

Combination of Paclitaxel and PXR Antagonist SPA70 Reverses Paclitaxel-Resistant Non-Small Cell Lung Cancer

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Cell Proliferation and IC50 Measurement
2.3. Colony Formation Assay
2.4. Wound Healing Assay and Invasion Assay
2.5. Intracellular Rh-123 Uptake
2.6. ChIP Assay
2.7. Flow Cytometric Measurement of Cell Cycle and Apoptosis
2.8. Immunofluorescence Staining
2.9. Real-Time Quantitative PCR
2.10. Western Blotting, GST-Pulldown and Co-IP
2.11. Animal Study
2.12. Immunohistochemistry
2.13. Synthesis of YM-1 and YM-2
2.14. Statistical Analysis
3. Results
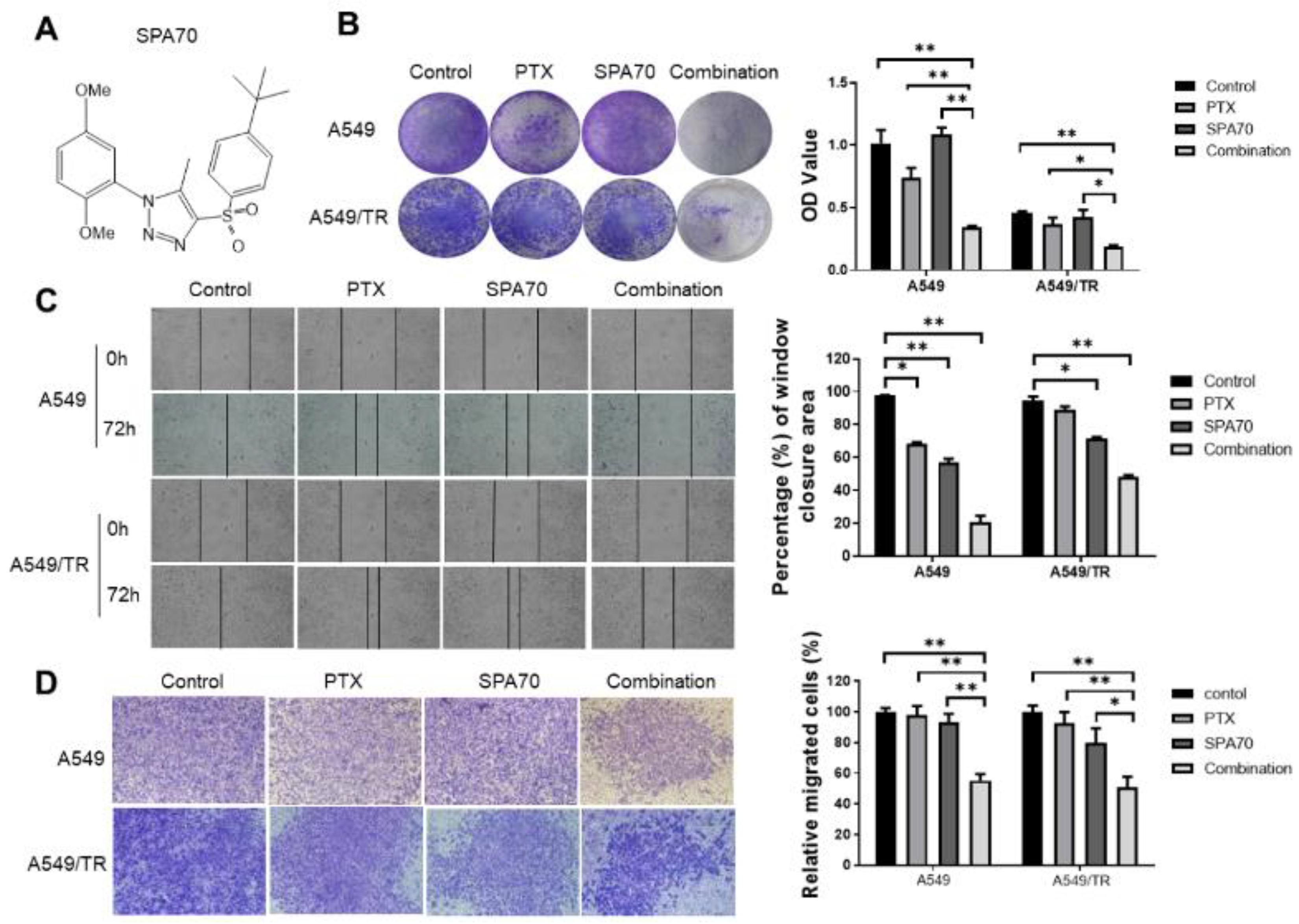
3.1. SPA70 Displays Potent Cytotoxicity and Synergistic Effect with PTX in NSCLC Cells Including PTX-Resistant Cell Line
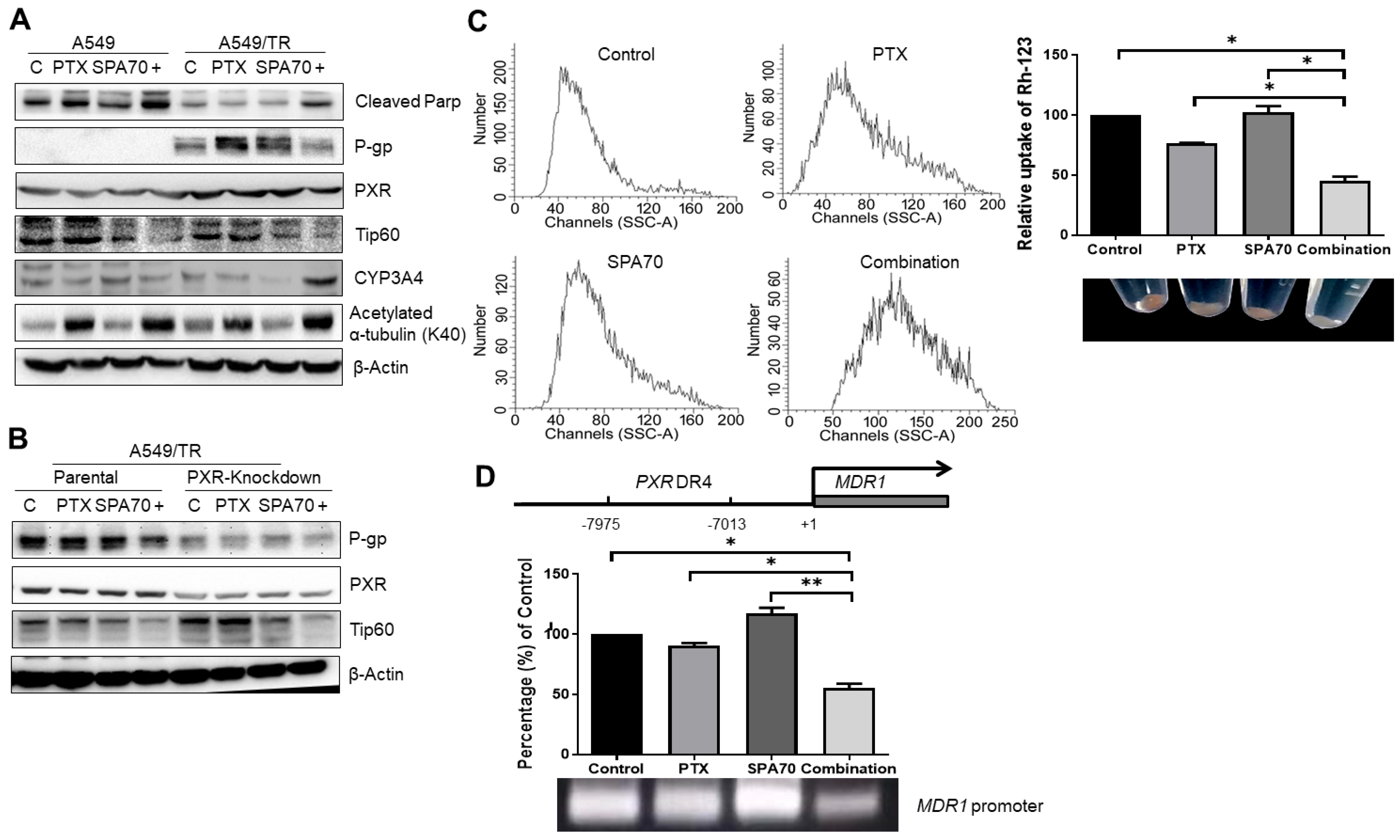
3.2. PTX with SPA70 Together Inhibits PXR-Mediated Transcriptional Regulation of P-gp
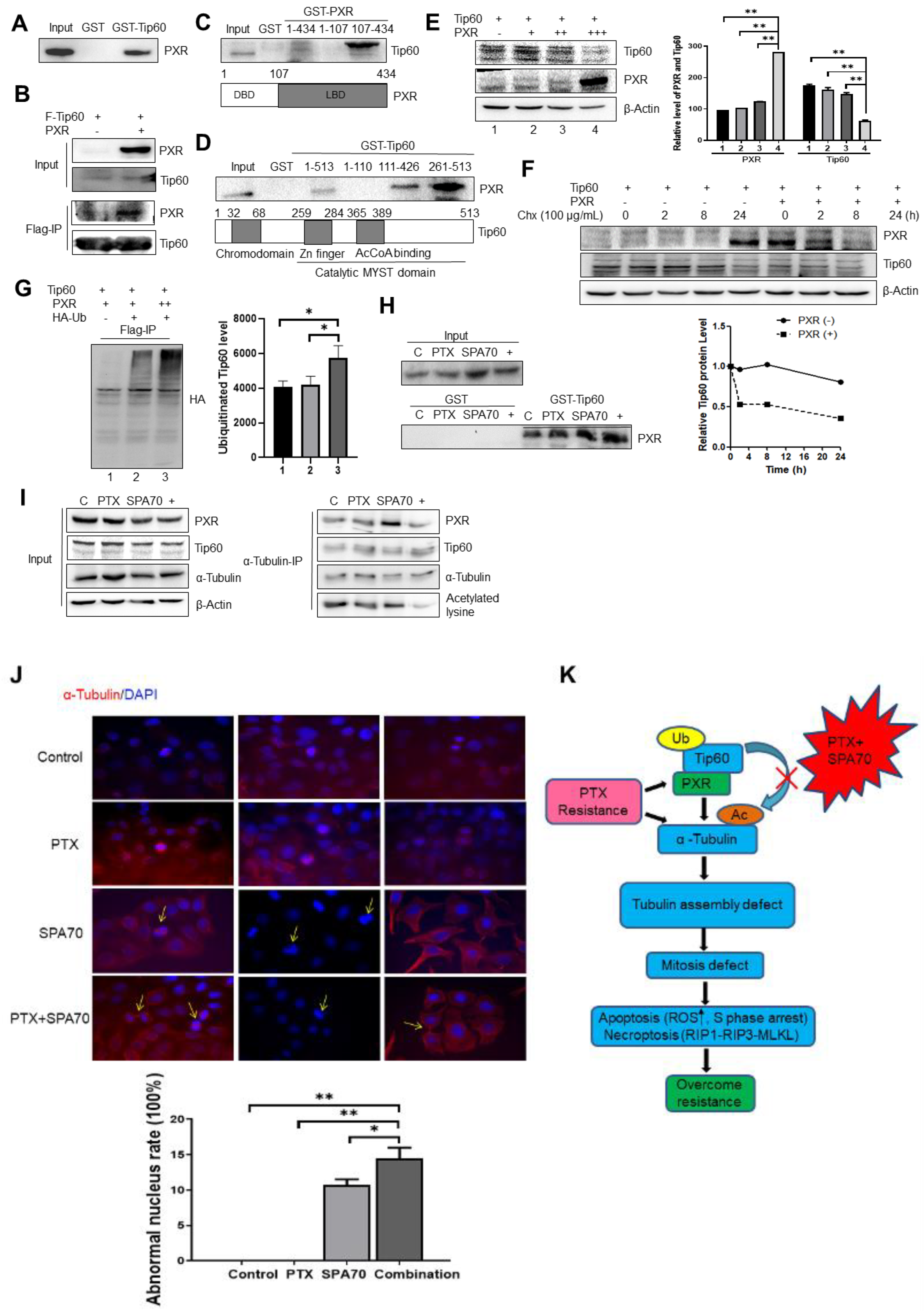
3.3. PXR Degraded Tip60 and Tip60-Acetylated α-Tubulin Was Diminished by the Combination Regimen
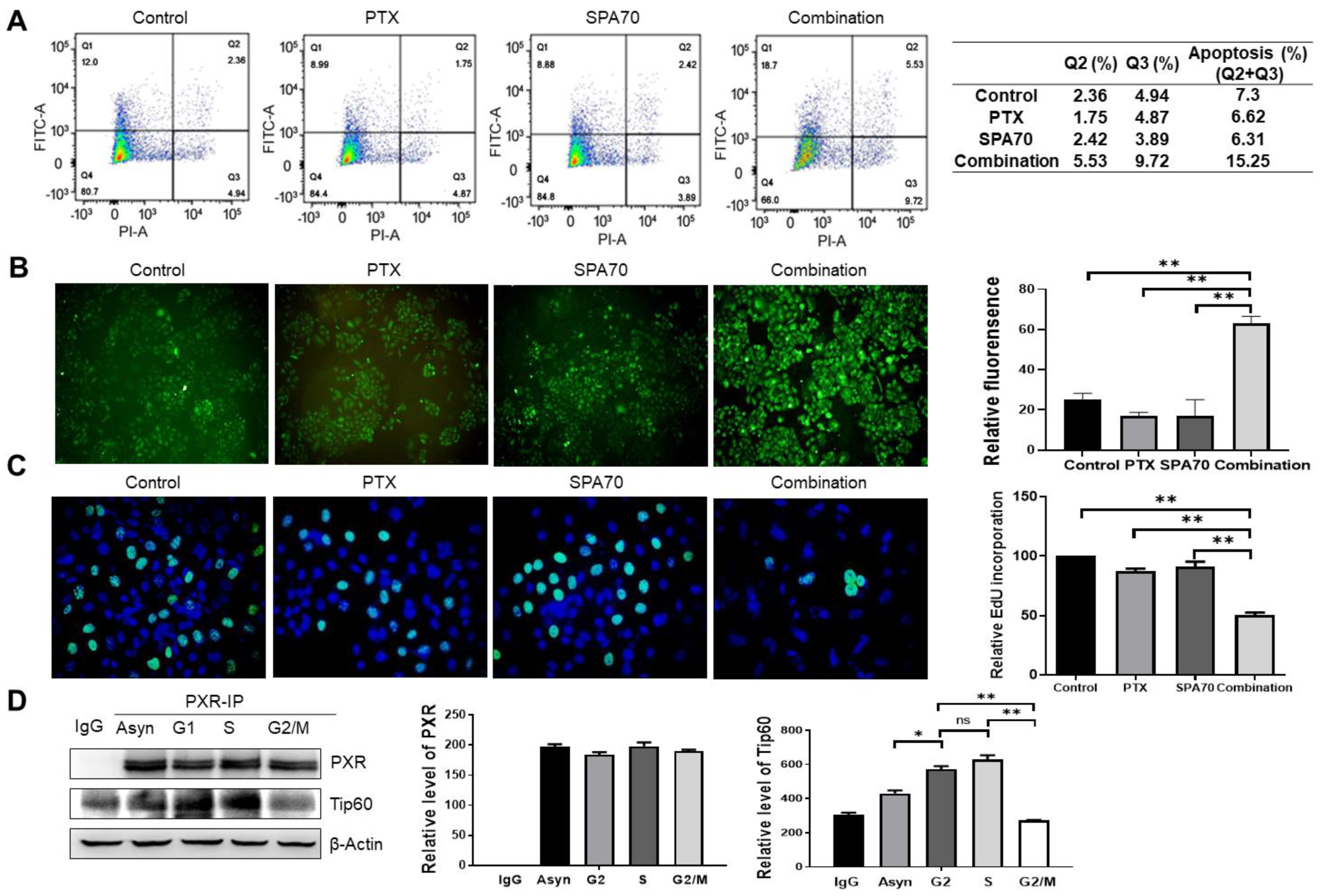
3.4. Combination Treatment Produces Apoptosis and Increases S-Phase Arrest in PTX-Resistant Cells
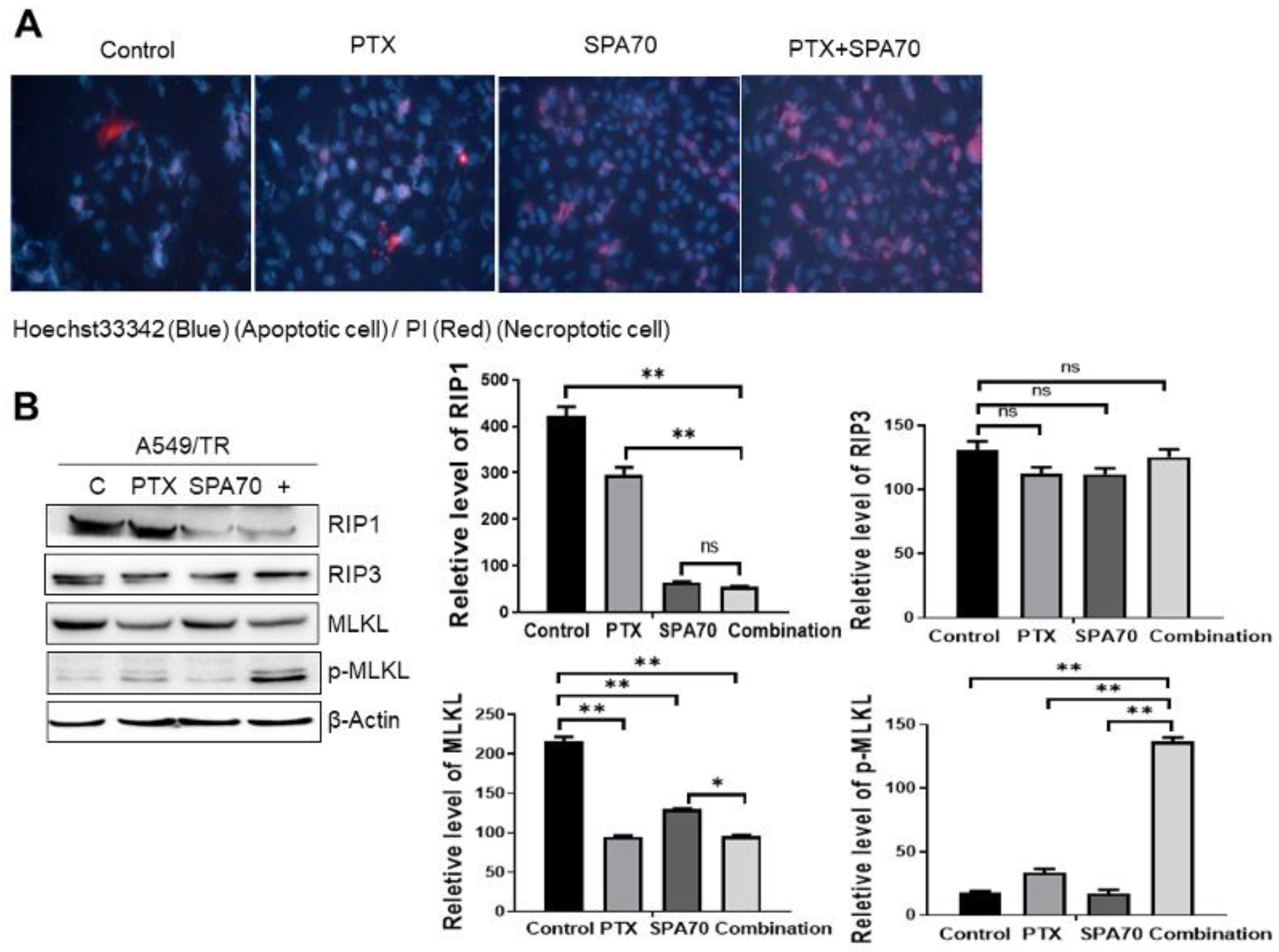
3.5. Combination Regimen Induces Necroptosis in Lung Cancer Cells
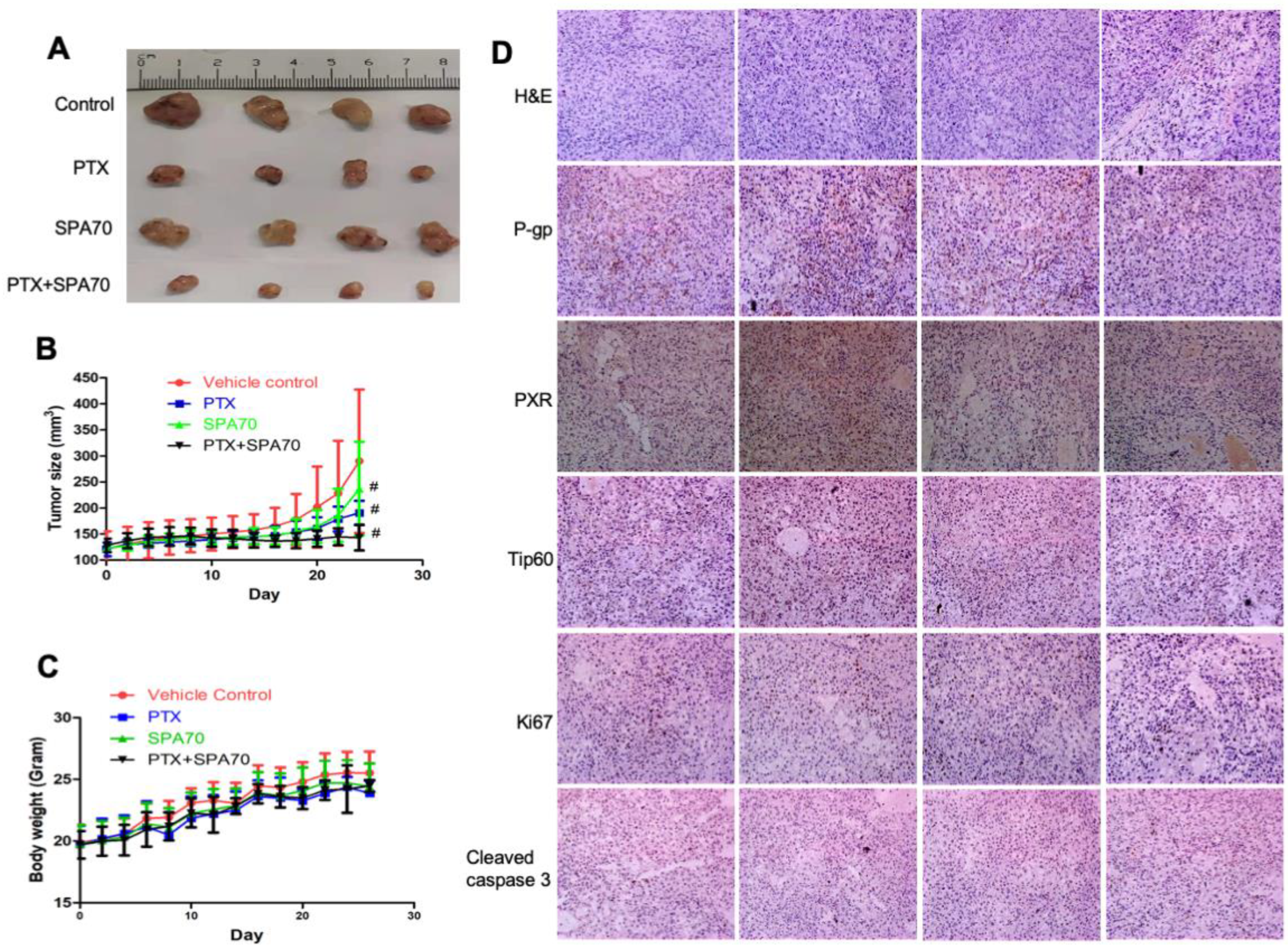
3.6. Combination of PTX and SPA70 Additively Suppresses PTX-Resistant Tumor Xenograft Growth In Vivo
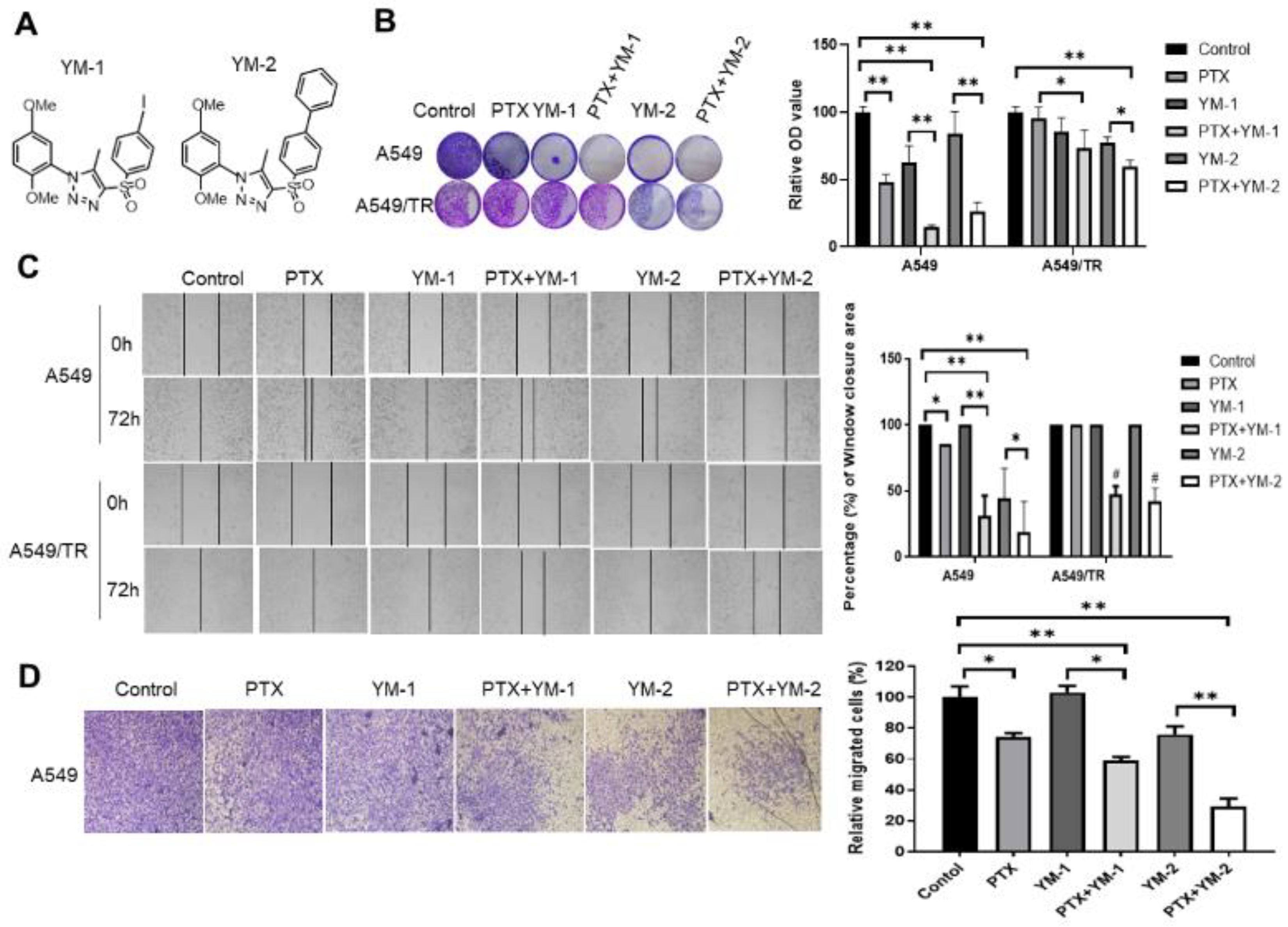
3.7. Two SPA70 Derivatives Also Have Synergistic Effects with PTX In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, S. Updates in non-small cell lung cancer. Clin. J. Oncol. Nurs. 2008, 12, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broderick, S.R. Adjuvant and Neoadjuvant Immunotherapy in Non-small Cell Lung Cancer. Thorac. Surg. Clin. 2020, 30, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Halliday, P.R.; Blakely, C.M.; Bivona, T.G. Emerging Targeted Therapies for the Treatment of Non-small Cell Lung Cancer. Curr. Oncol. Rep. 2019, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, M.; Koga, T.; Kiyota, H.; Horiguchi, T.; Shi, Q.W.; Hirose, K.; Uchida, T. Relationship between the structures of taxane derivatives and their microtubule polymerization activity. Biosci. Biotechnol. Biochem. 2012, 76, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Orth, M.; Unger, K.; Schoetz, U.; Belka, C.; Lauber, K. Taxane-mediated radiosensitization derives from chromosomal missegregation on tripolar mitotic spindles orchestrated by AURKA and TPX2. Oncogene 2018, 37, 52–62. [Google Scholar] [CrossRef]
- Mahmud, F.; Deng, S.; Chen, H.; Miller, D.D.; Li, W. Orally available tubulin inhibitor VERU-111 enhances antitumor efficacy in paclitaxel-resistant lung cancer. Cancer Lett. 2020, 495, 76–88. [Google Scholar] [CrossRef]
- Cui, H.; Arnst, K.; Miller, D.D.; Li, W. Recent Advances in Elucidating Paclitaxel Resistance Mechanisms in Non-small Cell Lung Cancer and Strategies to Overcome Drug Resistance. Curr. Med. Chem. 2020, 27, 6573–6595. [Google Scholar] [CrossRef]
- Aldonza, M.B.; Hong, J.Y.; Alinsug, M.V.; Song, J.; Lee, S.K. Multiplicity of acquired cross-resistance in paclitaxel-resistant cancer cells is associated with feedback control of TUBB3 via FOXO3a-mediated ABCB1 regulation. Oncotarget 2016, 7, 34395–34419. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, A.; Yang, H.; Cabral, F. Overexpression of mitotic centromere-associated Kinesin stimulates microtubule detachment and confers resistance to paclitaxel. Mol. Cancer Ther. 2011, 10, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, H.; Ren, Y.; Zou, S.; Fang, W.; Jiang, X.; Jia, L.; Li, M.; Liu, X.; Yuan, X.; et al. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis. 2016, 7, e2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarthi, J.; Elefant, F. dTip60 HAT activity controls synaptic bouton expansion at the Drosophila neuromuscular junction. PLoS ONE 2011, 6, e26202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmsen, S.; Meijerman, I.; Beijnen, J.H.; Schellens, J.H. Nuclear receptor mediated induction of cytochrome P450 3A4 by anticancer drugs: A key role for the pregnane X receptor. Cancer Chemother. Pharm. 2009, 64, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Huang, W.; Chen, F.; Hu, G.; Li, F.; Li, J.; Xuan, A. Pregnane X receptors regulate CYP2C8 and P-glycoprotein to impact on the resistance of NSCLC cells to Taxol. Cancer Med. 2016, 5, 3564–3571. [Google Scholar] [CrossRef]
- Yuan, T.; Sun, J.; Tian, J.; Hu, J.; Yin, H.; Yin, J. Involvement of ABC transporters in the detoxification of non-substrate nanoparticles in lung and cervical cancer cells. Toxicology 2021, 455, 152762. [Google Scholar] [CrossRef]
- Zhou, S.F. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef]
- Albermann, N.; Schmitz-Winnenthal, F.H.; Z’graggen, K.; Volk, C.; Hoffmann, M.M.; Haefeli, W.E.; Weiss, J. Expression of the drug transporters MDR1/ABCB1, MRP1/ABCC1, MRP2/ABCC2, BCRP/ABCG2, and PXR in peripheral blood mononuclear cells and their relationship with the expression in intestine and liver. Biochem. Pharmacol. 2005, 70, 949–958. [Google Scholar] [CrossRef]
- Bock, K.W. Functions and transcriptional regulation of adult human hepatic UDP-glucuronosyl-transferases (UGTs): Mechanisms responsible for interindividual variation of UGT levels. Biochem. Pharmacol. 2010, 80, 771–777. [Google Scholar] [CrossRef]
- Hayes, J.D.; Pulford, D.J. The glutathione S-Transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. 1995, 30, 445–600. [Google Scholar] [CrossRef]
