
Macrophage Phenotypes in Normal and Diabetic Wound Healing and Therapeutic Interventions


Abstract
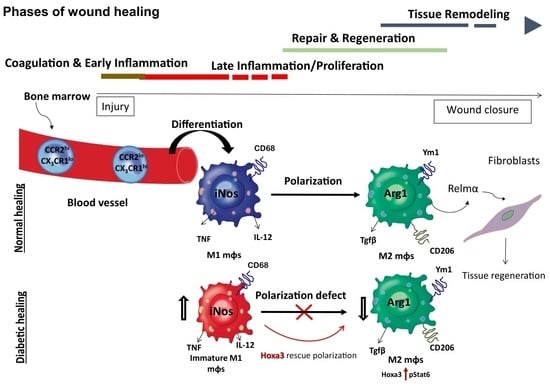
:
{kind=link}
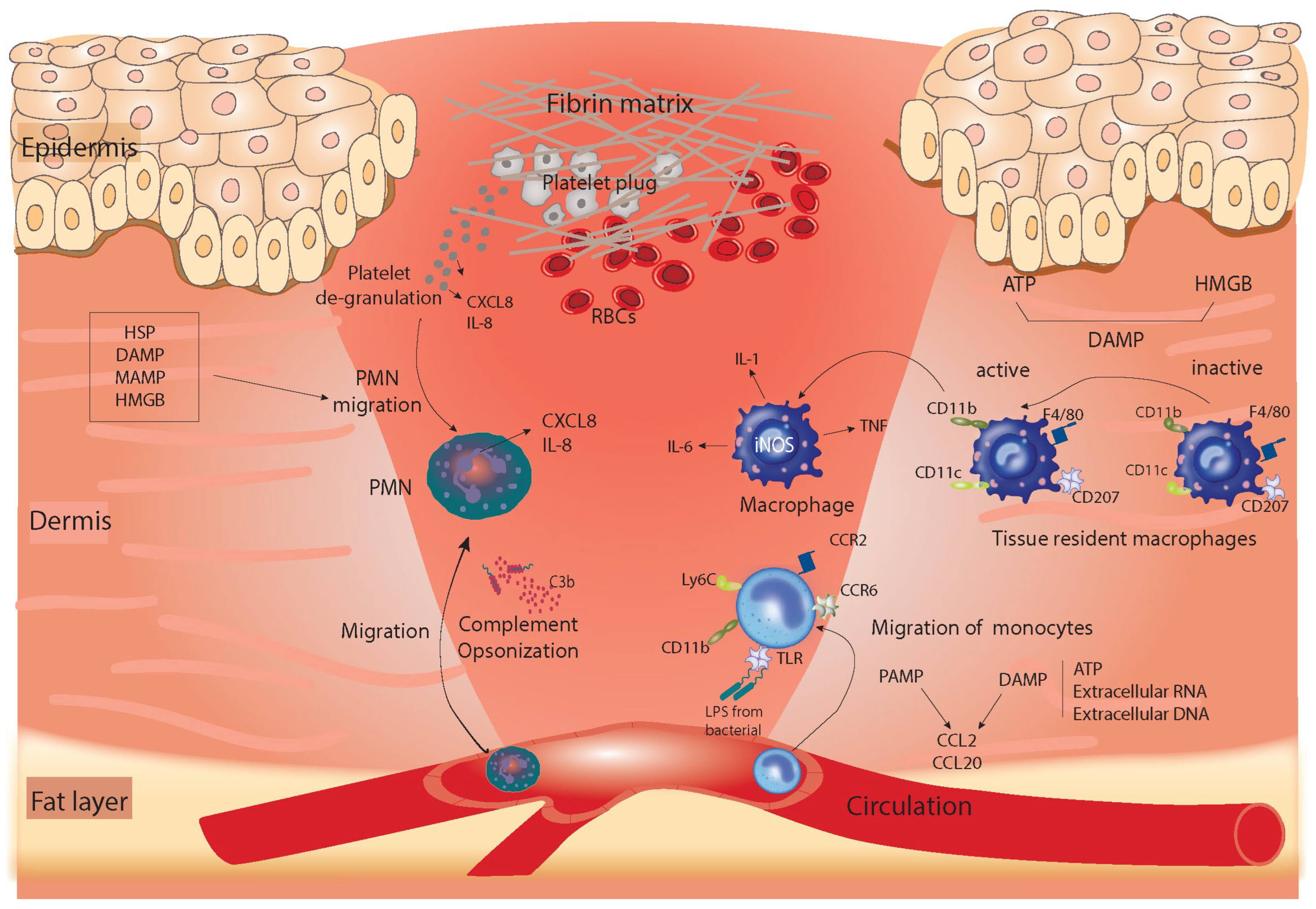{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Wound Healing
2.1. Hemostasis
2.2. Inflammatory Phase
2.3. Proliferation and Tissue-Regeneration Phase of Healing
2.4. Tissue-Remodeling Phase of Healing
3. Innate Immune Cells in Wound Healing
3.1. Monocytes
3.2. Macrophages
3.2.1. Macrophage Polarization: From Early Discoveries to Recent Updates
3.2.2. Macrophage Polarization in Wounds
4. Effect of Macrophage Dysregulation in Wound Healing
4.1. Diabetes
4.2. In Diabetic Models
Epigenetic Changes in Dysregulated Macrophages in Diabetes
5. Therapeutic Manipulation of Diabetic Macrophages
Therapeutic Use of Homeobox a3
6. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Reinke, J.M.; Sorg, H. Wound repair and regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.E. Function of the panniculus carnosus—A hypothesis. Vet. Rec. 2010, 167, 760. [Google Scholar] [CrossRef]
- Guo, S.; Dipietro, L.A. Factors affecting wound healing. J. Dent. Res. 2010, 89, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Scott Adzick, N.; Harrison, M.R.; Glick, P.L.; Beckstead, J.H.; Villa, R.L.; Scheuenstuhl, H.; Goodson, W.H. Comparison of fetal, newborn, and adult wound healing by histologic, enzyme-histochemical, and hydroxyproline determinations. J. Pediatric Surg. 1985, 20, 315–319. [Google Scholar] [CrossRef]
- Hopkinson-Woolley, J.; Hughes, D.; Gordon, S.; Martin, P. Macrophage recruitment during limb development and wound healing in the embryonic and foetal mouse. J. Cell Sci. 1994, 107, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Nunan, R.; Harding, K.G.; Martin, P. Clinical challenges of chronic wounds: Searching for an optimal animal model to recapitulate their complexity. Dis. Models Mech. 2014, 7, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr266. [Google Scholar] [CrossRef] [Green Version]
- Eming, S.A.; Hammerschmidt, M.; Krieg, T.; Roers, A. Interrelation of immunity and tissue repair or regeneration. Semin. Cell Dev. Biol. 2009, 20, 517–527. [Google Scholar] [CrossRef]
- Knipper, J.A.; Willenborg, S.; Brinckmann, J.; Bloch, W.; Maaß, T.; Wagener, R.; Krieg, T.; Sutherland, T.; Munitz, A.; Rothenberg, M.E.; et al. Interleukin-4 receptor α signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity 2015, 43, 803–816. [Google Scholar] [CrossRef] [Green Version]
- Lucas, T.; Waisman, A.; Ranjan, R.; Roes, J.; Krieg, T.; Müller, W.; Roers, A.; Eming, S.A. Differential roles of macrophages in diverse phases of skin repair. J. Immunol. 2010, 184, 3964–3977. [Google Scholar] [CrossRef] [Green Version]
- Nurden, A.T.; Nurden, P.; Sanchez, M.; Andia, I.; Anitua, E. Platelets and wound healing. Front. Biosci. 2008, 13, 3525–3548. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Han, G.; Owens, P.; Siddiqui, Y.; Li, A.G. Role of TGFβ-mediated inflammation in cutaneous wound healing. J. Investig. Dermatol. Symp. Proc. 2006, 11, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liarte, S.; Bernabé-García, Á.; Nicolás, F.J. Role of TGF-β in skin chronic wounds: A keratinocyte perspective. Cells 2020, 9, 306. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Ellis, S.; Lin, E.J.; Tartar, D. Immunology of wound healing. Curr. Dermatol. Rep. 2018, 7, 350–358. [Google Scholar] [CrossRef] [Green Version]
