
MicroRNAs and Their Big Therapeutic Impacts: Delivery Strategies for Cancer Intervention



Abstract
:1. Introduction
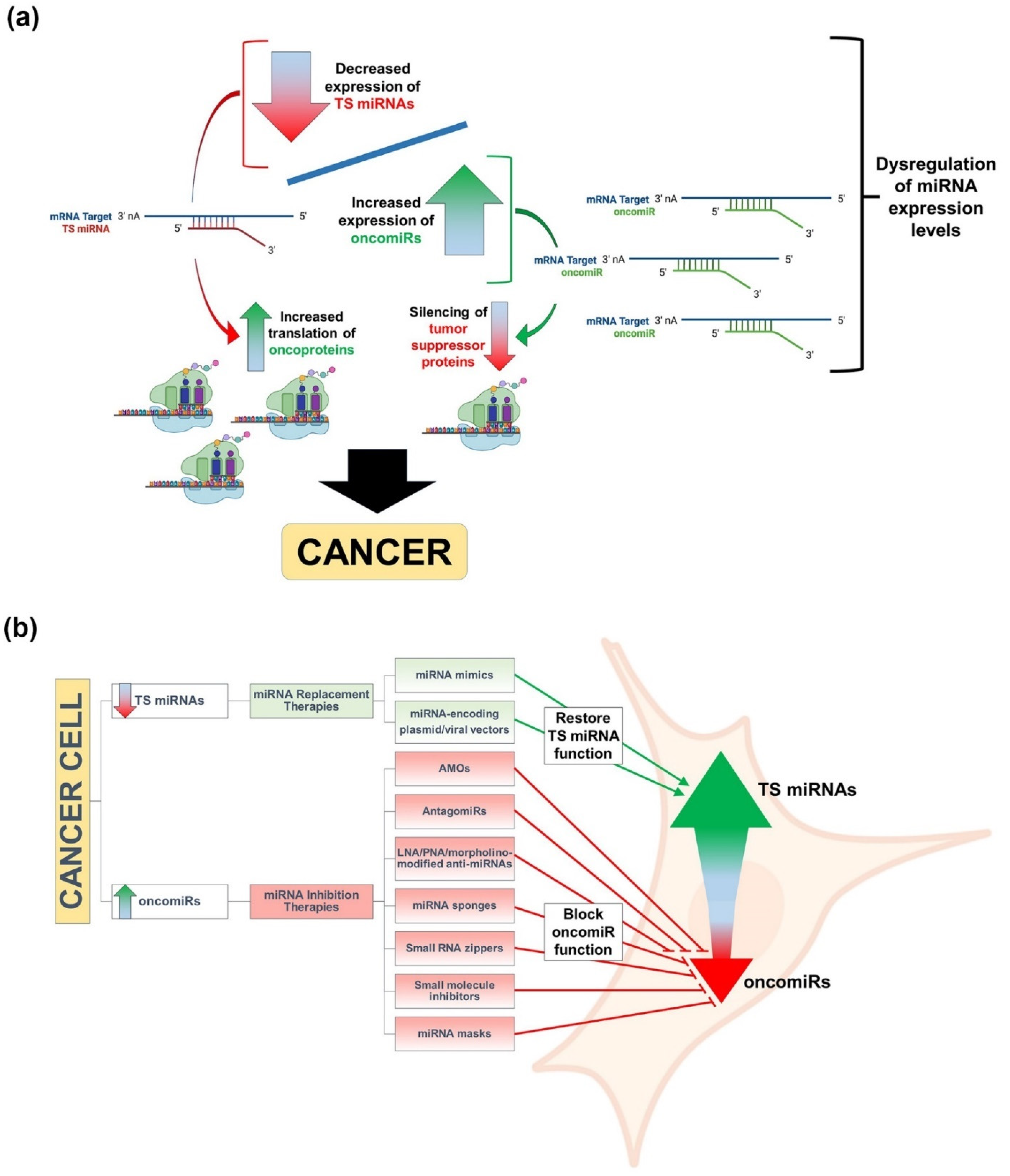
2. RNA Silencing and miRNAs
3. miRNAs, Cancer, and Therapeutic Approaches to miRNA Replacement/Inhibition
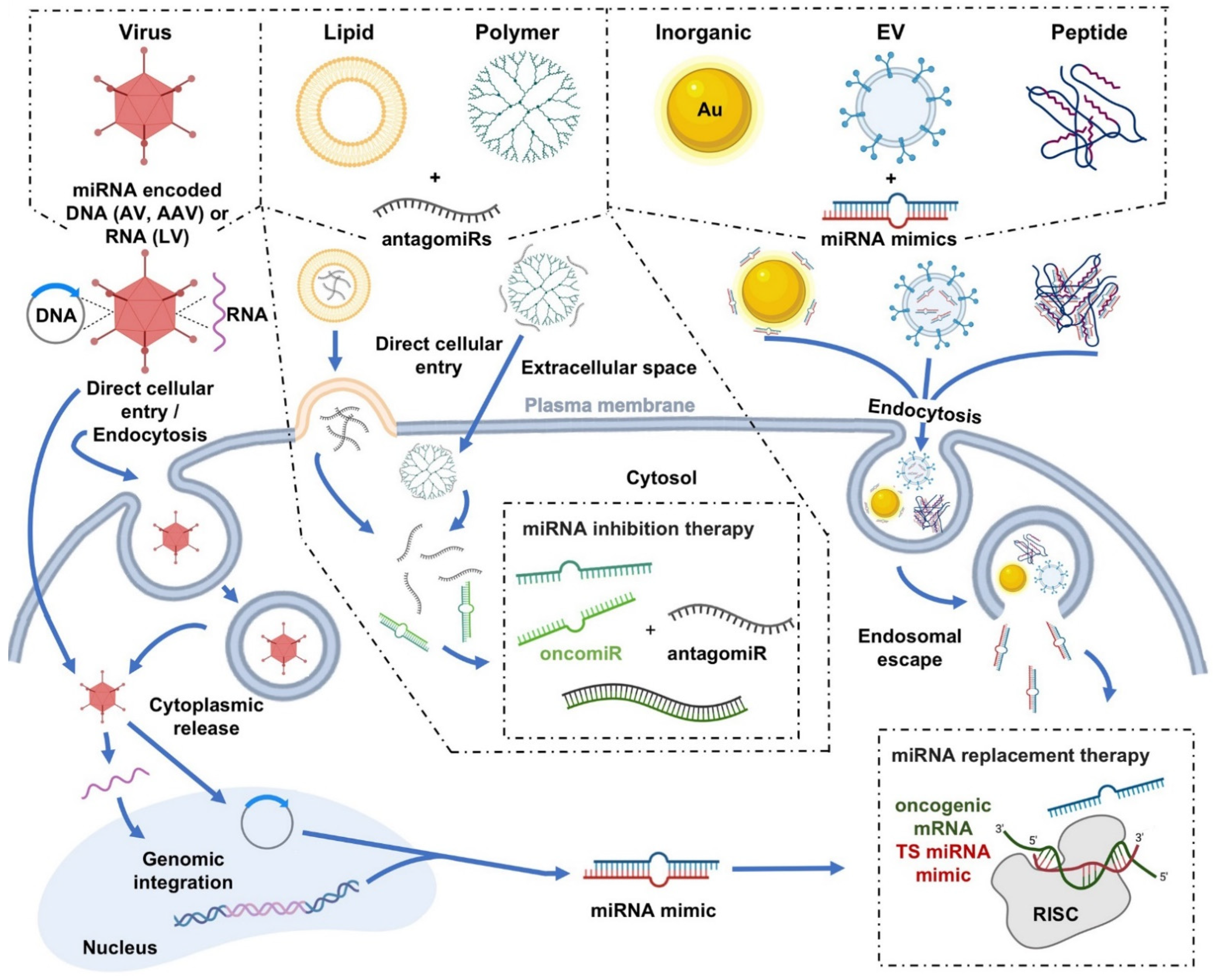
4. Delivery Platforms for miRNA-Based Cancer Therapeutics
4.1. Viral Delivery
4.2. Nonviral Delivery
4.2.1. Polymer Nanoparticles
Polyethylenimines
Polyamidoamine
Chitosan
Poly Lactic-Co-Gycolic Acid
4.2.2. Lipid-Based Nanoparticles
Cationic Liposomes
Neutral Liposomes
Ionizable Liposomes
4.2.3. Inorganic Nanoparticles
Calcium Phosphate
Silica
Gold
4.2.4. Extracellular Vesicles
4.2.5. Peptides
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. International Agency for Research on Cancer—GLOBOCAN 2020. Available online: https://gco.iarc.fr/ (accessed on 30 March 2021).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.-H.; Fu, S.-B.; Xiao, H.-S. Genome-wide analysis of microRNA and mRNA expression signatures in cancer. Acta Pharmacol. Sin. 2015, 36, 1200–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Otmani, K.; Lewalle, P. Tumor Suppressor miRNA in Cancer Cells and the Tumor Microenvironment: Mechanism of Deregulation and Clinical Implications. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar]
- Forterre, A.; Komuro, H.; Aminova, S.; Harada, M. A Comprehensive Review of Cancer MicroRNA Therapeutic Delivery Strategies. Cancers 2020, 12, 1852. [Google Scholar] [CrossRef]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, D.-Y.; Huang, L. In vivo delivery of miRNAs for cancer therapy: Challenges and strategies. Adv. Drug Deliv. Rev. 2014, 81, 128–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Cheng, Z.; Wang, Y.; Han, T. The Risks of miRNA Therapeutics: In a Drug Target Perspective. Drug Des. Dev. Ther. 2021, ume 15, 721–733. [Google Scholar] [CrossRef]
- Dasgupta, I.; Chatterjee, A. Recent Advances in miRNA Delivery Systems. Methods Protoc. 2021, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, M.S.; Sharp, P.A. Roles for MicroRNAs in Conferring Robustness to Biological Processes. Cell 2012, 149, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Michlewski, G.; Cáceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2018, 25, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebert, L.F.R.; Macrae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 287–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grässer, F.A.; Lenhof, H.-P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hücker, S.M.; Fehlmann, T.; Werno, C.; Weidele, K.; Lüke, F.; Schlenska-Lange, A.; Klein, C.A.; Keller, A.; Kirsch, S. Single-cell microRNA sequencing method comparison and application to cell lines and circulating lung tumor cells. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Macfarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [Green Version]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear Export of MicroRNA Precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [Green Version]
- Bohnsack, M.T.; Czaplinski, K.; Görlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Hutvágner, G.; Mclachlan, J.; Pasquinelli, A.E.; Bálint, É.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [Green Version]
- Grishok, A.; Pasquinelli, A.E.; Conte, D.; Li, N.; Parrish, S.; Ha, I.; Baillie, D.L.; Fire, A.; Ruvkun, G.; Mello, C.C. Genes and Mechanisms Related to RNA Interference Regulate Expression of the Small Temporal RNAs that Control C. elegans Developmental Timing. Cell 2001, 106, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Ketting, R.F.; Fischer, S.E.; Bernstein, E.; Sijen, T.; Hannon, G.J.; Plasterk, R.H. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 2001, 15, 2654–2659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, H.; Tomari, Y. RISC assembly: Coordination between small RNAs and Argonaute proteins. Biochim. Et Biophys. Acta (BBA)-Gene Regul. Mech. 2016, 1859, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, S.; Kobayashi, M.; Yoda, M.; Sakaguchi, Y.; Katsuma, S.; Suzuki, T.; Tomari, Y. Hsc70/Hsp90 Chaperone Machinery Mediates ATP-Dependent RISC Loading of Small RNA Duplexes. Mol. Cell 2010, 39, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Tomari, Y. Making RISC. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs Exhibit Strand Bias. Cell 2003, 115, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, D.S.; Hutvágner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the Assembly of the RNAi Enzyme Complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Katsura, A.; Yasuda, T.; Ueno, T.; Mano, H.; Sugimoto, K.; Miyazono, K. Small-RNA asymmetry is directly driven by mammalian Argonautes. Nat. Struct. Mol. Biol. 2015, 22, 512–521. [Google Scholar] [CrossRef]