- Niu, X.; Cui, H.; Gu, X.; Wu, T.; Sun, M.; Zhou, C.; Ma, M. Nuclear Receptor PXR Confers Irradiation Resistance by Promoting DNA Damage Response Through Stabilization of ATF3. Front. Oncol. 2022, 12, 837980. [Google Scholar] [CrossRef]
- Yuan, T.; Hu, J.; Zhu, X.; Yin, H.; Yin, J. Oxidative stress-mediated up-regulation of ABC transporters in lung cancer cells. J. Biochem. Mol. Toxicol. 2022, 36, e23095. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Xie, Y.; Lee, W.; Bookout, A.L.; Girard, L.; Raso, G.; Behrens, C.; Wistuba, I.I.; Gadzar, A.F.; Minna, J.D.; et al. Research resource: Diagnostic and therapeutic potential of nuclear receptor expression in lung cancer. Mol. Endocrinol. 2012, 26, 1443–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Wang, Y.M.; Chai, S.C.; Lv, L.; Zheng, J.; Wu, J.; Zhang, Q.; Wang, Y.D.; Griffin, P.R.; Chen, T. SPA70 is a potent antagonist of human pregnane X receptor. Nat. Commun. 2017, 8, 741. [Google Scholar] [CrossRef] [Green Version]
- Carnahan, V.E.; Redinbo, M.R. Structure and function of the human nuclear xenobiotic receptor PXR. Curr. Drug. Metab. 2005, 6, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Wu, T.; Li, G.; Gu, X.; Tian, Y.; Cui, H. Insights into the critical role of the PXR in preventing carcinogenesis and chemotherapeutic drug resistance. Int. J. Biol. Sci. 2022, 18, 742–759. [Google Scholar] [CrossRef]
- Kawana, K.; Ikuta, T.; Kobayashi, Y.; Gotoh, O.; Takeda, K.; Kawajiri, K. Molecular mechanism of nuclear translocation of an orphan nuclear receptor, SXR. Mol. Pharmacol. 2003, 63, 524–531. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, A.; Inagaki, K.; Igarashi-Migitaka, J.; Ozawa, Y.; Koibuchi, N. The endocrine disrupting chemical, diethylhexyl phthalate, activates MDR1 gene expression in human colon cancer LS174T cells. J. Endocrinol. 2006, 190, 897–902. [Google Scholar] [CrossRef] [Green Version]
