
PARP1 Might Substitute HSF1 to Reactivate Latent HIV-1 by Binding to Heat Shock Element

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Reagent
2.3. HSF1 Knockout Assay
2.4. HSF1 Knockdown Assay
2.5. Flow Cytometry
2.6. Western Blotting Analysis
2.7. Construction of 293T-HSE-Luc Cells
2.8. Luciferase Assays
2.9. RT-qPCR
2.10. Statistical Analysis
3. Results
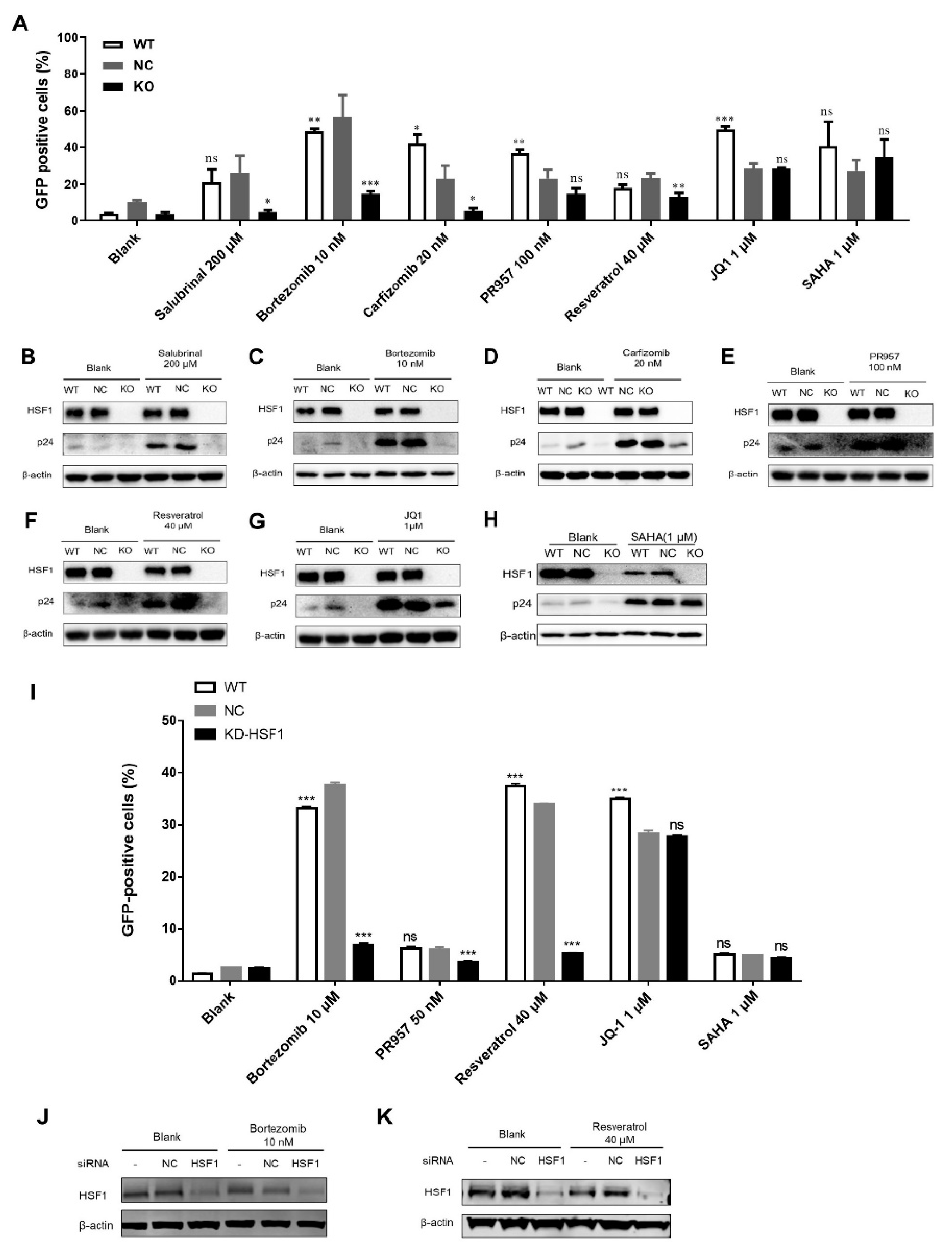
3.1. LRAs Reactivation Activity was Reversed after HSF1 Knockout
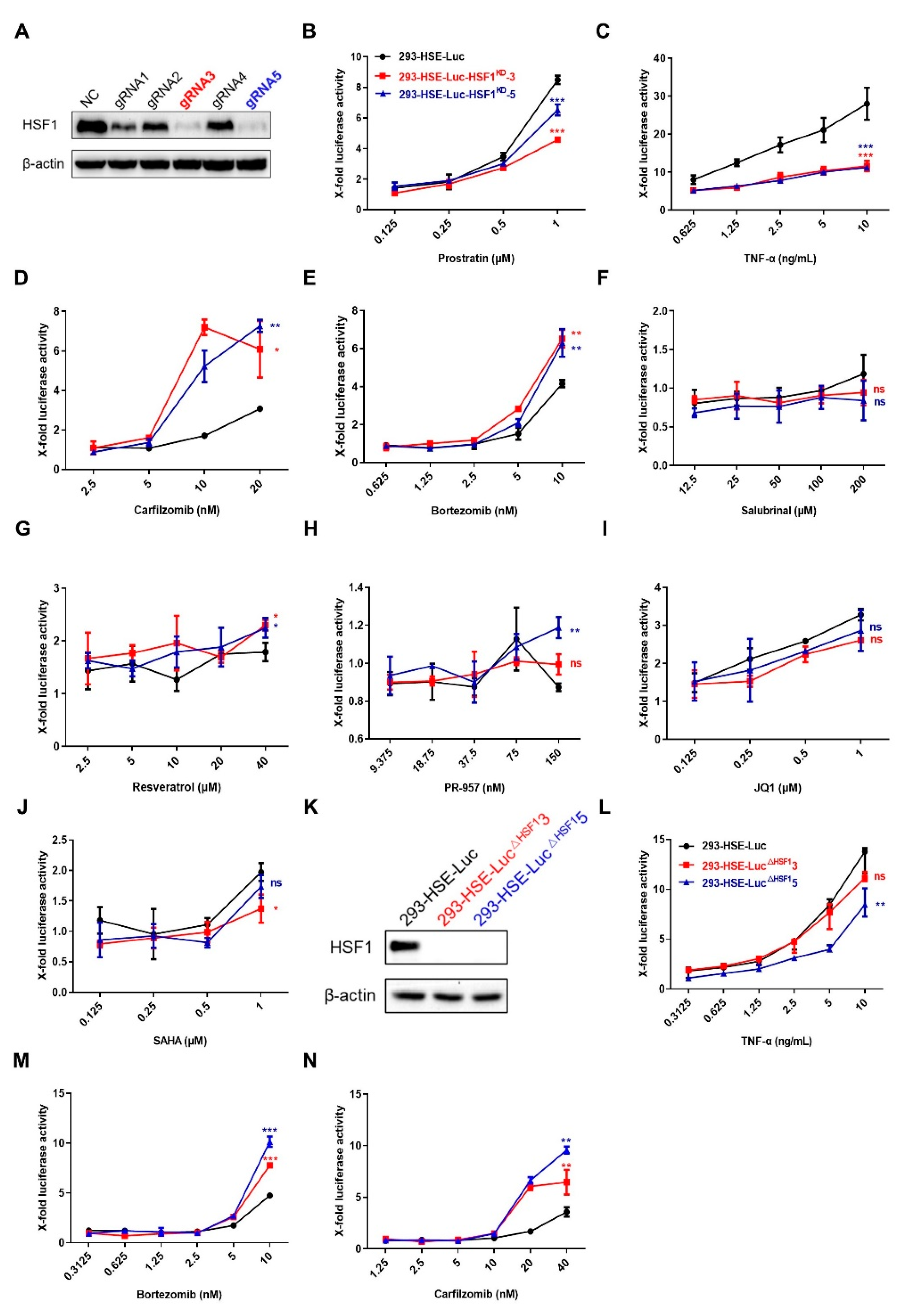
3.2. LRAs Promoted the Binding of HSF1 to HSE
3.3. LRAs Changed HSE Activity after HSF1 Deletion
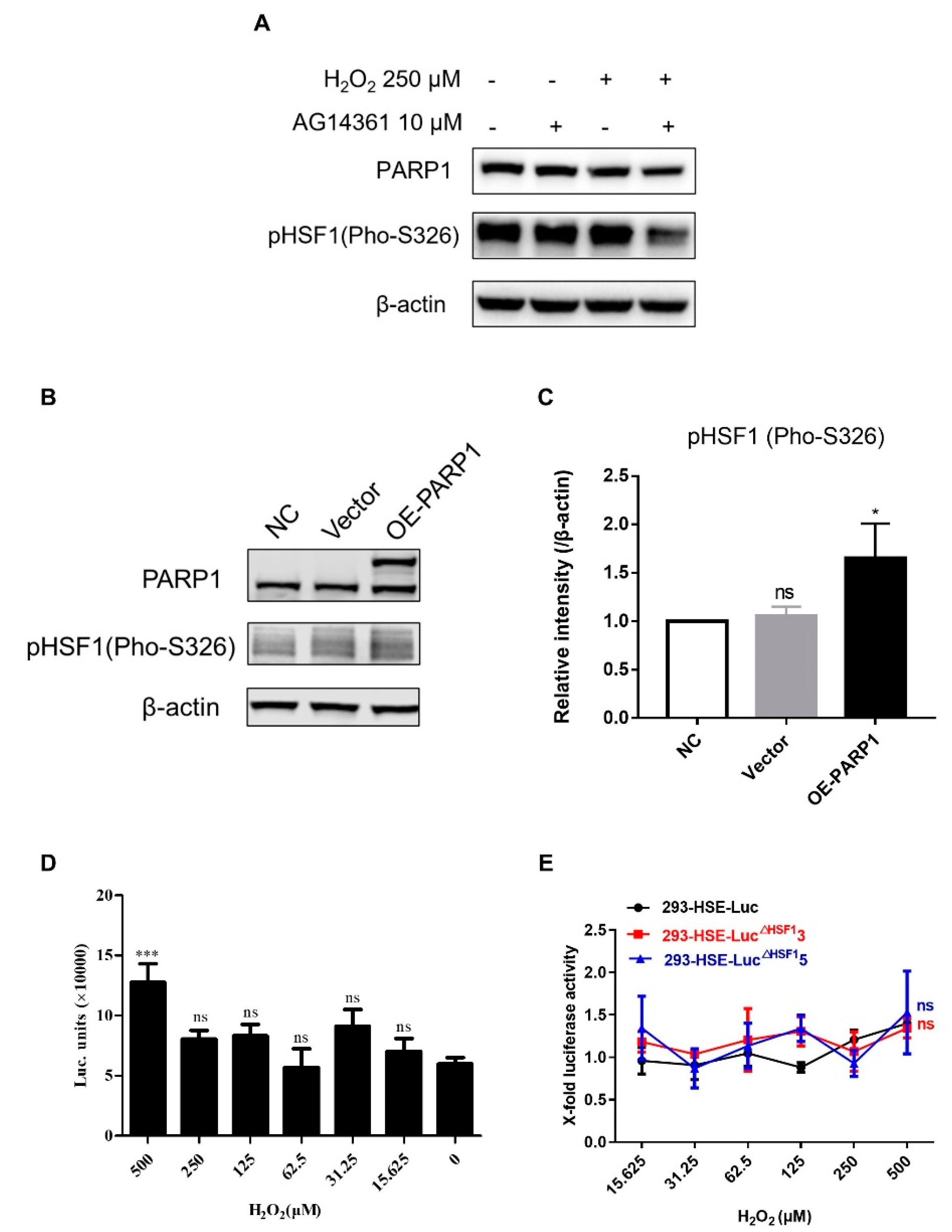
3.4. PARP1 Acted as HSF1 Potential Substitute Protein to Promote the Reactivation of HIV-1 Reservoir
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klaus, B.D.; Grodesky, M.J. HIV and HAART in 1997. Highly active antiretroviral therapy. Nurse Pract. 1997, 22, 139–142. [Google Scholar]
- Vandamme, A.M.; Van Vaerenbergh, K.; De Clercq, E. Anti-human immunodeficiency virus drug combination strategies. Antivir. Chem. Chemother. 1998, 9, 187–203. [Google Scholar] [CrossRef]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef]
- Zhao, M.; De Crignis, E.; Rokx, C.; Verbon, A.; van Gelder, T.; Mahmoudi, T.; Katsikis, P.D.; Mueller, Y.M. T cell toxicity of HIV latency reversing agents. Pharmacol. Res. 2019, 139, 524–534. [Google Scholar] [CrossRef]
- Delagreverie, H.M.; Delaugerre, C.; Lewin, S.R.; Deeks, S.G.; Li, J.Z. Ongoing Clinical Trials of Human Immunodeficiency Virus Latency-Reversing and Immunomodulatory Agents. Open Forum Infect. Dis. 2016, 3, ofw189. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Wang, S.; Guo, L.; Punekar, A.S.; Ladenstein, R.; Wang, D.C.; Liu, W. MD simulation of high-resolution X-ray structures reveals post-translational modification dependent conformational changes in HSF-DNA interaction. Protein Cell 2016, 7, 916–920. [Google Scholar] [CrossRef] [Green Version]
