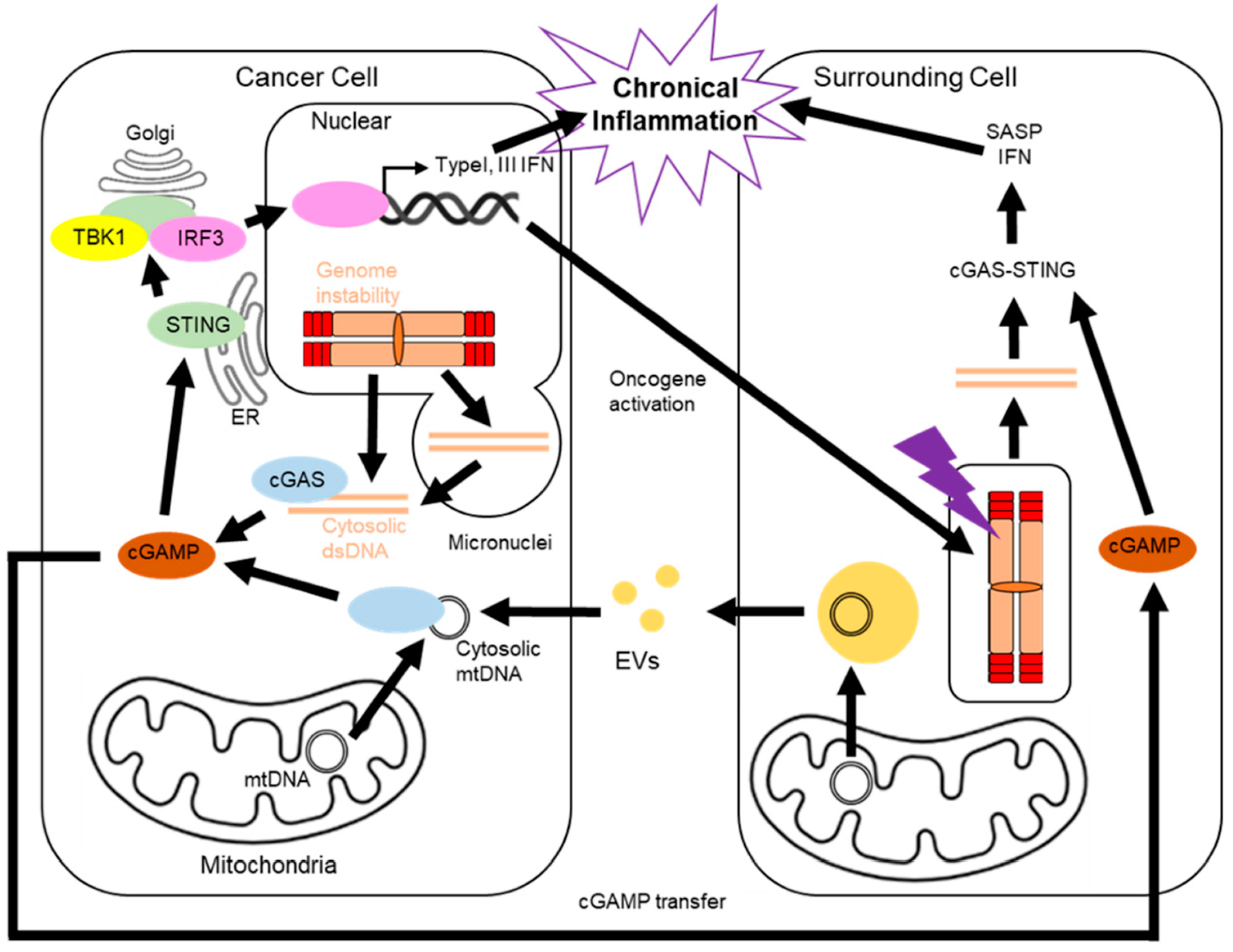
:1. Introduction
2. Telomere and the cGAS-STING Pathway
3. Cancer and Telomerase
4. Cancer and ALT
5. Cancer and the cGAS-STING Pathway
6. The cGAS-STING Pathway in Surrounding Cells
7. Future Perspectives/Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA Damage Is More Extensive and Persists Longer than Nuclear DNA Damage in Human Cells Following Oxidative Stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P.C. The Clonal Evolution of Tumor Cell Populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic Instability in Colorectal Cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef]
- Boveri, T. Concerning the Origin of Malignant Tumours by Theodor Boveri. Translated and Annotated by Henry Harris. J. Cell Sci. 2008, 121 (Suppl. 1), 1–84. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal Oxidative Damage to Mitochondrial and Nuclear DNA Is Extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [Green Version]
- Szatrowski, T.P.; Nathan, C.F. Production of Large Amounts of Hydrogen Peroxide by Human Tumor Cells1. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Morin, G.B. The Human Telomere Terminal Transferase Enzyme Is a Ribonucleoprotein That Synthesizes TTAGGG Repeats. Cell 1989, 59, 521–529. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere Elongation in Immortal Human Cells without Detectable Telomerase Activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar] [CrossRef]
- Haendeler, J.; Hoffmann, J.; Brandes, R.P.; Zeiher, A.M.; Dimmeler, S. Hydrogen Peroxide Triggers Nuclear Export of Telomerase Reverse Transcriptase via Src Kinase Family-Dependent Phosphorylation of Tyrosine 707. Mol. Cell. Biol. 2003, 23, 4598–4610. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase Therapy Provokes ALT and Mitochondrial Adaptive Mechanisms in Cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, B.D.; Benada, J.; Yung, P.Y.K.; Bell, R.A.V.; Pappas, G.; Urban, V.; Ahlskog, J.K.; Kuo, T.T.; Janscak, P.; Megeney, L.A.; et al. Cancer Cells Use Self-Inflicted DNA Breaks to Evade Growth Limits Imposed by Genotoxic Stress. Science 2022, 376, 476–483. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS Surveillance of Micronuclei Links Genome Instability to Innate Immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA Damage Is Irreparable and Causes Persistent DNA-Damage-Response Activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Rossiello, F.; Aguado, J.; Sepe, S.; Iannelli, F.; Nguyen, Q.; Pitchiaya, S.; Carninci, P.; d’Adda di Fagagna, F. DNA Damage Response Inhibition at Dysfunctional Telomeres by Modulation of Telomeric DNA Damage Response RNAs. Nat. Commun. 2017, 8, 13980. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.S.; Chen, Q.; Yatsunyk, L.A.; Nicoludis, J.M.; Garcia, M.S.; Kranaster, R.; Balasubramanian, S.; Monchaud, D.; Teulade-Fichou, M.-P.; Abramowitz, L.; et al. Rudimentary G-Quadruplex-Based Telomere Capping in Saccharomyces Cerevisiae. Nat. Struct. Mol. Biol. 2011, 18, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Bandaria, J.N.; Qureshi, M.H.; Yildiz, A.; Balci, H. G-Quadruplex Formation in Telomeres Enhances POT1/TPP1 Protection against RPA Binding. Proc. Natl. Acad. Sci. USA 2014, 111, 2990–2995. [Google Scholar] [CrossRef] [Green Version]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian Telomeres End in a Large Duplex Loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.Z.; Allsopp, R.C.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere End-Replication Problem and Cell Aging. J. Mol. Biol. 1992, 225, 951–960. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Loo, T.M.; Okada, R.; Kamachi, F.; Watanabe, Y.; Wakita, M.; Watanabe, S.; Kawamoto, S.; Miyata, K.; Barber, G.N.; et al. Downregulation of Cytoplasmic DNases Is Implicated in Cytoplasmic DNA Accumulation and SASP in Senescent Cells. Nat. Commun. 