- Frank, F.; Sonenberg, N.; Nagar, B. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature 2010, 465, 818–822. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.W.; Hannon, G.J.; Darnell, R.B. An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol. 2012, 19, 321–327. [Google Scholar] [CrossRef]
- Shin, C.; Nam, J.-W.; Farh, K.K.-H.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the MicroRNA Targeting Code: Functional Sites with Centered Pairing. Mol. Cell 2010, 38, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther.-Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.-H.; Ghoshal, K.; Villén, J.; Bartel, D.P. mRNA Destabilization Is the Dominant Effect of Mammalian MicroRNAs by the Time Substantial Repression Ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Gambari, R.; Brognara, E.; Spandidos, D.A.; Fabbri, E. Targeting oncomiRNAs and mimicking tumor suppressor miRNAs: New trends in the development of miRNA therapeutic strategies in oncology (Review). Int. J. Oncol. 2016, 49, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.W.; Ferland-McCollough, D.; Jackson, T.J.; Bushell, M. microRNAs in cancer management. Lancet Oncol. 2012, 13, e249–e258. [Google Scholar] [CrossRef]
- A Broderick, J.; Zamore, P.D. MicroRNA therapeutics. Gene Ther. 2011, 18, 1104–1110. [Google Scholar] [CrossRef]
- Bader, A.G.; Brown, D.; Winkler, M. The Promise of MicroRNA Replacement Therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander-Bryant, A.A.; Dumitriu, A.; Attaway, C.C.; Yu, H.; Jakymiw, A. Fusogenic-oligoarginine peptide-mediated silencing of the CIP2A oncogene suppresses oral cancer tumor growth in vivo. J. Control. Release 2015, 218, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daige, C.L.; Wiggins, J.F.; Priddy, L.; Nelligan-Davis, T.; Zhao, J.; Brown, D. Systemic Delivery of a miR34a Mimic as a Potential Therapeutic for Liver Cancer. Mol. Cancer Ther. 2014, 13, 2352–2360. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Feng, Y.; Zhang, R.; Zhang, W.; Shu, Y.; Zeng, Z.; Huang, S.; Zhang, L.; Huang, B.; Wu, D.; et al. A simplified system for the effective expression and delivery of functional mature microRNAs in mammalian cells. Cancer Gene Ther. 2019, 27, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Hosseinahli, N.; Aghapour, M.; Duijf, P.H.G.; Baradaran, B. Treating cancer with microRNA replacement therapy: A literature review. J. Cell. Physiol. 2018, 233, 5574–5588. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhu, Y.; Jing, A.; Wang, J.; Hu, F.; Feng, W.; Xiao, Z.; Chen, B. Cationic microRNA-delivering nanocarriers for efficient treatment of colon carcinoma in xenograft model. Gene Ther. 2016, 23, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Fei, J.; Lan, F.; Guo, M.; Li, Y.; Liu, Y. Inhibitory effects of anti-miRNA oligonucleotides (AMOs) on A549 cell growth. J. Drug Target. 2008, 16, 688–693. [Google Scholar] [CrossRef]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; A Weinberg, R. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef]
- Corsten, M.F.; Miranda, R.; Kasmieh, R.; Krichevsky, A.M.; Weissleder, R.; Shah, K. MicroRNA-21 Knockdown Disrupts Glioma Growth In vivo and Displays Synergistic Cytotoxicity with Neural Precursor Cell–Delivered S-TRAIL in Human Gliomas. Cancer Res. 2007, 67, 8994–9000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.N.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2014, 518, 107–110. [Google Scholar] [CrossRef] [Green Version]
- Chang, R.-M.; Xiao, S.; Lei, X.; Yang, H.; Fang, F.; Yang, L.-Y. miRNA-487a Promotes Proliferation and Metastasis in Hepatocellular Carcinoma. Clin. Cancer Res. 2017, 23, 2593–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, K.; Shi, Z.; Zhang, A.; Jia, Z.; Wang, G.; Pu, P.; Kang, C.; Han, L. A lentivirus-mediated miR-23b sponge diminishes the malignant phenotype of glioma cells in vitro and in vivo. Oncol. Rep. 2014, 31, 1573–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monroig, P.D.C.; Chen, L.; Zhang, S.; Calin, G.A. Small molecule compounds targeting miRNAs for cancer therapy. Adv. Drug Deliv. Rev. 2014, 81, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ors-Kumoglu, G.; Gulce-Iz, S.; Biray-Avci, C. Therapeutic microRNAs in human cancer. Cytotechnology 2019, 71, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Ju, J.; Hou, L.; Li, Z.; Xiao, D.; Li, Y.; Yao, J.; Wang, C.; Zhang, Y.; et al. Downregulation of miR-522 suppresses proliferation and metastasis of non-small cell lung cancer cells by directly targeting DENN/MADD domain containing 2D. Sci. Rep. 2016, 6, srep19346. [Google Scholar] [CrossRef] [Green Version]
- Miroshnichenko, S.; Patutina, O. Enhanced Inhibition of Tumorigenesis Using Combinations of miRNA-Targeted Therapeutics. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Liu, C.; Lü, J.; Zhao, Q.; Deng, S.; Wang, G.; Qiao, J.; Zhang, C.; Zhen, L.; Lu, Y.; et al. Small RNA zippers lock miRNA molecules and block miRNA function in mammalian cells. Nat. Commun. 2017, 8, 13964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kootstra, N.A.; Verma, I.M. Gene Therapy with Viral Vectors. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Donahue, N.D.; Acar, H.; Wilhelm, S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv. Drug Deliv. Rev. 2019, 143, 68–96. [Google Scholar] [CrossRef]