- Björk, J.; Hugli, T.E.; Smedegård, G. Microvascular effects of anaphylatoxins C3a and C5a. J. Immunol. 1985, 134, 1115–1119. [Google Scholar]
- Peerschke, E.I.B.; Yin, W.; Ghebrehiwet, B. Platelet mediated complement activation. Adv. Exp. Med. Biol. 2008, 632, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Sinno, H.; Prakash, S. Complements and the wound healing cascade: An updated review. Plast. Surg. Int. 2013, 2013, 146764. [Google Scholar] [CrossRef]
- Ng, M.F. The role of mast cells in wound healing. Int. Wound J. 2010, 7, 55–61. [Google Scholar] [CrossRef]
- Phillipson, M.; Kubes, P. The healing power of neutrophils. Trends Immunol. 2019, 40, 635–647. [Google Scholar] [CrossRef]
- Zarbock, A.; Ley, K. Mechanisms and consequences of neutrophil interaction with the endothelium. Am. J. Pathol. 2008, 172, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Broggi, A.; Granucci, F. Microbe- and danger-induced inflammation. Mol. Immunol. 2015, 63, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Vénéreau, E.; Ceriotti, C.; Bianchi, M.E. DAMPs from cell death to new life. Front. Immunol. 2015, 6, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [Green Version]
- O’Kane, S.; Ferguson, M.W.J. Transforming growth factor βs and wound healing. Int. J. Biochem. Cell Biol. 1997, 29, 63–78. [Google Scholar] [CrossRef]
- Shaw, T.J.; Martin, P. Wound repair at a glance. J. Cell Sci. 2009, 122, 3209–3213. [Google Scholar] [CrossRef] [Green Version]
- Merad, M.; Ginhoux, F.; Collin, M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat. Rev. Immunol. 2008, 8, 935–947. [Google Scholar] [CrossRef]
- Stojadinovic, O.; Yin, N.; Lehmann, J.; Pastar, I.; Kirsner, R.S.; Tomic-Canic, M. Increased number of Langerhans cells in the epidermis of diabetic foot ulcers correlates with healing outcome. Immunol. Res. 2013, 57, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley, J.M.; Brancato, S.K.; Thomay, A.A.; Reichner, J.S.; Albina, J.E. The phenotype of murine wound macrophages. J. Leukoc. Biol. 2010, 87, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicks, K.; Torbica, T.; Mace, K.A. Myeloid cell dysfunction and the pathogenesis of the diabetic chronic wound. Semin. Immunol. 2014, 26, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.M.; Bauer, R.J.; Velazquez, O.C. Angiogenesis, vasculogenesis, and induction of healing in chronic wounds. Vasc. Endovasc. Surg. 2005, 39, 293–306. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [Green Version]
- Willenborg, S.; Lucas, T.; van Loo, G.; Knipper, J.A.; Krieg, T.; Haase, I.; Brachvogel, B.; Hammerschmidt, M.; Nagy, A.; Ferrara, N.; et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 2012, 120, 613–625. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.E.; Wilgus, T.A. Vascular endothelial growth factor and angiogenesis in the regulation of cutaneous wound repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Bae, O.-N.; Ahn, S.; Jin, S.H.; Hong, S.H.; Lee, J.; Kim, E.-S.; Jeong, T.C.; Chun, Y.-J.; Lee, A.-Y.; Noh, M. Chemical allergens stimulate human epidermal keratinocytes to produce lymphangiogenic vascular endothelial growth factor. Toxicol. Appl. Pharmacol. 2015, 283, 147–155. [Google Scholar] [CrossRef]