2018, 9, 1249. [Google Scholar] [CrossRef] [PubMed]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF Induces Senescence and Apoptosis through Pathways Mediated by the Secreted Protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS Is Essential for Cellular Senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate Immune Sensing of Cytosolic Chromatin Fragments through cGAS Promotes Senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Loo, T.M.; Miyata, K.; Tanaka, Y.; Takahashi, A. Cellular Senescence and Senescence-Associated Secretory Phenotype via the cGAS-STING Signaling Pathway in Cancer. Cancer Sci. 2020, 111, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Abdisalaam, S.; Bhattacharya, S.; Mukherjee, S.; Sinha, D.; Srinivasan, K.; Zhu, M.; Akbay, E.A.; Sadek, H.A.; Shay, J.W.; Asaithamby, A. Dysfunctional Telomeres Trigger Cellular Senescence Mediated by Cyclic GMP-AMP Synthase. J. Biol. Chem. 2020, 295, 11144–11160. [Google Scholar] [CrossRef]
- Lv, N.; Zhao, Y.; Liu, X.; Ye, L.; Liang, Z.; Kang, Y.; Dong, Y.; Wang, W.; Kolliputi, N.; Shi, L. Dysfunctional Telomeres through Mitostress-Induced cGAS/STING Activation to Aggravate Immune Senescence and Viral Pneumonia. Aging Cell 2022, 21, e13594. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, X.; Xie, C.; Cai, S.; Li, M.; Jin, H.; Wu, S.; Cui, J.; Liu, H.; Zhao, Y. cGAS Guards against Chromosome End-to-End Fusions during Mitosis and Facilitates Replicative Senescence. Protein Cell 2022, 13, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Gutkin, A.; Uziel, O.; Beery, E.; Nordenberg, J.; Pinchasi, M.; Goldvaser, H.; Henick, S.; Goldberg, M.; Lahav, M. Tumor Cells Derived Exosomes Contain hTERT mRNA and Transform Nonmalignant Fibroblasts into Telomerase Positive Cells. Oncotarget 2016, 7, 59173–59188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compton, S.A.; Choi, J.-H.; Cesare, A.J.; Ozgür, S.; Griffith, J.D. Xrcc3 and Nbs1 Are Required for the Production of Extrachromosomal Telomeric Circles in Human Alternative Lengthening of Telomere Cells. Cancer Res. 2007, 67, 1513–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesare, A.J.; Reddel, R.R. Alternative Lengthening of Telomeres: Models, Mechanisms and Implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Shen, Y.-L.; Hsia, H.-Y.; Tiang, Y.-P.; Sung, T.-L.; Chen, L.-Y. Extrachromosomal Telomere Repeat DNA Is Linked to ALT Development via cGAS-STING DNA Sensing Pathway. Nat. Struct. Mol. Biol. 2017, 24, 1124–1131. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver Mutations in Histone H3.3 and Chromatin Remodelling Genes in Paediatric Glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered Telomeres in Tumors with ATRX and DAXX Mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [Green Version]
- Greider, C.W.; Blackburn, E.H. The Telomere Terminal Transferase of Tetrahymena Is a Ribonucleoprotein Enzyme with Two Kinds of Primer Specificity. Cell 1987, 51, 887–898. [Google Scholar] [CrossRef]
- Ten Hagen, K.G.; Gilbert, D.M.; Willard, H.F.; Cohen, S.N. Replication Timing of DNA Sequences Associated with Human Centromeres and Telomeres. Mol. Cell. Biol. 1990, 10, 6348–6355. [Google Scholar]
- Wright, W.E.; Tesmer, V.M.; Liao, M.L.; Shay, J.W. Normal Human Telomeres Are Not Late Replicating. Exp. Cell Res. 1999, 251, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.Y.; Kusdra, L.; Collins, K. Subnuclear Shuttling of Human Telomerase Induced by Transformation and DNA Damage. Nat. Cell Biol. 2002, 4, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Aalfs, C.M.; van den Berg, H.; Barth, P.G.; Hennekam, R.C. The Hoyeraal-Hreidarsson Syndrome: The Fourth Case of a Separate Entity with Prenatal Growth Retardation, Progressive Pancytopenia and Cerebellar Hypoplasia. Eur. J. Pediatr. 1995, 154, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Wood, E.; Collins, K. A Telomerase Component Is Defective in the Human Disease Dyskeratosis Congenita. Nature 1999, 402, 551–555. [Google Scholar] [CrossRef]
- Wang, C.; Meier, U.T. Architecture and Assembly of Mammalian H/ACA Small Nucleolar and Telomerase Ribonucleoproteins. EMBO J. 2004, 23, 1857–1867. [Google Scholar] [CrossRef] [Green Version]
- Fujii, Y.; Zhao, F.X.; Fu, S.C.; Nakai, N.; Lai, C.Y. Stable Preparation of Aldose Reductase Isoenzymes from Human Placenta. Protein Expr. Purif. 1991, 2, 420–425. [Google Scholar] [CrossRef]
- Dragon, F.; Pogacić, V.; Filipowicz, W. In Vitro Assembly of Human H/ACA Small Nucleolar RNPs Reveals Unique Features of U17 and Telomerase RNAs. Mol. Cell. Biol. 2000, 20, 3037–3048. [Google Scholar] [CrossRef] [Green Version]