- Rawat, P.; Mitra, D. Cellular heat shock factor 1 positively regulates human immunodeficiency virus-1 gene expression and replication by two distinct pathways. Nucleic Acids Res. 2011, 39, 5879–5892. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.Y.; Zhao, W.; Wang, C.Y.; Lin, J.; Zeng, X.Y.; Ren, R.X.; Wang, K.; Xun, T.R.; Shai, Y.; Liu, S.W. Heat Shock Protein 90 Facilitates Latent HIV Reactivation through Maintaining the Function of Positive Transcriptional Elongation Factor b (p-TEFb) under Proteasome Inhibition. J. Biol. Chem. 2016, 291, 26177–26187. [Google Scholar] [CrossRef] [Green Version]
- Vicenzi, E.; Poli, G. Ultraviolet irradiation and cytokines as regulators of HIV latency and expression. Chem. Biol. Interact. 1994, 91, 101–109. [Google Scholar] [CrossRef]
- Iordanskiy, S.; Duyne, R.V.; Sampey, G.C.; Woodson, C.M.; Fry, K.; Saifuddin, M.; Guo, J.; Wu, Y.T.; Romerio, F.; Kashanchi, F. Therapeutic doses of irradiation activate viral transcription and induce apoptosis in HIV-1 infected cells. Virology 2015, 485, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ortner, V.; Ludwig, A.; Riegel, E.; Dunzinger, S.; Czerny, T. An artificial HSE promoter for efficient and selective detection of heat shock pathway activity. Cell Stress Chaperones 2015, 20, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, X.; Lu, W.; Xu, X.; Pan, X.; Liang, T.; Duan, S.; Chen, Y.; Li, L.; Liu, S. PR-957, a selective immunoproteasome inhibitor, reactivates latent HIV-1 through p-TEFb activation mediated by HSF-1. Biochem. Pharmacol. 2018, 156, 511–523. [Google Scholar] [CrossRef]