- Geick, A.; Eichelbaum, M.; Burk, O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001, 276, 14581–14587. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.H.; Min, H.Y.; Chung, H.J.; Song, J.; Park, H.J.; Kim, S.; Lee, S.K. Anti-proliferative activity and suppression of P-glycoprotein by (-)-antofine, a natural phenanthroindolizidine alkaloid, in paclitaxel-resistant human lung cancer cells. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2012, 50, 1060–1065. [Google Scholar] [CrossRef]
- Kim, S.W.; Md, H.; Cho, M.; Kim, N.H.; Choi, H.Y.; Han, J.W.; Park, H.J.; Oh, J.W.; Shin, J.G. Role of 14-3-3 sigma in over-expression of P-gp by rifampin and paclitaxel stimulation through interaction with PXR. Cell. Signal. 2017, 31, 124–134. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Y.; Wang, M.T.; Zeng, S.; Nie, D. Human pregnane x receptor and resistance to chemotherapy in prostate cancer. Cancer Res. 2007, 67, 10361–10367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuyama, H.; Nakamura, K.; Nobumoto, E.; Hiramatsu, Y. Inhibition of pregnane X receptor pathway contributes to the cell growth inhibition and apoptosis of anticancer agents in ovarian cancer cells. Int. J. Oncol. 2016, 49, 1211–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.S. Overcoming drug resistance by regulating nuclear receptors. Adv. Drug Deliver Rev. 2010, 62, 1257–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, M.; Dash, A.K.; Ponnusamy, K.; Tyagi, R.K. Nuclear localization signal region in nuclear receptor PXR governs the receptor association with mitotic chromatin. Chromosome Res. 2018, 26, 255–276. [Google Scholar] [CrossRef]
- Umesono, K.; Evans, R.M. Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell 1989, 57, 1139–1146. [Google Scholar] [CrossRef]