- Metz, C.N. Fibrocytes: A unique cell population implicated in wound healing. Cell. Mol. Life Sci. CMLS 2003, 60, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Donnelly, S.C.; Peng, T.; Bucala, R.; Metz, C.N. Peripheral blood fibrocytes: Differentiation pathway and migration to wound sites. J. Immunol. 2001, 166, 7556–7562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, K.J.; McKenzie, I.A.; Mill, P.; Smith, K.M.; Akhavan, M.; Barnabé-Heider, F.; Biernaskie, J.; Junek, A.; Kobayashi, N.R.; Toma, J.G.; et al. A dermal niche for multipotent adult skin-derived precursor cells. Nat. Cell Biol. 2004, 6, 1082–1093. [Google Scholar] [CrossRef]
- Achterberg, V.F.; Buscemi, L.; Diekmann, H.; Smith-Clerc, J.; Schwengler, H.; Meister, J.-J.; Wenck, H.; Gallinat, S.; Hinz, B. The nano-scale mechanical properties of the extracellular matrix regulate dermal fibroblast function. J. Investig. Dermatol. 2014, 134, 1862–1872. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.Y.; Sneddon, J.B.; Alizadeh, A.A.; Sood, R.; West, R.B.; Montgomery, K.; Chi, J.-T.; van de Rijn, M.; Botstein, D.; Brown, P.O. Gene expression signature of fibroblast serum response predicts human cancer progression: Similarities between tumors and wounds. PLoS Biol. 2004, 2, E7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDougall, S.; Dallon, J.; Sherratt, J.; Maini, P. Fibroblast migration and collagen deposition during dermal wound healing: Mathematical modelling and clinical implications. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2006, 364, 1385–1405. [Google Scholar] [CrossRef]
- Faler, B.J.; Macsata, R.A.; Plummer, D.; Mishra, L.; Sidawy, A.N. Transforming growth factor-beta and wound healing. Perspect. Vasc. Surg. Endovasc. Ther. 2006, 18, 55–62. [Google Scholar] [CrossRef]
- Shaw, T.J.; Martin, P. Wound repair: A showcase for cell plasticity and migration. Curr. Opin. Cell Biol. 2016, 42, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Varga, J.; Rosenbloom, J.; Jimenez, S.A. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem. J. 1987, 247, 597–604. [Google Scholar] [CrossRef]
- Cordeiro, M.F.; Bhattacharya, S.S.; Schultz, G.S.; Khaw, P.T. TGF-beta1, -beta2, and -beta3 in vitro: Biphasic effects on Tenon’s fibroblast contraction, proliferation, and migration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 756–763. [Google Scholar]
- Freedberg, I.M.; Tomic-Canic, M.; Komine, M.; Blumenberg, M. Keratins and the keratinocyte activation cycle. J. Investig. Dermatol. 2001, 116, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisse, T.S.; King, B.L.; Rieger, S. Comparative transcriptomic profiling of hydrogen peroxide signaling networks in zebrafish and human keratinocytes: Implications toward conservation, migration and wound healing. Sci. Rep. 2016, 6, 20328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riding, A.; Pullar, C.E. ATP release and P2Y receptor signaling are essential for keratinocyte galvanotaxis. J. Cell Physiol. 2016, 231, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Ferrer, M.; Afshar-Sherif, A.-R.; Uwamariya, C.; de Crombrugghe, B.; Davidson, J.M.; Bhowmick, N.A. Dermal transforming growth factor-beta responsiveness mediates wound contraction and epithelial closure. Am. J. Pathol. 2010, 176, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Pastar, I.; Stojadinovic, O.; Krzyzanowska, A.; Barrientos, S.; Stuelten, C.; Zimmerman, K.; Blumenberg, M.; Brem, H.; Tomic-Canic, M. Attenuation of the transforming growth factor beta-signaling pathway in chronic venous ulcers. Mol. Med. 2010, 16, 92–101. [Google Scholar] [CrossRef]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Jackson, C.J. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef] [Green Version]