- Ang, L.; Guo, L.; Wang, J.; Huang, J.; Lou, X.; Zhao, M. Oncolytic virotherapy armed with an engineered interfering lncRNA exhibits antitumor activity by blocking the epithelial mesenchymal transition in triple-negative breast cancer. Cancer Lett. 2020, 479, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Tan, G.; Jiang, X.; Han, P.; Zhai, B.; Dong, X.; Qiao, H.; Jiang, H.; Sun, X. An artificial lncRNA targeting multiple miRNAs overcomes sorafenib resistance in hepatocellular carcinoma cells. Oncotarget 2016, 7, 73257–73269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Q.; Basnet, S.; Dai, Z.; Li, S.; Zhang, Z.; Ge, H. A novel E1B55kDa-deleted oncolytic adenovirus carrying microRNA-143 exerts specific antitumor efficacy on colorectal cancer cells. Am. J. Transl. Res. 2016, 8, 3822–3830. [Google Scholar] [PubMed]
- Callegari, E.; Elamin, B.K.; D’Abundo, L.; Falzoni, S.; Donvito, G.; Moshiri, F.; Milazzo, M.; Altavilla, G.; Giacomelli, L.; Fornari, F.; et al. Anti-Tumor Activity of a miR-199-dependent Oncolytic Adenovirus. PLoS ONE 2013, 8, e73964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, W.; Chen, Q.; Ma, L.; Liu, J.; Yang, Z.; Shen, J.; Cui, Y.; Bian, X.-W.; Qian, C. Oncolytic adenovirus co-expressing miRNA-34a and IL-24 induces superior antitumor activity in experimental tumor model. Klin. Wochenschr. 2013, 91, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Komoll, R.-M.; Hu, Q.; Olarewaju, O.; von Döhlen, L.; Yuan, Q.; Xie, Y.; Tsay, H.-C.; Daon, J.; Qin, R.; Manns, M.P.; et al. MicroRNA-342-3p is a potent tumour suppressor in hepatocellular carcinoma. J. Hepatol. 2020, 74, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.-W.; Chang, T.-C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhere, D.; Arghiani, N.; Lechtich, E.R.; Yao, Y.; Alsaab, S.; Bei, F.; Matin, M.M.; Shah, K. Simultaneous downregulation of miR-21 and upregulation of miR-7 has anti-tumor efficacy. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasar, S.; Salerno, E.; Yuan, Y.; Underbayev, C.; Vollenweider, D.; Laurindo, M.F.; Fernandes, H.; Bonci, D.; Addario, A.; Mazzella, F.; et al. Systemic in vivo lentiviral delivery of miR-15a/16 reduces malignancy in the NZB de novo mouse model of chronic lymphocytic leukemia. Genes Immun. 2011, 13, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lai, L.; Chen, Q.; Song, Y.; Xu, S.; Ma, F.; Wang, X.; Wang, J.; Yu, H.; Cao, X.; et al. MicroRNA-494 Is Required for the Accumulation and Functions of Tumor-Expanded Myeloid-Derived Suppressor Cells via Targeting of PTEN. J. Immunol. 2012, 188, 5500–5510. [Google Scholar] [CrossRef]
- Fernandez, C.A.; Rice, K.G. Engineered Nanoscaled Polyplex Gene Delivery Systems. Mol. Pharm. 2009, 6, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.I.v.D.; Yun, C.-O.; Schiffelers, R.M.; Hennink, W.E. Polymeric delivery systems for nucleic acid therapeutics: Approaching the clinic. J. Control. Release 2021, 331, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Höbel, S.; Aigner, A. Polyethylenimines for siRNA and miRNA delivery in vivo. WIREs Nanomed. Nanobiotechnology 2013, 5, 484–501. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.F.; Weirauch, U.; Thomas, M.; Grünweller, A.; Hartmann, R.K.; Aigner, A. MicroRNA Replacement Therapy for miR-145 and miR-33a Is Efficacious in a Model of Colon Carcinoma. Cancer Res. 2011, 71, 5214–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Liu, T.; Fang, O.; Dong, W.; Zhang, F.; Leach, L.; Hu, X.; Luo, Z. MicroRNA-708-5p acts as a therapeutic agent against metastatic lung cancer. Oncotarget 2015, 7, 2417–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Wang, K.; Sun, X.; Li, Y.; Fu, Q.; Liang, T.; Tang, G. A redox-sensitive, oligopeptide-guided, self-assembling, and efficiency-enhanced (ROSE) system for functional delivery of microRNA therapeutics for treatment of hepatocellular carcinoma. Biomaterials 2016, 104, 192–200. [Google Scholar] [CrossRef]
- Biray Avcı, Ç.; Özcan, İ.; Balcı, T.; Özer, Ö.; Gündüz, C. Design of polyethylene glycol-polyethylenimine nanocomplexes as non-viral carriers: Mir-150 delivery to chronic myeloid leukemia cells. Cell Biol. Int. 2013, 37, 1205–1214. [Google Scholar] [CrossRef]
- Talekar, M.; Trivedi, M.; Shah, P.; Ouyang, Q.; Oka, A.; Gandham, S.; Amiji, M.M. Combination wt-p53 and MicroRNA-125b Transfection in a Genetically Engineered Lung Cancer Model Using Dual CD44/EGFR-targeting Nanoparticles. Mol. Ther. 2016, 24, 759–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-P.; Chien, Y.; Chiou, G.-Y.; Cherng, J.-Y.; Wang, M.-L.; Lo, W.-L.; Chang, Y.-L.; Huang, P.-I.; Chen, Y.-W.; Shih, Y.-H.; et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials 2012, 33, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Dorrance, A.M.; Neviani, P.; Ferenchak, G.J.; Huang, X.; Nicolet, D.; Maharry, K.S.; Ozer, H.G.; Hoellarbauer, P.; Khalife, J.; Hill, E.B.; et al. Targeting leukemia stem cells in vivo with antagomiR-126 nanoparticles in acute myeloid leukemia. Leukemia 2015, 29, 2143–2153. [Google Scholar] [CrossRef] [Green Version]