- Pavek, P. Pregnane X Receptor (PXR)-Mediated Gene Repression and Cross-Talk of PXR with Other Nuclear Receptors via Coactivator Interactions. Front. Pharmacol. 2016, 7, 456. [Google Scholar] [CrossRef] [Green Version]
- Pondugula, S.R.; Pavek, P.; Mani, S. Pregnane X Receptor and Cancer: Context-Specificity is Key. Nucl. Recept. Res. 2016, 3, 101198. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y. Epigenetic regulation of pregnane X receptor activity. Drug Metab. Rev. 2013, 45, 166–172. [Google Scholar] [CrossRef]
- Rosenfeld, M.G.; Lunyak, V.V.; Glass, C.K. Sensors and signals: A coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006, 20, 1405–1428. [Google Scholar] [CrossRef] [Green Version]
- Bakshi, K.; Ranjitha, B.; Dubey, S.; Jagannadham, J.; Jaiswal, B.; Gupta, A. Novel complex of HAT protein TIP60 and nuclear receptor PXR promotes cell migration and adhesion. Sci. Rep. 2017, 7, 3635. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Wang, Q.; Miller, D.D.; Li, W. The Tubulin Inhibitor VERU-111 in Combination With Vemurafenib Provides an Effective Treatment of Vemurafenib-Resistant A375 Melanoma. Front. Pharmacol. 2021, 12, 637098. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Guo, M.; Xu, D.; Ding, Z.C.; Zhou, G.; Ding, H.F.; Zhang, J.; Tang, Y.; Yan, C. The stress-responsive gene ATF3 regulates the histone acetyltransferase Tip60. Nat. Commun. 2015, 6, 6752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, W. Possible mechanisms of paclitaxel-induced apoptosis. Biochem. Pharmacol. 1999, 57, 1215–1221. [Google Scholar]
- Shan, B.; Pan, H.; Najafov, A.; Yuan, J. Necroptosis in development and diseases. Genes Dev. 2018, 32, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Huang, M.; Yang, X.; Hou, J. MLKL: Functions beyond serving as the Executioner of Necroptosis. Theranostics 2021, 11, 4759–4769. [Google Scholar] [CrossRef]