- Pogacić, V.; Dragon, F.; Filipowicz, W. Human H/ACA Small Nucleolar RNPs and Telomerase Share Evolutionarily Conserved Proteins NHP2 and NOP10. Mol. Cell. Biol. 2000, 20, 9028–9040. [Google Scholar] [CrossRef] [Green Version]
- Tycowski, K.T.; Shu, M.-D.; Kukoyi, A.; Steitz, J.A. A Conserved WD40 Protein Binds the Cajal Body Localization Signal of scaRNP Particles. Mol. Cell 2009, 34, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Venteicher, A.S.; Abreu, E.B.; Meng, Z.; McCann, K.E.; Terns, R.M.; Veenstra, T.D.; Terns, M.P.; Artandi, S.E. A Human Telomerase Holoenzyme Protein Required for Cajal Body Localization and Telomere Synthesis. Science 2009, 323, 644–648. [Google Scholar] [CrossRef] [Green Version]
- Masutomi, K.; Kaneko, S.; Hayashi, N.; Yamashita, T.; Shirota, Y.; Kobayashi, K.; Murakami, S. Telomerase Activity Reconstituted in Vitro with Purified Human Telomerase Reverse Transcriptase and Human Telomerase RNA Component. J. Biol. Chem. 2000, 275, 22568–22573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, D.; Collins, K. Distinct Biogenesis Pathways for Human Telomerase RNA and H/ACA Small Nucleolar RNAs. Mol. Cell 2003, 11, 1361–1372. [Google Scholar] [CrossRef]
- Vogan, J.M.; Zhang, X.; Youmans, D.T.; Regalado, S.G.; Johnson, J.Z.; Hockemeyer, D.; Collins, K. Minimized Human Telomerase Maintains Telomeres and Resolves Endogenous Roles of H/ACA Proteins, TCAB1, and Cajal Bodies. Elife 2016, 5, e18221. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-K.; Theimer, C.A.; Mitchell, J.R.; Collins, K.; Feigon, J. Effect of Pseudouridylation on the Structure and Activity of the Catalytically Essential P6.1 Hairpin in Human Telomerase RNA. Nucleic Acids Res. 2010, 38, 6746–6756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafontaine, D.L.J.; Bousquet-Antonelli, C.; Henry, Y.; Caizergues-Ferrer, M.; Tollervey, D. The Box H+ACA snoRNAs Carry Cbf5p, the Putative rRNA Pseudouridine Synthase. Genes Dev. 1998, 12, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Zebarjadian, Y.; King, T.; Fournier, M.J.; Clarke, L.; Carbon, J. Point Mutations in Yeast CBF5 Can Abolish in Vivo Pseudouridylation of rRNA. Mol. Cell. Biol. 1999, 19, 7461–7472. [Google Scholar] [CrossRef] [Green Version]
- Jády, B.E.; Richard, P.; Bertrand, E.; Kiss, T. Cell Cycle-Dependent Recruitment of Telomerase RNA and Cajal Bodies to Human Telomeres. Mol. Biol. Cell 2006, 17, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, R.L.; Ziegler, T.D.; Supakorndej, T.; Terns, R.M.; Terns, M.P. Cell Cycle-Regulated Trafficking of Human Telomerase to Telomeres. Mol. Biol. Cell 2006, 17, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Zhong, F.; Savage, S.A.; Shkreli, M.; Giri, N.; Jessop, L.; Myers, T.; Chen, R.; Alter, B.P.; Artandi, S.E. Disruption of Telomerase Trafficking by TCAB1 Mutation Causes Dyskeratosis Congenita. Genes Dev. 2011, 25, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Stern, J.L.; Zyner, K.G.; Pickett, H.A.; Cohen, S.B.; Bryan, T.M. Telomerase Recruitment Requires Both TCAB1 and Cajal Bodies Independently. Mol. Cell. Biol. 2012, 32, 2384–2395. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; He, Y.; Wang, Y.; Song, H.; Zhou, Z.H.; Feigon, J. Structure of Active Human Telomerase with Telomere Shelterin Protein TPP1. Nature 2022, 604, 578–583. [Google Scholar] [CrossRef]
- Ghanim, G.E.; Fountain, A.J.; van Roon, A.-M.M.; Rangan, R.; Das, R.; Collins, K.; Nguyen, T.H.D. Structure of Human Telomerase Holoenzyme with Bound Telomeric DNA. Nature 2021, 593, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Bacchetti, S. A Survey of Telomerase Activity in Human Cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Akiyama, M.; Hideshima, T.; Hayashi, T.; Tai, Y.-T.; Mitsiades, C.S.; Mitsiades, N.; Chauhan, D.; Richardson, P.; Munshi, N.C.; Anderson, K.C. Nuclear Factor-kappaB p65 Mediates Tumor Necrosis Factor Alpha-Induced Nuclear Translocation of Telomerase Reverse Transcriptase Protein. Cancer Res. 2003, 63, 18–21. [Google Scholar]