- Greenhalgh, D.G. The role of apoptosis in wound healing. Int. J. Biochem. Cell Biol. 1998, 30, 1019–1030. [Google Scholar] [CrossRef]
- Chalmers, R.L. The evidence for the role of transforming growth factor-beta in the formation of abnormal scarring. Int. Wound J. 2011, 8, 218–223. [Google Scholar] [CrossRef]
- Shah, M.; Foreman, D.M.; Ferguson, M.W. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J. Cell Sci. 1995, 108, 985–1002. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Zhou, L.; Ochoa, S.; Hasan, A.; Badiavas, E.; Falanga, V. TGF-β3 stimulates and regulates collagen synthesis through TGF-β1-dependent and independent mechanisms. J. Investig. Dermatol. 1997, 108, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Nunan, R. Cellular and molecular mechanisms of repair in acute and chronic wound healing. Br. J. Dermatol. 2015, 173, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Pakyari, M.; Farrokhi, A.; Maharlooei, M.K.; Ghahary, A. Critical role of transforming growth factor beta in different phases of wound healing. Adv. Wound Care 2013, 2, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Khanna, S.; Biswas, S.; Shang, Y.; Collard, E.; Azad, A.; Kauh, C.; Bhasker, V.; Gordillo, G.M.; Sen, C.K.; Roy, S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS ONE 2010, 5, e9539. [Google Scholar] [CrossRef] [Green Version]
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Barman, P.K.; Pang, J.; Urao, N.; Koh, T.J. Skin wounding-induced monocyte expansion in mice is not abrogated by IL-1 receptor 1 deficiency. J. Immunol. 2019, 202, 2720–2727. [Google Scholar] [CrossRef]
- Ishida, Y.; Gao, J.-L.; Murphy, P.M. Chemokine receptor CX3CR1 mediates skin wound healing by promoting macrophage and fibroblast accumulation and function. J. Immunol. 2008, 180, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Brazil, J.C.; Quiros, M.; Nusrat, A.; Parkos, C.A. Innate immune cell-epithelial crosstalk during wound repair. J. Clin. Investig. 2019, 129, 2983–2993. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, M.; Wicks, K.; Gardasevic, M.; Mace, K.A. Cx3CR1 expression identifies distinct macrophage populations that contribute differentially to inflammation and repair. ImmunoHorizons 2019, 3, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; D’Souza, D.; Martin, J.; Grose, R.; Cooper, L.; Maki, R.; McKercher, S.R. Wound healing in the PU.1 null mouse—Tissue repair is not dependent on inflammatory cells. Curr. Biol. 2003, 13, 1122–1128. [Google Scholar] [CrossRef] [Green Version]
- Leibovich, S.J.; Ross, R. The role of the macrophage in wound repair: A study with hydrocortisone and antimacrophage serum. Am. J. Pathol. 1975, 78, 71–100. [Google Scholar]
- Goren, I.; Allmann, N.; Yogev, N.; Schürmann, C.; Linke, A.; Holdener, M.; Waisman, A.; Pfeilschifter, J.; Frank, S. A transgenic mouse model of inducible macrophage depletion: Effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am. J. Pathol. 2009, 175, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Mirza, R.; DiPietro, L.A.; Koh, T.J. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am. J. Pathol. 2009, 175, 2454–2462. [Google Scholar] [CrossRef] [Green Version]