- Zeng, X.; Pan, X.; Xu, X.; Lin, J.; Que, F.; Tian, Y.; Li, L.; Liu, S. Resveratrol Reactivates Latent HIV through Increasing Histone Acetylation and Activating Heat Shock Factor 1. J. Agric. Food Chem. 2017, 65, 4384–4394. [Google Scholar] [CrossRef]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Chen, T.; Peng, Z.; Wang, Z.; Liu, J.; Yang, T.; Wu, L.; Liu, G.; Zhou, M.; Tong, M.; et al. Histone chaperone CAF-1 promotes HIV-1 latency by leading the formation of phase-separated suppressive nuclear bodies. EMBO J. 2021, 40, e106632. [Google Scholar] [CrossRef]
- Timmons, A.; Fray, E.; Kumar, M.; Wu, F.; Dai, W.; Bullen, C.K.; Kim, P.; Hetzel, C.; Yang, C.; Beg, S.; et al. HSF1 inhibition attenuates HIV-1 latency reversal mediated by several candidate LRAs In Vitro and Ex Vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 15763–15771. [Google Scholar] [CrossRef]
- Platt, E.J.; Wehrly, K.; Kuhmann, S.E.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 Cell Surface Concentrations on Infections by Macrophagetropic Isolates of Human Immunodeficiency Virus Type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar] [CrossRef] [Green Version]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [Green Version]
- Fossati, S.; Formentini, L.; Wang, Z.Q.; Moroni, F.; Chiarugi, A. Poly(ADP-ribosyl)ation regulates heat shock factor-1 activity and the heat shock response in murine fibroblasts. Biochem. Cell Biol. 2006, 84, 703–712. [Google Scholar] [CrossRef]