- Duncan, R.; Izzo, L. Dendrimer biocompatibility and toxicity. Adv. Drug Deliv. Rev. 2005, 57, 2215–2237. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, X.; Guo, C.; Ren, D.; Zhao, Y.; Xiao, W.; Jiao, W. Aptamer-Dendrimer Bioconjugates for Targeted Delivery of miR-34a Expressing Plasmid and Antitumor Effects in Non-Small Cell Lung Cancer Cells. PLoS ONE 2015, 10, e0139136. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, B.; Zhou, L.; Shi, Y.; Li, Z.; Xia, Y.; Tian, J. Imaging Dendrimer-Grafted Graphene Oxide Mediated Anti-miR-21 Delivery With an Activatable Luciferase Reporter. ACS Appl. Mater. Interfaces 2016, 8, 9014–9021. [Google Scholar] [CrossRef] [PubMed]
- Ravi Kumar, M.N.V. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46, 1–27. [Google Scholar] [CrossRef]
- Martirosyan, A.; Olesen, M.J.; Howard, K.A. Chitosan-Based Nanoparticles for Mucosal Delivery of RNAi Therapeutics. Adv. Genet. 2014, 88, 325–352. [Google Scholar] [CrossRef]
- Kaban, K.; Salva, E.; Akbuga, J. The effects of chitosan/miR-200c nanoplexes on different stages of cancers in breast cancer cell lines. Eur. J. Pharm. Sci. 2016, 95, 103–110. [Google Scholar] [CrossRef]
- Kaban, K.; Salva, E.; Akbuga, J. In Vitro Dose Studies on Chitosan Nanoplexes for microRNA Delivery in Breast Cancer Cells. Nucleic Acid Ther. 2017, 27, 45–55. [Google Scholar] [CrossRef]
- Gaur, S.; Wen, Y.; Song, J.H.; Parikh, N.U.; Mangala, L.S.; Blessing, A.M.; Ivan, C.; Wu, S.Y.; Varkaris, A.; Shi, Y.; et al. Chitosan nanoparticle-mediated delivery of miRNA-34a decreases prostate tumor growth in the bone and its expression induces non-canonical autophagy. Oncotarget 2015, 6, 29161–29177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.J.; Saltzman, W.M. Polymer Nanoparticle-Mediated Delivery of MicroRNA Inhibition and Alternative Splicing. Mol. Pharm. 2012, 9, 1481–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosco, D.; Cilurzo, F.; Maiuolo, J.; Federico, C.; Di Martino, M.T.; Cristiano, M.C.; Tassone, P.; Fresta, M.; Paolino, D. Delivery of miR-34a by chitosan/PLGA nanoplexes for the anticancer treatment of multiple myeloma. Sci. Rep. 2015, 5, 17579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devulapally, R.; Foygel, K.; Sekar, T.V.; Willmann, J.K.; Paulmurugan, R. Gemcitabine and Antisense-microRNA Co-encapsulated PLGA–PEG Polymer Nanoparticles for Hepatocellular Carcinoma Therapy. ACS Appl. Mater. Interfaces 2016, 8, 33412–33422. [Google Scholar] [CrossRef] [Green Version]
- Devulapally, R.; Sekar, N.M.; Sekar, T.V.; Foygel, K.; Massoud, T.F.; Willmann, J.K.; Paulmurugan, R. Polymer Nanoparticles Mediated Codelivery of AntimiR-10b and AntimiR-21 for Achieving Triple Negative Breast Cancer Therapy. ACS Nano 2015, 9, 2290–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devulapally, R.; Sekar, T.V.; Paulmurugan, R. Formulation of Anti-miR-21 and 4-Hydroxytamoxifen Co-loaded Biodegradable Polymer Nanoparticles and Their Antiproliferative Effect on Breast Cancer Cells. Mol. Pharm. 2015, 12, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-Y.; Choe, J.W.; Pu, K.; Devulapally, R.; Bachawal, S.; Machtaler, S.; Chowdhury, S.M.; Luong, R.; Tian, L.; Khuri-Yakub, B.; et al. Ultrasound-guided delivery of microRNA loaded nanoparticles into cancer. J. Control. Release 2015, 203, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewert, K.K.; Scodeller, P.; Simón-Gracia, L.; Steffes, V.M.; Wonder, E.A.; Teesalu, T.; Safinya, C.R. Cationic Liposomes as Vectors for Nucleic Acid and Hydrophobic Drug Therapeutics. Pharmaceutics 2021, 13, 1365. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Mislick, K.; Baldeschwieler, J.D. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc. Natl. Acad. Sci. USA 1996, 93, 12349–12354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Lee, R.J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yan, X.; Jacobson, O.; Sun, W.; Wang, Z.; Tong, X.; Xia, Y.; Ling, D.; Chen, X. Improved Tumor Uptake by Optimizing Liposome Based RES Blockade Strategy. Theranostics 2017, 7, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, J.; Le, V.M.; Gong, Q.; Li, S.; Gao, F.; Ni, L.; Liu, J.; Liang, X. EpCAM Aptamer-Functionalized Cationic Liposome-Based Nanoparticles Loaded with miR-139-5p for Targeted Therapy in Colorectal Cancer. Mol. Pharm. 2019, 16, 4696–4710. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Liang, H.; Yao, W.; Wang, N.; Zhang, S.; Yan, X.; Feng, H.; Pang, W.; Wang, Y.; Wang, X.; et al. MiR-143 and MiR-145 Regulate IGF1R to Suppress Cell Proliferation in Colorectal Cancer. PLoS ONE 2014, 9, e114420. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.; Takigawa, N.; Ito, S.; Kashihara, H.; Ichihara, E.; Yasuda, T.; Shimizu, K.; Tanimoto, M.; Kiura, K. Liposomal Delivery of MicroRNA-7–Expressing Plasmid Overcomes Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor-Resistance in Lung Cancer Cells. Mol. Cancer Ther. 2011, 10, 1720–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Crawford, M.; Mao, Y.; Lee, R.J.; Davis, I.C.; Elton, T.S.; Lee, L.J.; Nana-Sinkam, S.P. Therapeutic Delivery of MicroRNA-29b by Cationic Lipoplexes for Lung Cancer. Mol. Ther.-Nucleic Acids 2013, 2, e84. [Google Scholar] [CrossRef]