- Mackaness, G.B. The immunological basis of acquired cellular resistance. J. Exp. Med. 1964, 120, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Dalton, D.K.; Pitts-Meek, S.; Keshav, S.; Figari, I.S.; Bradley, A.; Stewart, T.A. Multiple defects of immune cell function in mice with disrupted interferon-γ genes. Science 1993, 259, 1739–1742. [Google Scholar] [CrossRef]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Rőszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, C.J.; Leibovich, S.J. Regulation of macrophage polarization and wound healing. Adv. Wound Care 2012, 1, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [Green Version]
- Fleetwood, A.J.; Lawrence, T.; Hamilton, J.A.; Cook, A.D. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: Implications for CSF blockade in inflammation. J. Immunol. 2007, 178, 5245–5252. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Br. J. Pharmacol. 2009, 158, 638–651. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [Green Version]
- Raes, G.; De Baetselier, P.; Noël, W.; Beschin, A.; Brombacher, F.; Hassanzadeh Gh, G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J. Leukoc. Biol. 2002, 71, 597–602. [Google Scholar]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannon, P.; Wood, S.; Restivo, T.; Campbell, L.; Hardman, M.J.; Mace, K.A. Diabetes induces stable intrinsic changes to myeloid cells that contribute to chronic inflammation during wound healing in mice. Dis. Models Mech. 2013, 6, 1434–1447. [Google Scholar] [CrossRef] [Green Version]
- Mirza, R.; Koh, T.J. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 2011, 56, 256–264. [Google Scholar] [CrossRef]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Salmon-Ehr, V.; Ramont, L.; Godeau, G.; Birembaut, P.; Guenounou, M.; Bernard, P.; Maquart, F.-X. Implication of interleukin-4 in wound healing. Lab. Investig. 2000, 80, 1337–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minutti, C.M.; Jackson-Jones, L.H.; García-Fojeda, B.; Knipper, J.A.; Sutherland, T.E.; Logan, N.; Ringqvist, E.; Guillamat-Prats, R.; Ferenbach, D.A.; Artigas, A.; et al. Local amplifiers of IL-4Rα-mediated macrophage activation promote repair in lung and liver. Science 2017, 356, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, S.-Y.; Wang, X.; Vijayan, S.; Tang, Y.; Kim, Y.O.; Padberg, K.; Regen, T.; Molokanova, O.; Chen, T.; Bopp, T.; et al. IL-4 receptor alpha signaling through macrophages differentially regulates liver fibrosis progression and reversal. EBioMedicine 2018, 29, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Nair, M.G. Macrophages in wound healing: Activation and plasticity. Immunol. Cell Biol. 2019, 97, 258–267. [Google Scholar] [CrossRef]
- Sutherland, T.E.; Rückerl, D.; Logan, N.; Duncan, S.; Wynn, T.A.; Allen, J.E. Ym1 induces RELMα and rescues IL-4Rα deficiency in lung repair during nematode infection. PLoS Pathog. 2018, 14, e1007423. [Google Scholar] [CrossRef] [Green Version]
- Wållberg, M.; Cooke, A. Immune mechanisms in type 1 diabetes. Trends Immunol. 2013, 34, 583–591. [Google Scholar] [CrossRef]
- Alberti, K.G.M.M.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus. Provisional report of a WHO Consultation. Diabet. Med. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Artis, D. Immune regulation of metabolic homeostasis in health and disease. Cell 2015, 161, 146–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I.; Ruslami, R. Type 2 diabetes and its impact on the immune system. Curr. Diabetes Rev. 2020, 16, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef]