- Fujimoto, M.; Takii, R.; Katiyar, A.; Srivastava, P.; Nakai, A. Poly(ADP-Ribose) Polymerase 1 Promotes the Human Heat Shock Response by Facilitating Heat Shock Transcription Factor 1 Binding to DNA. Mol. Cell Biol. 2018, 38, e00051-18. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, M.; Takii, R.; Takaki, E.; Katiyar, A.; Nakato, R.; Shirahige, K.; Nakai, A. The HSF1-PARP13-PARP1 complex facilitates DNA repair and promotes mammary tumorigenesis. Nat. Commun. 2017, 8, 1638. [Google Scholar] [CrossRef]
- Yu, D.; Liu, R.; Yang, G.; Zhou, Q. The PARP1-Siah1 Axis Controls HIV-1 Transcription and Expression of Siah1 Substrates. Cell Rep. 2018, 23, 3741–3749. [Google Scholar] [CrossRef]
- Li, Z.; Wu, J.; Chavez, L.; Hoh, R.; Deeks, S.G.; Pillai, S.K.; Zhou, Q. Reiterative Enrichment and Authentication of CRISPRi Targets (REACT) identifies the proteasome as a key contributor to HIV-1 latency. PLoS Pathog. 2019, 15, e1007498. [Google Scholar] [CrossRef] [Green Version]
- Pang, W.; Zhang, G.; Jiang, J.; Zheng, H.; Zhang, L.; Zhang, X.; Song, J.; Zhang, M.; Zhu, J.; Lei, A.; et al. HIV-1 can infect northern pig-tailed macaques (Macaca leonina) and form viral reservoirs in vivo. Sci. Bull. 2017, 62, 1315–1324. [Google Scholar] [CrossRef]
- Archin, N.M.; Bateson, R.; Tripathy, M.K.; Crooks, A.M.; Yang, K.H.; Dahl, N.P.; Kearney, M.F.; Anderson, E.M.; Coffin, J.M.; Strain, M.C.; et al. HIV-1 expression within resting CD4+ T cells after multiple doses of vorinostat. J. Infect. Dis. 2014, 210, 728–735. [Google Scholar] [CrossRef]
- Ke, R.; Lewin, S.R.; Elliott, J.H.; Perelson, A.S. Modeling the Effects of Vorinostat In Vivo Reveals both Transient and Delayed HIV Transcriptional Activation and Minimal Killing of Latently Infected Cells. PLoS Pathog. 2015, 11, e1005237. [Google Scholar] [CrossRef]
- Lindquist, S. The heat-shock response. Annu. Rev. Biochem. 1986, 55, 1151–1191. [Google Scholar] [CrossRef]