- Naspinski, C.; Gu, X.; Zhou, G.D.; Mertens-Talcott, S.U.; Donnelly, K.C.; Tian, Y. Pregnane X receptor protects HepG2 cells from BaP-induced DNA damage. Toxicol. Sci. 2008, 104, 67–73. [Google Scholar] [CrossRef]
- Cui, H.; Gu, X.; Chen, J.; Xie, Y.; Ke, S.; Wu, J.; Golovko, A.; Morpurgo, B.; Yan, C.; Phillips, T.D.; et al. Pregnane X receptor regulates the AhR/Cyp1A1 pathway and protects liver cells from benzo-[α]-pyrene-induced DNA damage. Toxicol. Lett. 2017, 275, 67–76. [Google Scholar] [CrossRef]
- Kim, E.S. Chemotherapy Resistance in Lung Cancer. Adv. Exp. Med. Biol. 2016, 893, 189–209. [Google Scholar]
- Bhoumik, A.; Singha, N.; O’Connell, M.J.; Ronai, Z.A. Regulation of TIP60 by ATF2 modulates ATM activation. J. Biol. Chem. 2008, 283, 17605–17614. [Google Scholar] [CrossRef] [Green Version]
- Sugatani, J.; Noguchi, Y.; Hattori, Y.; Yamaguchi, M.; Yamazaki, Y.; Ikari, A. Threonine-408 Regulates the Stability of Human Pregnane X Receptor through Its Phosphorylation and the CHIP/Chaperone-Autophagy Pathway. Drug Metab. Dispos. Biol. Fate Chem. 2016, 44, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Moreau, F.; Chadee, K. PPARγ is an E3 ligase that induces the degradation of NFκB/p65. Nat. Commun. 2012, 3, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassi, C.; Li, Y.T.; Khu, K.; Mateo, F.; Baniasadi, P.S.; Elia, A.; Mason, J.; Stambolic, V.; Pujana, M.A.; Mak, T.W.; et al. The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell Death Differ. 2016, 23, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Guo, F.; Xu, H.; Liang, W.; Wang, C.; Yang, X.D. Combination Therapy using Co-encapsulated Resveratrol and Paclitaxel in Liposomes for Drug Resistance Reversal in Breast Cancer Cells in vivo. Sci. Rep. 2016, 6, 22390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhou, Y.; Zheng, Z.; Li, J.; Yan, Y.; Wu, W. Sulforaphane metabolites reduce resistance to paclitaxel via microtubule disruption. Cell Death Dis. 2018, 9, 1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchman, C.D.; Chai, S.C.; Chen, T. A current structural perspective on PXR and CAR in drug metabolism. Expert Opin Drug Metab. Toxicol. 2018, 14, 635–647. [Google Scholar] [CrossRef]