- Ding, D.; Xi, P.; Zhou, J.; Wang, M.; Cong, Y.-S. Human Telomerase Reverse Transcriptase Regulates MMP Expression Independently of Telomerase Activity via NF-κB-Dependent Transcription. FASEB J. 2013, 27, 4375–4383. [Google Scholar] [CrossRef]
- Liu, N.; Ding, D.; Hao, W.; Yang, F.; Wu, X.; Wang, M.; Xu, X.; Ju, Z.; Liu, J.-P.; Song, Z.; et al. hTERT Promotes Tumor Angiogenesis by Activating VEGF via Interactions with the Sp1 Transcription Factor. Nucleic Acids Res. 2016, 44, 8693–8703. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; Van Houten, B. Mitochondrial hTERT Exacerbates Free-Radical-Mediated mtDNA Damage. Aging Cell 2004, 3, 399–411. [Google Scholar] [CrossRef]
- Santos, J.H.; Meyer, J.N.; Van Houten, B. Mitochondrial Localization of Telomerase as a Determinant for Hydrogen Peroxide-Induced Mitochondrial DNA Damage and Apoptosis. Hum. Mol. Genet. 2006, 15, 1757–1768. [Google Scholar] [CrossRef]
- Ahmed, S.; Passos, J.F.; Birket, M.J.; Beckmann, T.; Brings, S.; Peters, H.; Birch-Machin, M.A.; von Zglinicki, T.; Saretzki, G. Telomerase Does Not Counteract Telomere Shortening but Protects Mitochondrial Function under Oxidative Stress. J. Cell Sci. 2008, 121, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Singhapol, C.; Pal, D.; Czapiewski, R.; Porika, M.; Nelson, G.; Saretzki, G.C. Mitochondrial Telomerase Protects Cancer Cells from Nuclear DNA Damage and Apoptosis. PLoS ONE 2013, 8, e52989. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Reyes, A.; Green, P.; Caron, M.J.; Bonini, M.G.; Gordon, D.M.; Holt, I.J.; Santos, J.H. Human Telomerase Acts as a hTR-Independent Reverse Transcriptase in Mitochondria. Nucleic Acids Res. 2012, 40, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haendeler, J.; Dröse, S.; Büchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial Telomerase Reverse Transcriptase Binds to and Protects Mitochondrial DNA and Function from Damage. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indran, I.R.; Hande, M.P.; Pervaiz, S. hTERT Overexpression Alleviates Intracellular ROS Production, Improves Mitochondrial Function, and Inhibits ROS-Mediated Apoptosis in Cancer Cells. Cancer Res. 2011, 71, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Green, P.D.; Sharma, N.K.; Santos, J.H. Telomerase Impinges on the Cellular Response to Oxidative Stress Through Mitochondrial ROS-Mediated Regulation of Autophagy. Int. J. Mol. Sci. 2019, 20, 1509. [Google Scholar] [CrossRef] [Green Version]
- Martens, A.; Schmid, B.; Akintola, O.; Saretzki, G. Telomerase Does Not Improve DNA Repair in Mitochondria upon Stress but Increases MnSOD Protein under Serum-Free Conditions. Int. J. Mol. Sci. 2019, 21, 27. [Google Scholar] [CrossRef] [Green Version]
- Ebata, H.; Shima, T.; Iizuka, R.; Uemura, S. Accumulation of TERT in Mitochondria Shows Two Opposing Effects on Apoptosis. bioRxiv 2021, 2021, 1–22. [Google Scholar]
- Kilian, A.; Bowtell, D.D.; Abud, H.E.; Hime, G.R.; Venter, D.J.; Keese, P.K.; Duncan, E.L.; Reddel, R.R.; Jefferson, R.A. Isolation of a Candidate Human Telomerase Catalytic Subunit Gene, Which Reveals Complex Splicing Patterns in Different Cell Types. Hum. Mol. Genet. 1997, 6, 2011–2019. [Google Scholar] [CrossRef]
- Wick, M.; Zubov, D.; Hagen, G. Genomic Organization and Promoter Characterization of the Gene Encoding the Human Telomerase Reverse Transcriptase (hTERT). Gene 1999, 232, 97–106. [Google Scholar] [CrossRef]
- Hisatomi, H.; Ohyashiki, K.; Ohyashiki, J.H.; Nagao, K.; Kanamaru, T.; Hirata, H.; Hibi, N.; Tsukada, Y. Expression Profile of a Gamma-Deletion Variant of the Human Telomerase Reverse Transcriptase Gene. Neoplasia 2003, 5, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Saebøe-Larssen, S.; Fossberg, E.; Gaudernack, G. Characterization of Novel Alternative Splicing Sites in Human Telomerase Reverse Transcriptase (hTERT): Analysis of Expression and Mutual Correlation in mRNA Isoforms from Normal and Tumour Tissues. BMC Mol. Biol. 2006, 7, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrdlicková, R.; Nehyba, J.; Bose, H.R., Jr. Alternatively Spliced Telomerase Reverse Transcriptase Variants Lacking Telomerase Activity Stimulate Cell Proliferation. Mol. Cell. Biol. 2012, 32, 4283–4296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Ludlow, A.T.; Min, J.; Robin, J.D.; Stadler, G.; Mender, I.; Lai, T.