- Hattori, Y.; Suzuki, S.; Kawakami, S.; Yamashita, F.; Hashida, M. The role of dioleoylphosphatidylethanolamine (DOPE) in targeted gene delivery with mannosylated cationic liposomes via intravenous route. J. Control. Release 2005, 108, 484–495. [Google Scholar] [CrossRef]
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine 2011, 6, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Thewalt, J.L.; Bloom, M. Phosphatidylcholine: Cholesterol phase diagrams. Biophys. J. 1992, 63, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Semple, S.C.; Chonn, A.; Cullis, P.R. Influence of Cholesterol on the Association of Plasma Proteins with Liposomes. Biochemistry 1996, 35, 2521–2525. [Google Scholar] [CrossRef]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic Delivery of Tumor Suppressor microRNA Mimics Using a Neutral Lipid Emulsion Inhibits Lung Tumors in Mice. Mol. Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2016, 35, 180–188. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Cortez, M.A.; Valdecanas, D.; Zhang, X.; Zhan, Y.; Bhardwaj, V.; Calin, G.A.; Komaki, R.; Giri, D.K.; Quini, C.C.; Wolfe, T.; et al. Therapeutic Delivery of miR-200c Enhances Radiosensitivity in Lung Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1494–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Antonellis, P.; Liguori, L.; Falanga, A.; Carotenuto, M.; Ferrucci, V.; Andolfo, I.; Marinaro, F.; Scognamiglio, I.; Virgilio, A.; De Rosa, G.; et al. MicroRNA 199b-5p delivery through stable nucleic acid lipid particles (SNALPs) in tumorigenic cell lines. Naunyn-Schmiedebergs Arch. fur Exp. Pathol. und Pharmakol. 2013, 386, 287–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labatut, A.E.; Mattheolabakis, G. Non-viral based miR delivery and recent developments. Eur. J. Pharm. Biopharm. 2018, 128, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Epple, M. Inorganic Nanoparticles as Carriers of Nucleic Acids into Cells. Angew. Chem. Int. Ed. 2008, 47, 1382–1395. [Google Scholar] [CrossRef]
- Morimoto, Y.; Mizushima, T.; Wu, X.; Okuzaki, D.; Yokoyama, Y.; Inoue, A.; Hata, T.; Hirose, H.; Qian, Y.; Wang, J.; et al. miR-4711-5p regulates cancer stemness and cell cycle progression via KLF5, MDM2 and TFDP1 in colon cancer cells. Br. J. Cancer 2020, 122, 1037–1049. [Google Scholar] [CrossRef]
- Hiraki, M.; Nishimura, J.; Takahashi, H.; Wu, X.; Takahashi, Y.; Miyo, M.; Nishida, N.; Uemura, M.; Hata, T.; Takemasa, I.; et al. Concurrent Targeting of KRAS and AKT by MiR-4689 Is a Novel Treatment Against Mutant KRAS Colorectal Cancer. Mol. Ther.-Nucleic Acids 2015, 4, e231. [Google Scholar] [CrossRef]
- Inoue, A.; Mizushima, T.; Wu, X.; Okuzaki, D.; Kambara, N.; Ishikawa, S.; Wang, J.; Qian, Y.; Hirose, H.; Yokoyama, Y.; et al. A miR-29b Byproduct Sequence Exhibits Potent Tumor-Suppressive Activities via Inhibition of NF-κB Signaling in KRAS-Mutant Colon Cancer Cells. Mol. Cancer Ther. 2018, 17, 977–987. [Google Scholar] [CrossRef] [Green Version]
- Mamaeva, V.; Sahlgren, C.; Lindén, M. Mesoporous silica nanoparticles in medicine—Recent advances. Adv. Drug Deliv. Rev. 2013, 65, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Slowing, I.I.; Vivero-Escoto, J.L.; Wu, C.-W.; Lin, V.S.-Y. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv. Drug Deliv. Rev. 2008, 60, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, A.; Prasetyanto, E.A.; Septiadi, D.; Manicardi, A.; Brognara, E.; Gambari, R.; Corradini, R.; De Cola, L. Combined Delivery of Temozolomide and Anti-miR221 PNA Using Mesoporous Silica Nanoparticles Induces Apoptosis in Resistant Glioma Cells. Small 2015, 11, 5687–5695. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Chen, L.; Zhang, J.; Han, L.; Zhang, A.; Zhang, C.; Zheng, Y.; Jiang, T.; Pu, P.; Kang, C. Downregulation of miR-221/222 sensitizes glioma cells to temozolomide by regulating apoptosis independently of p53 status. Oncol. Rep. 2011, 27, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.-C.; Ballard, T.E.; Ackerson, C.J.; Feldheim, D.L.; Margolis, D.M.; Melander, C. Inhibition of HIV Fusion with Multivalent Gold Nanoparticles. J. Am. Chem. Soc. 2008, 130, 6896–6897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, J.A.; Overton, K.W.; Speight, M.E.; Oldenburg, C.N.; Loo, L.; Robarge, W.; Franzen, S.; Feldheim, D.L. Cellular Uptake of Gold Nanoparticles Passivated with BSA−SV40 Large T Antigen Conjugates. Anal. Chem. 2007, 79, 9150–9159. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Saha, K.; Kim, C.; Rotello, V.M. The Role of Surface Functionality in Determining Nanoparticle Cytotoxicity. Accounts Chem. Res. 2013, 46, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chompoosor, A.; Saha, K.; Ghosh, P.S.; Macarthy, D.J.; Miranda, O.R.; Zhu, Z.-J.; Arcaro, K.F.; Rotello, V.M. The Role of Surface Functionality on Acute Cytotoxicity, ROS Generation and DNA Damage by Cationic Gold Nanoparticles. Small 2010, 6, 2246–2249. [Google Scholar] [CrossRef] [Green Version]