- Cheng, X.; Belshan, M.; Ratner, L. Hsp40 facilitates nuclear import of the human immunodeficiency virus type 2 Vpx-mediated preintegration complex. J. Virol. 2008, 82, 1229–1237. [Google Scholar] [CrossRef] [Green Version]
- Low, J.S.; Fassati, A. Hsp90: A chaperone for HIV-1. Parasitology 2014, 141, 1192–1202. [Google Scholar] [CrossRef]
- Wyzewski, Z.; Gregorczyk, K.P.; Szczepanowska, J.; Szulc-Dabrowska, L. Functional role of Hsp60 as a positive regulator of human viral infection progression. Acta Virol. 2018, 62, 33–40. [Google Scholar] [CrossRef]
- Nekongo, E.E.; Ponomarenko, A.I.; Dewal, M.B.; Butty, V.L.; Browne, E.P.; Shoulders, M.D. HSF1 Activation Can Restrict HIV Replication. ACS Infect. Dis. 2020, 6, 1659–1666. [Google Scholar] [CrossRef]
- Peng, W.; Hong, Z.; Chen, X.; Gao, H.; Dai, Z.; Zhao, J.; Liu, W.; Li, D.; Deng, K. Thiostrepton Reactivates Latent HIV-1 through the p-TEFb and NF-kappaB Pathways Mediated by Heat Shock Response. Antimicrob. Agents Chemother. 2020, 64, e02328-19. [Google Scholar] [CrossRef]
- Bonner, J.J.; Chen, D.; Storey, K.; Tushan, M.; Lea, K. Structural analysis of yeast HSF by site-specific crosslinking. J. Mol. Biol. 2000, 302, 581–592. [Google Scholar] [CrossRef]
- Jaeger, A.M.; Pemble, C.W.T.; Sistonen, L.; Thiele, D.J. Structures of HSF2 reveal mechanisms for differential regulation of human heat-shock factors. Nat. Struct. Mol. Biol. 2016, 23, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Nakai, A.; Tanabe, M.; Kawazoe, Y.; Inazawa, J.; Morimoto, R.I.; Nagata, K. HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol. Cell Biol. 1997, 17, 469–481. [Google Scholar] [CrossRef] [Green Version]