-P.; Zhang, N.; Wright, W.E.; Shay, J.W. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect-Over Long Distances (TPE-OLD): Implications for Aging and Cancer. PLoS Biol. 2016, 14, e2000016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Listerman, I.; Sun, J.; Gazzaniga, F.S.; Lukas, J.L.; Blackburn, E.H. The Major Reverse Transcriptase-Incompetent Splice Variant of the Human Telomerase Protein Inhibits Telomerase Activity but Protects from Apoptosis. Cancer Res. 2013, 73, 2817–2828. [Google Scholar] [CrossRef] [Green Version]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an Alternative Mechanism for Maintaining Telomere Length in Human Tumors and Tumor-Derived Cell Lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef]
- McEachern, M.J.; Blackburn, E.H. Cap-Prevented Recombination between Terminal Telomeric Repeat Arrays (telomere CPR) Maintains Telomeres in Kluyveromyces Lactis Lacking Telomerase. Genes Dev. 1996, 10, 1822–1834. [Google Scholar] [CrossRef] [Green Version]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere Maintenance by Recombination in Human Cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef]
- Hakin-Smith, V.; Jellinek, D.A.; Levy, D.; Carroll, T.; Teo, M.; Timperley, W.R.; McKay, M.J.; Reddel, R.R.; Royds, J.A. Alternative Lengthening of Telomeres and Survival in Patients with Glioblastoma Multiforme. Lancet 2003, 361, 836–838. [Google Scholar] [CrossRef]
- Henson, J.D.; Hannay, J.A.; McCarthy, S.W.; Royds, J.A.; Yeager, T.R.; Robinson, R.A.; Wharton, S.B.; Jellinek, D.A.; Arbuckle, S.M.; Yoo, J.; et al. A Robust Assay for Alternative Lengthening of Telomeres in Tumors Shows the Significance of Alternative Lengthening of Telomeres in Sarcomas and Astrocytomas. Clin. Cancer Res. 2005, 11, 217–225. [Google Scholar] [CrossRef]
- Costa, A.; Daidone, M.G.; Daprai, L.; Villa, R.; Cantù, S.; Pilotti, S.; Mariani, L.; Gronchi, A.; Henson, J.D.; Reddel, R.R.; et al. Telomere Maintenance Mechanisms in Liposarcomas: Association with Histologic Subtypes and Disease Progression. Cancer Res. 2006, 66, 8918–8924. [Google Scholar] [CrossRef] [Green Version]
- Jeyapalan, J.N.; Mendez-Bermudez, A.; Zaffaroni, N.; Dubrova, Y.E.; Royle, N.J. Evidence for Alternative Lengthening of Telomeres in Liposarcomas in the Absence of ALT-Associated PML Bodies. Int. J. Cancer 2008, 122, 2414–2421. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.-H.; Jiang, W.-Q.; Cesare, A.J.; Neumann, A.A.; Wadhwa, R.; Reddel, R.R. Disruption of Telomere Maintenance by Depletion of the MRE11/RAD50/NBS1 Complex in Cells That Use Alternative Lengthening of Telomeres. J. Biol. Chem. 2007, 282, 29314–29322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.-Q.; Zhong, Z.-H.; Henson Jeremy, D.; Neumann Axel, A.; Chang Andy, C.-M.; Reddel Roger, R. Suppression of Alternative Lengthening of Telomeres by Sp100-Mediated Sequestration of the MRE11/RAD50/NBS1 Complex. Mol. Cell. Biol. 2005, 25, 2708–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Paull, T.T. Direct Activation of the ATM Protein Kinase by the Mre11/Rad50/Nbs1 Complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA Damage Activates ATM through Intermolecular Autophosphorylation and Dimer Dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Wang, H.; Shi, L.Z.; Wong, C.C.L.; Han, X.; Hwang, P.Y.-H.; Truong, L.N.; Zhu, Q.; Shao, Z.; Chen, D.J.; Berns, M.W.; et al. The Interaction of CtIP and Nbs1 Connects CDK and ATM to Regulate HR-Mediated Double-Strand Break Repair. PLoS Genet. 2013, 9, e1003277. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Wan, L.; Jiang, G.; Cao, L.; Xia, F.; Tian, T.; Zhu, X.; Wu, M.; Huen, M.S.Y.; Wang, Y.; et al. ATM Controls the Extent of DNA End Resection by Eliciting Sequential Posttranslational Modifications of CtIP. Proc. Natl. Acad. Sci. USA 2021, 118, e2022600118. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP Promotes DNA End Resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.N.M.; Iwasaki, H.; Russell, P.; et al. Nbs1 Flexibly Tethers Ctp1 and Mre11-Rad50 to Coordinate DNA Double-Strand Break Processing and Repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Karlseder, J.; Hoke, K.; Mirzoeva, O.K.; Bakkenist, C.; Kastan, M.B.; Petrini, J.H.J.; de Lange, T. The Telomeric Protein TRF2 Binds the ATM Kinase and Can Inhibit the ATM-Dependent DNA Damage Response. PLoS Biol. 2004, 2, E240. [Google Scholar] [CrossRef] [PubMed]