- De Jong, W.H.; Hagens, W.I.; Krystek, P.; Burger, M.C.; Sips, A.J.; Geertsma, R.E. Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomaterials 2008, 29, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Perrett, S.; Nie, G. Understanding the Particokinetics of Engineered Nanomaterials for Safe and Effective Therapeutic Applications. Small 2012, 9, 1619–1634. [Google Scholar] [CrossRef]
- Daniel, M.-C.; Astruc, D. Gold Nanoparticles: Assembly, Supramolecular Chemistry, Quantum-Size-Related Properties, and Applications toward Biology, Catalysis, and Nanotechnology. Chem. Rev. 2003, 104, 293–346. [Google Scholar] [CrossRef]
- Xue, H.-Y.; Liu, Y.; Liao, J.-Z.; Lin, J.-S.; Li, B.; Yuan, W.-G.; Lee, R.J.; Li, L.; Xu, C.-R.; He, X.-X. Gold nanoparticles delivered miR-375 for treatment of hepatocellular carcinoma. Oncotarget 2016, 7, 86675–86686. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Singh, L.C.; Shohet, J.M.; Gunaratne, P.H. A gold nanoparticle platform for the delivery of functional microRNAs into cancer cells. Biomaterials 2012, 34, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Gilam, A.; Conde, J.; Weissglas-Volkov, D.; Oliva, N.; Friedman, E.; Artzi, N.; Shomron, N. Local microRNA delivery targets Palladin and prevents metastatic breast cancer. Nat. Commun. 2016, 7, 12868. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Huo, M.; Li, B.; Wang, W.; Piao, H.; Wang, Y.; Zhu, Z.; Li, D.; Wang, T.; Liu, K. The Role of Exosomes and Exosomal MicroRNA in Cardiovascular Disease. Front. Cell Dev. Biol. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- De Jong, O.G.; Kooijmans, S.A.A.; Murphy, D.E.; Jiang, L.; Evers, M.J.W.; Sluijter, J.P.G.; Vader, P.; Schiffelers, R.M. Drug Delivery with Extracellular Vesicles: From Imagination to Innovation. Acc. Chem. Res. 2019, 52, 1761–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef]
- Escrevente, C.; Keller, S.; Altevogt, P.; Costa, J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer 2011, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Akao, Y.; Iio, A.; Itoh, T.; Noguchi, S.; Itoh, Y.; Ohtsuki, Y.; Naoe, T. Microvesicle-mediated RNA Molecule Delivery System Using Monocytes/Macrophages. Mol. Ther. 2011, 19, 395–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Ríos, A.J.; Molina-Crespo, Á.; Bouzo, B.L.; López-López, R.; Moreno-Bueno, G.; De La Fuente, M. Exosome-mimetic nanoplatforms for targeted cancer drug delivery. J. Nanobiotechnol. 2019, 17, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Sawada, K.; Miyamoto, M.; Shimizu, A.; Yamamoto, M.; Kinose, Y.; Nakamura, K.; Kawano, M.; Kodama, M.; Hashimoto, K.; et al. Exploring the potential of engineered exosomes as delivery systems for tumor-suppressor microRNA replacement therapy in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 527, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.-I.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Langel, Ü. Cell-Penetrating Peptides; Springer: New York, NY, USA, 2015; Volume 1324. [Google Scholar]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, W.B.; Fuselier, T.; He, J.; Wimley, W.C. Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci. 2015, 40, 749–764. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.C.; Zhang, H.; Jakymiw, A. Peptide carriers to the rescue: Overcoming the barriers to siRNA delivery for cancer treatment. Transl. Res. 2019, 214, 92–104. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Saleh, M.N.; Infante, J.R.; Goel, S.; Falchook, G.S.; Shapiro, G.; Chung, K.Y.; Conry, R.M.; Hong, D.S.; Wang, J.S.-Z.; et al. Phase I trial of a novel stapled peptide ALRN-6924 disrupting MDMX- and MDM2-mediated inhibition of WT p53 in patients with solid tumors and lymphomas. J. Clin. Oncol. 2017, 35, 2505. [Google Scholar] [CrossRef]