- Harakeh, S.; Kalamegam, G.; Pushparaj, P.N.; Al-Hejin, A.; Alfadul, S.M.; Al Amri, T.; Barnawi, S.; Al Sadoun, H.; Mirza, A.A.; Azhar, E. Chemokines and their association with body mass index among healthy Saudis. Saudi J. Biol. Sci. 2020, 27, 6–11. [Google Scholar] [CrossRef]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [Green Version]
- Miao, M.; Niu, Y.; Xie, T.; Yuan, B.; Qing, C.; Lu, S. Diabetes-impaired wound healing and altered macrophage activation: A possible pathophysiologic correlation. Wound Repair Regen. 2012, 20, 203–213. [Google Scholar] [CrossRef]
- Crane, M.J.; Daley, J.M.; van Houtte, O.; Brancato, S.K.; Henry, W.L., Jr.; Albina, J.E. The monocyte to macrophage transition in the murine sterile wound. PLoS ONE 2014, 9, e86660. [Google Scholar] [CrossRef] [Green Version]
- Little, M.C.; Hurst, R.J.M.; Else, K.J. Dynamic changes in macrophage activation and proliferation during the development and resolution of intestinal inflammation. J. Immunol. 2014, 193, 4684–4695. [Google Scholar] [CrossRef] [Green Version]
- Alrdahe, S.; Al Sadoun, H.; Torbica, T.; McKenzie, E.A.; Bowling, F.L.; Boulton, A.J.M.; Mace, K.A. Dysregulation of macrophage development and phenotype in diabetic human macrophages can be rescued by Hoxa3 protein transduction. PLoS ONE 2019, 14, e0223980. [Google Scholar] [CrossRef]
- Molawi, K.; Sieweke, M.H. Transcriptional control of macrophage identity, self-renewal, and function. Adv. Immunol. 2013, 120, 269–300. [Google Scholar] [CrossRef] [PubMed]
- Wicks, K.; Torbica, T.; Umehara, T.; Amin, S.; Bobola, N.; Mace, K.A. Diabetes inhibits Gr-1+ myeloid cell maturation via Cebpa deregulation. Diabetes 2015, 64, 4184–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef] [Green Version]
- Torbica, T.; Wicks, K.; Umehara, T.; Gungordu, L.; Alrdahe, S.; Wemyss, K.; Grainger, J.R.; Mace, K.A. Chronic inflammation in response to injury: Retention of myeloid cells in injured tissue is driven by myeloid cell intrinsic factors. J. Investig. Dermatol. 2019, 139, 1583–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brown, Z.K.; Van Nostrand, E.L.; Higgins, J.P.; Kim, S.K. The Inflammatory Transcription Factors NFκB, STAT1 and STAT3 Drive Age-Associated Transcriptional Changes in the Human Kidney. PLoS Genet. 2015, 11, e1005734. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B.; Park, S.H. Epigenetic Regulation of Myeloid Cells. Microbiol. Spectr. 2016, 4, 10–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Ivashkiv, L.B. Cross-regulation of signaling pathways by interferon-gamma: Implications for immune responses and autoimmune diseases. Immunity 2009, 31, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Mashili, F.; Chibalin, A.V.; Krook, A.; Zierath, J.R. Constitutive STAT3 phosphorylation contributes to skeletal muscle insulin resistance in type 2 diabetes. Diabetes 2013, 62, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Louiselle, A.E.; Niemiec, S.M.; Zgheib, C.; Liechty, K.W. Macrophage polarization and diabetic wound healing. Transl. Res. 2021, 236, 109–116. [Google Scholar] [CrossRef]