- Stroik, S.; Kurtz, K.; Hendrickson, E.A. CtIP Is Essential for Telomere Replication. Nucleic Acids Res. 2019, 47, 8927–8940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM Phosphorylates Histone H2AX in Response to DNA Double-Strand Breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmink, B.A.; Tubbs, A.T.; Dorsett, Y.; Bednarski, J.J.; Walker, L.M.; Feng, Z.; Sharma, G.G.; McKinnon, P.J.; Zhang, J.; Bassing, C.H.; et al. H2AX Prevents CtIP-Mediated DNA End Resection and Aberrant Repair in G1-Phase Lymphocytes. Nature 2011, 469, 245–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, S.M.; Coackley, C.; Bristow, R.G. ATM-Dependent Phosphorylation of 53BP1 in Response to Genomic Stress in Oxic and Hypoxic Cells. Radiother. Oncol. 2011, 99, 307–312. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous Occurrence of Telomeric DNA Damage Response in the Absence of Chromosome Fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef]
- Kakarougkas, A.; Ismail, A.; Klement, K.; Goodarzi, A.A.; Conrad, S.; Freire, R.; Shibata, A.; Lobrich, M.; Jeggo, P.A. Opposing Roles for 53BP1 during Homologous Recombination. Nucleic Acids Res. 2013, 41, 9719–9731. [Google Scholar] [CrossRef] [Green Version]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal Homology Searches Drive Directional ALT Telomere Movement and Synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Oza, P.; Jaspersen, S.L.; Miele, A.; Dekker, J.; Peterson, C.L. Mechanisms That Regulate Localization of a DNA Double-Strand Break to the Nuclear Periphery. Genes Dev. 2009, 23, 912–927. [Google Scholar] [CrossRef] [Green Version]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-Strand Breaks in Heterochromatin Move outside of a Dynamic HP1a Domain to Complete Recombinational Repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miné-Hattab, J.; Rothstein, R. Increased Chromosome Mobility Facilitates Homology Search during Recombination. Nat. Cell Biol. 2012, 14, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere Dysfunction Induces Metabolic and Mitochondrial Compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient Control of Glucose Homeostasis through a Complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of poly(ADP-Ribose) Polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.M.; Hande, M.P.; Lansdorp, P.M.; Wang, Z.Q. DNA Strand Break-Sensing Molecule poly(ADP-Ribose) Polymerase Cooperates with p53 in Telomere Function, Chromosome Stability, and Tumor Suppression. Mol. Cell. Biol. 2001, 21, 4046–4054. [Google Scholar] [CrossRef] [Green Version]
- Palacios, J.A.; Herranz, D.; De Bonis, M.L.; Velasco, S.; Serrano, M.; Blasco, M.A. SIRT1 Contributes to Telomere Maintenance and Augments Global Homologous Recombination. J. Cell Biol. 2010, 191, 1299–1313. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS Produces a 2′-5′-Linked Cyclic Dinucleotide Second Messenger That Activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.-C.; Zhang, X. Cryo-EM Structures of STING Reveal Its Mechanism of Activation by Cyclic GMP–AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Du, F.; Xu, P.; Shu, C.; Sankaran, B.; Bell, S.L.; Liu, M.; Lei, Y.; Gao, X.; Fu, X.; et al. A Conserved PLPLRT/SD Motif of STING Mediates the Recruitment and Activation of TBK1. Nature 2019, 569, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a Controls dsDNA-Driven Dynamic Translocation of STING and the Innate Immune Response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbs, N.; Burnaevskiy, N.; Chen, D.; Gonugunta, V.K.; Alto, N.M.; Yan, N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 2015, 18, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING Requires Palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.-C.; Chen, Z.J. Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [Green Version]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural Mechanism of Cytosolic DNA Sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Wu, F.