- Søgaard, C.K.; Blindheim, A.; Røst, L.M.; Petrović, V.; Nepal, A.; Bachke, S.; Liabakk, N.-B.; Gederaas, O.A.; Viset, T.; Arum, C.-J.; et al. “Two hits - one stone”; increased efficacy of cisplatin-based therapies by targeting PCNA’s role in both DNA repair and cellular signaling. Oncotarget 2018, 9, 32448–32465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrouss, I.; Nemati, F.; Roncal, F.; Wislez, M.; Dorgham, K.; Vallerand, D.; Rabbe, N.; Karboul, N.; Carlotti, F.; Bravo, J.; et al. Specific Targeting of Caspase-9/PP2A Interaction as Potential New Anti-Cancer Therapy. PLoS ONE 2013, 8, e60816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lulla, R.R.; Goldman, S.; Yamada, T.; Beattie, C.W.; Bressler, L.; Pacini, M.; Pollack, I.F.; Fisher, P.G.; Packer, R.J.; Dunkel, I.J.; et al. Phase I trial of p28 (NSC745104), a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in pediatric patients with recurrent or progressive central nervous system tumors: A Pediatric Brain Tumor Consortium Study. Neuro-Oncology 2016, 18, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Bennett, G.; Lutz, R.; Park, P.; Harrison, H.; Lee, K. Abstract 1167: Development of BT1718, a novel Bicycle Drug Conjugate for the treatment of lung cancer. Cancer Res. 2017, 77, 1167. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Vunk, B.; Langel, Ü. Status update in the use of cell-penetrating peptides for the delivery of macromolecular therapeutics. Expert Opin. Biol. Ther. 2020, 21, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Srimanee, A.; Arvanitidou, M.; Kim, K.; Hällbrink, M.; Langel, Ü. Cell-penetrating peptides for siRNA delivery to glioblastomas. Peptides 2018, 104, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Lehto, T.; Ezzat, K.; Wood, M.J.A.; EL Andaloussi, S. Peptides for nucleic acid delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, X.; Wang, Y.; Yu, T.; Huang, L.; Chen, L.; Li, J.; Zhang, C.; Zhang, Y. Multi-Functional Peptide-MicroRNA Nanocomplex for Targeted MicroRNA Delivery and Function Imaging. Chem.–A Eur. J. 2018, 24, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhao, Y.; Gong, A.; Miao, W.; Yan, L.; Nie, P.; Wang, Z. Improved Cellular Delivery of Antisense Oligonucleotide for miRNA-21 Imaging In Vivo Using Cell-Penetrating Peptide-Based Nanoprobes. Mol. Pharm. 2021, 18, 787–795. [Google Scholar] [CrossRef]
- Suh, J.S.; Lee, J.Y.; Choi, Y.S.; Chong, P.C.; Park, Y.J. Peptide-mediated intracellular delivery of miRNA-29b for osteogenic stem cell differentiation. Biomaterials 2013, 34, 4347–4359. [Google Scholar] [CrossRef] [PubMed]
- Urgard, E.; Lorents, A.; Klaas, M.; Padari, K.; Viil, J.; Runnel, T.; Langel, K.; Kingo, K.; Tkaczyk, E.; Langel, Ü.; et al. Pre-administration of PepFect6-microRNA-146a nanocomplexes inhibits inflammatory responses in keratinocytes and in a mouse model of irritant contact dermatitis. J. Control. Release 2016, 235, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Cantini, L.; Attaway, C.C.; Butler, B.; Andino, L.M.; Sokolosky, M.L.; Jakymiw, A. Fusogenic-Oligoarginine Peptide-Mediated Delivery of siRNAs Targeting the CIP2A Oncogene into Oral Cancer Cells. PLoS ONE 2013, 8, e73348. [Google Scholar] [CrossRef] [Green Version]
- Alexander-Bryant, A.A.; Zhang, H.; Attaway, C.C.; Pugh, W.; Eggart, L.; Sansevere, R.M.; Andino, L.M.; Dinh, L.; Cantini, L.P.; Jakymiw, A. Dual peptide-mediated targeted delivery of bioactive siRNAs to oral cancer cells in vivo. Oral Oncol. 2017, 72, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Holjencin, C.E.; Feinberg, C.R.; Hedrick, T.; Halsey, G.; Williams, R.D.; Patel, P.V.; Biles, E.; Cummings, J.C.; Wagner, C.; Vyavahare, N.; et al. Advancing peptide siRNA-carrier designs through L/D-amino acid stereochemical modifications to enhance gene silencing. Mol. Ther.-Nucleic Acids 2021, 24, 462–476. [Google Scholar] [CrossRef]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; Von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef]
- Lehmann, M.J.; Sherer, N.M.; Marks, C.B.; Pypaert, M.; Mothes, W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 2005, 170, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Lidke, D.S.; Lidke, K.A.; Rieger, B.; Jovin, T.M.; Arndt-Jovin, D.J. Reaching out for signals. J. Cell Biol. 2005, 170, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Rehman, Z.U.; Sjollema, K.A.; Kuipers, J.; Hoekstra, D.; Zuhorn, I.S. Nonviral Gene Delivery Vectors Use Syndecan-Dependent Transport Mechanisms in Filopodia To Reach the Cell Surface. ACS Nano 2012, 6, 7521–7532. [Google Scholar] [CrossRef]
- Romero, S.; Grompone, G.; Carayol, N.; Mounier, J.; Guadagnini, S.; Prevost, M.-C.; Sansonetti, P.J.; Van Nhieu, G.T. ATP-Mediated Erk1/2 Activation Stimulates Bacterial Capture by Filopodia, which Precedes Shigella Invasion of Epithelial Cells. Cell Host Microbe 2011, 9, 508–519. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Dorantes, M.; Romero-Cordoba, S.L.; Peralta-Zaragoza, O.; Salido-Guadarrama, I.; Hidalgo-Miranda, A. MicroRNAs transported by exosomes in body fluids as mediators of intercellular communication in cancer. OncoTargets Ther. 2014, 7, 1327–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stalder, L.; Heusermann, W.; Sokol, L.; Trojer, D.; Wirz, J.; Hean, J.; Fritzsche, A.; Aeschimann, F.; Pfanzagl, V.; Basselet, P.; et al. The rough endoplasmatic reticulum is a central nucleation site of siRNA-mediated RNA silencing. EMBO J. 2013, 32, 1115–1127. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Liu, L.; Zhuang, X.; Yu, Y.; Liu, X.; Cui, X.; Ji, L.; Pan, Z.; Cao, X.; Mo, B.; et al. MicroRNAs Inhibit the Translation of Target mRNAs on the Endoplasmic Reticulum in Arabidopsis. Cell 2013, 153, 562–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, G.; Kao, S.C.; Pavlakis, N.; Brahmbhatt, H.; MacDiarmid, J.; Clarke, S.; Boyer, M.; van Zandwijk, N. Clinical development of TargomiRs, a miRNA mimic-based treatment for patients with recurrent thoracic cancer. Epigenomics 2016, 8, 1079–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, S.C.; Fulham, M.; Wong, K.; Cooper, W.; Brahmbhatt, H.; MacDiarmid, J.; Pattison, S.; Sagong, J.O.; Huynh, Y.; Leslie, F.; et al. A Significant Metabolic and Radiological Response after a Novel Targeted MicroRNA-based Treatment Approach in Malignant Pleural Mesothelioma. Am. J. Respir. Crit. Care Med. 2015, 191, 1467–1469. [Google Scholar] [CrossRef] [Green Version]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug | miRNA Therapeutic | Delivery System | Disease | Status * | Manufacturer |
|---|---|---|---|---|---|
| Lademirsen (RG-012/SAR339375) | anti-miR-21 | AMO a inhibitor | Alport Syndrome | Phase II (active) NCT02855268 | Regulus Therapeutics; Genzyme |
| AZD4076/RG-125 | anti-miR-103/107 | GalNAc-conjugated antimiR | Type 2 diabetes; nonalcoholic fatty liver diseases | Phase I (active) NCT02612662; Phase I/IIa (completed) NCT02826525 | AstraZeneca; Regulus Therapeutics |
| Remlarsen (MRG-201) | miR-29 mimic | Cholesterol-conjugated miRNA duplex | Keloid | Phase II (completed) NCT03601052 | miRagen Therapeutics |
| Cobomarsen (MRG-106) | anti-miR-155 | LNA-modified antimiR | Lymphomas; Leukemias | Phase I (completed) NCT02580552; Phase II (terminated) NCT03837457 NCT03713320 | miRagen Therapeutics |
| MRG-110 | anti-miR-92a | LNA-modified antimiR | Skin wound | Phase I (completed) NCT03603431 | miRagen Therapeutics |
| MesomiR-1/ TargomiR | miR-16 mimic | EDVs-nonliving bacterial minicells | Mesothelioma; non-small cell lung cancer | Phase I (completed) NCT02369198 | EnGeneIC; Asbestos Diseases Research Foundation |
| Miravirsen | anti-miR-122 | LNA-modified antimiR | Hepatitis C | Phase I and II (completed) NCT01646489 NCT01200420 NCT01872936 NCT02031133 NCT02508090 | Santaris Pharma A/S; Hoffmann-La Roche |
| RGLS4326 | anti-miR-17 | AMO inhibitor | Polycystic kidney disease | Phase I (completed) NCT04536688 | Regulus Therapeutics |
| RG-101 | anti-miR-122 | GalNAc-conjugated antimiR | Hepatitis C | Phase I and II (discontinued) | Regulus Therapeutics |
| MRX34 | miR-34 mimic | Lipid-based nanoparticle (liposome) | Cancer | Phase I (terminated) NCT01829971 NCT02862145 | Mirna Therapeutics |
| pSil-miR200c and PMIS miR200a | Plasmid DNAs encoding miR-200c and a miRNA inhibitor targeting miR-200a | Biodegradable sponge | Tooth extraction | Phase I (withdrawn) NCT02579187 | University of Iowa |
| miRNA Delivery Platforms for Cancer Therapeutics | ||||||
|---|---|---|---|---|---|---|
| Advantages | Viral | Nonviral | ||||
| Virus | Lipid | Polymer | Inorganic | EV | Peptide | |
| • Highly efficacious gene delivery | • Non- immunogenic • Control of size, lipid composition and functional groups, and drug loading • Co-delivery of multiple drugs | • Non- immunogenic • Control of size, polymer composition and functional groups, and drug loading • Co-delivery of multiple drugs | • Non- immunogenic • Ease of production • Control of size, composition and functional groups, and drug loading | • Non- immunogenic • Control of functional groups and drug loading • Co-delivery of multiple drugs • Tissue/organ- specific delivery | • Ease/cost of production • Control of physiochemical properties and functions • Tissue/organ- specific delivery | |
| Disadvantages | • Immunogenic • Biosafety concerns | • Non-specific delivery • Low in vivo efficacy • Cytotoxicity | • Non-specific delivery • Low in vivo efficacy • Cytotoxicity | • Low in vivo efficacy | • Lack of experimental data/studies • Inherent diverse composition of EV cargos • Cost of production | • Lack of experimental data/studies |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holjencin, C.; Jakymiw, A. MicroRNAs and Their Big Therapeutic Impacts: Delivery Strategies for Cancer Intervention. Cells 2022, 11, 2332. https://doi.org/10.3390/cells11152332
Holjencin C, Jakymiw A. MicroRNAs and Their Big Therapeutic Impacts: Delivery Strategies for Cancer Intervention. Cells. 2022; 11(15):2332. https://doi.org/10.3390/cells11152332
Chicago/Turabian StyleHoljencin, Charles, and Andrew Jakymiw. 2022. "MicroRNAs and Their Big Therapeutic Impacts: Delivery Strategies for Cancer Intervention" Cells 11, no. 15: 2332. https://doi.org/10.3390/cells11152332
APA StyleHoljencin, C., & Jakymiw, A. (2022). MicroRNAs and Their Big Therapeutic Impacts: Delivery Strategies for Cancer Intervention. Cells, 11(15), 2332. https://doi.org/10.3390/cells11152332