- Huang, S.-M.; Wu, C.-S.; Chiu, M.-H.; Wu, C.-H.; Chang, Y.-T.; Chen, G.-S.; Lan, C.-C.E. High glucose environment induces M1 macrophage polarization that impairs keratinocyte migration via TNF-α: An important mechanism to delay the diabetic wound healing. J. Dermatol. Sci. 2019, 96, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Morey, M.; O’Gaora, P.; Pandit, A.; Hélary, C. Hyperglycemia acts in synergy with hypoxia to maintain the pro-inflammatory phenotype of macrophages. PLoS ONE 2019, 14, e0220577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzler, C.; Kämpfer, H.; Stallmeyer, B.; Pfeilschifter, J.; Frank, S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: Prolonged persistence of neutrophils and macrophages during the late phase of repair. J. Investig. Dermatol. 2000, 115, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, H.D.; Longaker, M.T.; Werner, S. Reduced expression of PDGF and PDGF receptors during impaired wound healing. J. Investig. Dermatol. 1997, 109, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, S.; Breeden, M.; Hübner, G.; Greenhalgh, D.G.; Longaker, M.T. Induction of keratinocyte growth factor expression is reduced and delayed during wound healing in the genetically diabetic mouse. J. Investig. Dermatol. 1994, 103, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Carson, W.F.T.; Cavassani, K.A.; Soares, E.M.; Hirai, S.; Kittan, N.A.; Schaller, M.A.; Scola, M.M.; Joshi, A.; Matsukawa, A.; Aronoff, D.M.; et al. The STAT4/MLL1 Epigenetic Axis Regulates the Antimicrobial Functions of Murine Macrophages. J. Immunol. 2017, 199, 1865–1874. [Google Scholar] [CrossRef] [Green Version]
- Robert, I.; Aussems, M.; Keutgens, A.; Zhang, X.; Hennuy, B.; Viatour, P.; Vanstraelen, G.; Merville, M.P.; Chapelle, J.P.; de Leval, L.; et al. Matrix Metalloproteinase-9 gene induction by a truncated oncogenic NF-κB2 protein involves the recruitment of MLL1 and MLL2 H3K4 histone methyltransferase complexes. Oncogene 2009, 28, 1626–1638. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhu, K.; Li, S.; Liao, Y.; Du, R.; Zhang, X.; Shu, H.B.; Guo, A.Y.; Li, L.; Wu, M. MLL1, a H3K4 methyltransferase, regulates the TNFα-stimulated activation of genes downstream of NF-κB. J. Cell Sci. 2012, 125, 4058–4066. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, A.; Sukumar, S.; Hajela, A.; Mukherjee, S.; Dutta, P.; Bhadada, S.K.; Bhansali, A. The microbiology of diabetic foot infections in patients recently treated with antibiotic therapy: A prospective study from India. J. Diabetes Its Complicat. 2017, 31, 407–412. [Google Scholar] [CrossRef]
- Davis, F.M.; denDekker, A.; Kimball, A.; Joshi, A.D.; El Azzouny, M.; Wolf, S.J.; Obi, A.T.; Lipinski, J.; Gudjonsson, J.E.; Xing, X.; et al. Epigenetic Regulation of TLR4 in Diabetic Macrophages Modulates Immunometabolism and Wound Repair. J. Immunol. 2020, 204, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Kimball, A.S.; Davis, F.M.; denDekker, A.; Joshi, A.D.; Schaller, M.A.; Bermick, J.; Xing, X.; Burant, C.F.; Obi, A.T.; Nysz, D.; et al. The Histone Methyltransferase Setdb2 Modulates Macrophage Phenotype and Uric Acid Production in Diabetic Wound Repair. Immunity 2019, 51, 258–271.e255. [Google Scholar] [CrossRef] [PubMed]
- Kroetz, D.N.; Allen, R.M.; Schaller, M.A.; Cavallaro, C.; Ito, T.; Kunkel, S.L. Type I Interferon Induced Epigenetic Regulation of Macrophages Suppresses Innate and Adaptive Immunity in Acute Respiratory Viral Infection. PLoS Pathog. 2015, 11, e1005338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishio, H.; Saita, Y.; Kobayashi, Y.; Takaku, T.; Fukusato, S.; Uchino, S.; Wakayama, T.; Ikeda, H.; Kaneko, K. Platelet-rich plasma promotes recruitment of macrophages in the process of tendon healing. Regen. Ther. 2020, 14, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Al Sadoun, H.; Burgess, M.; Hentges, K.E.; Mace, K.A. Enforced expression of Hoxa3 inhibits classical and promotes alternative activation of macrophages in vitro and in vivo. J. Immunol. 2016, 197, 872–884. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Yin, H.; Yuan, B.; Liu, T.; Luo, L.; Huang, P.; Dai, L.; Zeng, K. IL-33 improves wound healing through enhanced M2 macrophage polarization in diabetic mice. Mol. Immunol. 2017, 90, 42–49. [Google Scholar] [CrossRef]