-H.; Wang, X.; Wang, L.; Siedow, J.N.; Zhang, W.; Pei, Z.-M. Molecular Evolutionary and Structural Analysis of the Cytosolic DNA Sensor cGAS and STING. Nucleic Acids Res. 2014, 42, 8243–8257. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic Progression Following DNA Damage Enables Pattern Recognition within Micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA Exonuclease Trex1 Regulates Radiotherapy-Induced Tumour Immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid. Med. Cell. Longev. 2017, 2017, 8060949. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.S.; Baty, J.W.; Dong, L.-F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and Transfer of Mitochondrial DNA via Exosomes Regulate Escape from Dormancy in Hormonal Therapy-Resistant Breast Cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [Green Version]
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernández-Delgado, I.; Latorre-Pellicer, A.; Acín-Pérez, R.; Martín-Cófreces, N.B.; Jaso-Tamame, Á.L.; Iborra, S.; Jorge, I.; et al. Priming of Dendritic Cells by DNA-Containing Extracellular Vesicles from Activated T Cells through Antigen-Driven Contacts. Nat. Commun. 2018, 9, 2658. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-Induced Senescence Is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Ohtani, N.; Zebedee, Z.; Huot, T.J.; Stinson, J.A.; Sugimoto, M.; Ohashi, Y.; Sharrocks, A.D.; Peters, G.; Hara, E. Opposing Effects of Ets and Id Proteins on p16INK4a Expression during Cellular Senescence. Nature 2001, 409, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Chandrasekaran, C.; Gordon, J.I.; Beach, D.; Jacks, T.; Hannon, G.J. Radiation-Induced Cell Cycle Arrest Compromised by p21 Deficiency. Nature 1995, 377, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Maehara, K.; Yamakoshi, K.; Ohtani, N.; Kubo, Y.; Takahashi, A.; Arase, S.; Jones, N.; Hara, E. Reduction of Total E2F/DP Activity Induces Senescence-like Cell Cycle Arrest in Cancer Cells Lacking Functional pRB and p53. J. Cell Biol. 2005, 168, 553–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.-I.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic Signalling and the p16INK4a-Rb Pathway Cooperate to Enforce Irreversible Cellular Senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Takahashi, A.; Hanyu, A.; Hori, S.; Sato, S.; Naka, K.; Hirao, A.; Ohtani, N.; Hara, E. Crosstalk between the Rb Pathway and AKT Signaling Forms a Quiescence-Senescence Switch. Cell Rep. 2014, 7, 194–207. [Google Scholar] [CrossRef] [Green Version]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 Loss Is a Senescence-Associated Biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 Retrotransposition Activity in Human Genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 Drives IFN in Senescent Cells and Promotes Age-Associated Inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- Simon, M.; Van Meter, M.; Ablaeva, J.; Ke, Z.; Gonzalez, R.S.; Taguchi, T.; De Cecco, M.; Leonova, K.I.; Kogan, V.; Helfand, S.L.; et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab. 2019, 29, 871–885.e5. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-Induced Gut Microbial Metabolite Promotes Liver Cancer through Senescence Secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes Maintain Cellular Homeostasis by Excreting Harmful DNA from Cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [Green Version]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small Extracellular Vesicles Secreted from Senescent Cells Promote Cancer Cell Proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef]