- Rabindran, S.K.; Giorgi, G.; Clos, J.; Wu, C. Molecular cloning and expression of a human heat shock factor, HSF1. Proc. Natl. Acad. Sci. USA 1991, 88, 6906–6910. [Google Scholar] [CrossRef] [Green Version]
- Kroeger, P.E.; Sarge, K.D.; Morimoto, R.I. Mouse heat shock transcription factors 1 and 2 prefer a trimeric binding site but interact differently with the HSP70 heat shock element. Mol. Cell Biol. 1993, 13, 3370–3383. [Google Scholar] [CrossRef]
- Yamamoto, N.; Takemori, Y.; Sakurai, M.; Sugiyama, K.; Sakurai, H. Differential recognition of heat shock elements by members of the heat shock transcription factor family. FEBS J. 2009, 276, 1962–1974. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Lin, Y.; Zeng, X.; Yang, C.; Duan, S.; Ding, L.; Lu, W.; Lin, J.; Pan, X.; Ma, X.; et al. PARP1 Might Substitute HSF1 to Reactivate Latent HIV-1 by Binding to Heat Shock Element. Cells 2022, 11, 2331. https://doi.org/10.3390/cells11152331
Xu X, Lin Y, Zeng X, Yang C, Duan S, Ding L, Lu W, Lin J, Pan X, Ma X, et al. PARP1 Might Substitute HSF1 to Reactivate Latent HIV-1 by Binding to Heat Shock Element. Cells. 2022; 11(15):2331. https://doi.org/10.3390/cells11152331
Chicago/Turabian StyleXu, Xinfeng, Yingtong Lin, Xiaoyun Zeng, Chan Yang, Siqin Duan, Liqiong Ding, Wanzhen Lu, Jian Lin, Xiaoyan Pan, Xiancai Ma, and et al. 2022. "PARP1 Might Substitute HSF1 to Reactivate Latent HIV-1 by Binding to Heat Shock Element" Cells 11, no. 15: 2331. https://doi.org/10.3390/cells11152331
APA StyleXu, X., Lin, Y., Zeng, X., Yang, C., Duan, S., Ding, L., Lu, W., Lin, J., Pan, X., Ma, X., & Liu, S. (2022). PARP1 Might Substitute HSF1 to Reactivate Latent HIV-1 by Binding to Heat Shock Element. Cells, 11(15), 2331. https://doi.org/10.3390/cells11152331