- Cash, J.L.; Bass, M.D.; Campbell, J.; Barnes, M.; Kubes, P.; Martin, P. Resolution mediator chemerin15 reprograms the wound microenvironment to promote repair and reduce scarring. Curr. Biol. 2014, 24, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Jetten, N.; Roumans, N.; Gijbels, M.J.; Romano, A.; Post, M.J.; de Winther, M.P.J.; van der Hulst, R.R.W.J.; Xanthoulea, S. Wound administration of M2-polarized macrophages does not improve murine cutaneous healing responses. PLoS ONE 2014, 9, e102994. [Google Scholar] [CrossRef] [Green Version]
- Mace, K.A.; Hansen, S.L.; Myers, C.; Young, D.M.; Boudreau, N. HOXA3 induces cell migration in endothelial and epithelial cells promoting angiogenesis and wound repair. J. Cell Sci. 2005, 118, 2567–2577. [Google Scholar] [CrossRef] [Green Version]
- Mace, K.A.; Restivo, T.E.; Rinn, J.L.; Paquet, A.C.; Chang, H.Y.; Young, D.M.; Boudreau, N.J. HOXA3 modulates injury-induced mobilization and recruitment of bone marrow-derived cells. Stem Cells 2009, 27, 1654–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the antennapedia homeodomain is receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahdipour, E.; Charnock, J.C.; Mace, K.A. Hoxa3 promotes the differentiation of hematopoietic progenitor cells into proangiogenic Gr-1+CD11b+ myeloid cells. Blood 2011, 117, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Dupont, E.; Prochiantz, A.; Joliot, A. Identification of a signal peptide for unconventional secretion. J. Biol. Chem. 2007, 282, 8994–9000. [Google Scholar] [CrossRef] [Green Version]
- Klinkert, K.; Whelan, D.; Clover, A.J.P.; Leblond, A.-L.; Kumar, A.H.S.; Caplice, N.M. Selective M2 macrophage depletion leads to prolonged inflammation in surgical wounds. Eur. Surg. Res. 2017, 58, 109–120. [Google Scholar] [CrossRef]
- DiPietro, L.A.; Wilgus, T.A.; Koh, T.J. Macrophages in healing wounds: Paradoxes and paradigms. Int. J. Mol. Sci. 2021, 22, 950. [Google Scholar] [CrossRef]
- Kugel, S.; Mostoslavsky, R. Chromatin and beyond: The multitasking roles for SIRT6. Trends Biochem. Sci. 2014, 39, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Koo, J.-H.; Jang, H.-Y.; Lee, Y.; Moon, Y.J.; Bae, E.J.; Yun, S.-K.; Park, B.-H. Myeloid cell-specific sirtuin 6 deficiency delays wound healing in mice by modulating inflammation and macrophage phenotypes. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Sadoun, H. Macrophage Phenotypes in Normal and Diabetic Wound Healing and Therapeutic Interventions. Cells 2022, 11, 2430. https://doi.org/10.3390/cells11152430
Al Sadoun H. Macrophage Phenotypes in Normal and Diabetic Wound Healing and Therapeutic Interventions. Cells. 2022; 11(15):2430. https://doi.org/10.3390/cells11152430
Chicago/Turabian StyleAl Sadoun, Hadeel. 2022. "Macrophage Phenotypes in Normal and Diabetic Wound Healing and Therapeutic Interventions" Cells 11, no. 15: 2430. https://doi.org/10.3390/cells11152430
APA StyleAl Sadoun, H. (2022). Macrophage Phenotypes in Normal and Diabetic Wound Healing and Therapeutic Interventions. Cells, 11(15), 2430. https://doi.org/10.3390/cells11152430