- Miyata, K.; Imai, Y.; Hori, S.; Nishio, M.; Loo, T.M.; Okada, R.; Yang, L.; Nakadai, T.; Maruyama, R.; Fujii, R.; et al. Pericentromeric Noncoding RNA Changes DNA Binding of CTCF and Inflammatory Gene Expression in Senescence and Cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2025647118. [Google Scholar] [CrossRef]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell Intrinsic Immunity Spreads to Bystander Cells via the Intercellular Transfer of cGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef] [Green Version]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.N.Y.; Schmitt, C.A. Genotoxic Stress-Induced Senescence. In Cellular Senescence: Methods and Protocols; Demaria, M., Ed.; Springer: New York, NY, USA, 2019; pp. 93–105. ISBN 9781493989317. [Google Scholar]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch, E.; Christov, K.; Roninson, I.B. A Senescence-like Phenotype Distinguishes Tumor Cells That Undergo Terminal Proliferation Arrest after Exposure to Anticancer Agents. Cancer Res. 1999, 59, 3761–3767. [Google Scholar]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-Hydroxydaunomycin, a New Antitumor Antibiotic from S. Peucetius Var. Caesius. Biotechnol. Bioeng. 1969, 11, 1101–1110. [Google Scholar] [CrossRef]
- Bielak-Zmijewska, A.; Wnuk, M.; Przybylska, D.; Grabowska, W.; Lewinska, A.; Alster, O.; Korwek, Z.; Cmoch, A.; Myszka, A.; Pikula, S.; et al. A Comparison of Replicative Senescence and Doxorubicin-Induced Premature Senescence of Vascular Smooth Muscle Cells Isolated from Human Aorta. Biogerontology 2014, 15, 47–64. [Google Scholar] [CrossRef] [Green Version]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of Microtubule Assembly in Vitro by Taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Scheule, R.K.; Perkins, R.C.; Hamilton, R.; Holian, A. Bleomycin Stimulation of Cytokine Secretion by the Human Alveolar Macrophage. Am. J. Physiol. 1992, 262, L386–L391. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hornsby, P.J. Senescent Human Fibroblasts Increase the Early Growth of Xenograft Tumors via Matrix Metalloproteinase Secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Long, Q.; Fu, D.; Zhu, D.; Ji, Y.; Han, L.; Zhang, B.; Xu, Q.; Liu, B.; Li, Y.; et al. Targeting SPINK1 in the Damaged Tumour Microenvironment Alleviates Therapeutic Resistance. Nat. Commun. 2018, 9, 4315. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ Heel of Senescent Cells: From Transcriptome to Senolytic Drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of Senescent Cells by ABT263 Rejuvenates Aged Hematopoietic Stem Cells in Mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Saleh, T.; Carpenter, V.J.; Tyutyunyk-Massey, L.; Murray, G.; Leverson, J.D.; Souers, A.J.; Alotaibi, M.R.; Faber, A.C.; Reed, J.; Harada, H.; et al. Clearance of Therapy-Induced Senescent Tumor Cells by the Senolytic ABT-263 via Interference with BCL-XL -BAX Interaction. Mol. Oncol. 2020, 14, 2504–2519. [Google Scholar] [CrossRef]
- Shahbandi, A.; Rao, S.G.; Anderson, A.Y.; Frey, W.D.; Olayiwola, J.O.; Ungerleider, N.A.; Jackson, J.G. BH3 Mimetics Selectively Eliminate Chemotherapy-Induced Senescent Cells and Improve Response in TP53 Wild-Type Breast Cancer. Cell Death Differ. 2020, 27, 3097–3116. [Google Scholar] [CrossRef]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET Family Protein Degrader Provokes Senolysis by Targeting NHEJ and Autophagy in Senescent Cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR Regulates the pro-Tumorigenic Senescence-Associated Secretory Phenotype by Promoting IL1A Translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Fu, D.; Xu, Q.; Cong, X.; Wu, C.; Zhong, X.; Ma, Y.; Lv, Z.; Chen, F.; Han, L.; et al. The Senescence-Associated Secretory Phenotype Is Potentiated by Feedforward Regulatory Mechanisms Involving Zscan4 and TAK1. Nat. Commun. 2018, 9, 1723. [Google Scholar] [CrossRef] [PubMed]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell Surface-Bound IL-1alpha Is an Upstream Regulator of the Senescence-Associated IL-6/IL-8 Cytokine Network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebata, H.; Loo, T.M.; Takahashi, A. Telomere Maintenance and the cGAS-STING Pathway in Cancer. Cells 2022, 11, 1958. https://doi.org/10.3390/cells11121958
Ebata H, Loo TM, Takahashi A. Telomere Maintenance and the cGAS-STING Pathway in Cancer. Cells. 2022; 11(12):1958. https://doi.org/10.3390/cells11121958
Chicago/Turabian StyleEbata, Hiroshi, Tze Mun Loo, and Akiko Takahashi. 2022. "Telomere Maintenance and the cGAS-STING Pathway in Cancer" Cells 11, no. 12: 1958. https://doi.org/10.3390/cells11121958
APA StyleEbata, H., Loo, T. M., & Takahashi, A. (2022). Telomere Maintenance and the cGAS-STING Pathway in Cancer. Cells, 11(12), 1958. https://doi.org/10.3390/cells11121958