- Chai, S.C.; Wright, W.C.; Chen, T. Strategies for developing pregnane X receptor antagonists: Implications from metabolism to cancer. Med. Res. Rev. 2020, 40, 1061–1083. [Google Scholar] [CrossRef] [PubMed]
- Sugar, A.M.; Alsip, S.G.; Galgiani, J.N.; Graybill, J.R.; Dismukes, W.E.; Cloud, G.A.; Craven, P.C.; Stevens, D.A. Pharmacology and toxicity of high-dose ketoconazole. Antimicrob. Agents Chemother. 1987, 31, 1874–1878. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PTX IC50 (μM) | SPA70 IC50 (μM) | |
|---|---|---|
| A549 | 0.01 ± 0.00 | 2.41 ± 0.13 |
| A549/TR | 0.50 ± 0.22 | 5.62 ± 0.53 |
| H460 | 0.01 ± 0.00 | 4.63 ± 3.99 |
| H460/TR | 0.80 ± 0.20 | 7.10 ± 2.63 |
| RI (A549/TR/A549) | 100.4 | 2.33 |
| RI (H460/TR/H460) | 80 | 1.53 |
| A549 | H460 | A549/TR | H460/TR | |||||
|---|---|---|---|---|---|---|---|---|
| CI ED50 | CI ED50 | CI ED50 | CI ED75 | CI ED90 | CI ED50 | CI ED75 | CI ED90 | |
| Combination (PTX + SPA70) | 0.91 | 0.12 | 0.03 | 0.01 | 0.004 | 0.99 | 0.05 | 0.002 |
| Treatment Group | TGI (100%) |
|---|---|
| Vehicle control | - |
| PTX | 58.4 ± 9.0 # |
| SPA70 | 32.0 ± 7.8 # |
| PTX + SPA70 | 89.5 ± 7.2 #,* |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, X.; Wu, T.; Yin, Q.; Gu, X.; Li, G.; Zhou, C.; Ma, M.; Su, L.; Tang, S.; Tian, Y.; et al. Combination of Paclitaxel and PXR Antagonist SPA70 Reverses Paclitaxel-Resistant Non-Small Cell Lung Cancer. Cells 2022, 11, 3094. https://doi.org/10.3390/cells11193094
Niu X, Wu T, Yin Q, Gu X, Li G, Zhou C, Ma M, Su L, Tang S, Tian Y, et al. Combination of Paclitaxel and PXR Antagonist SPA70 Reverses Paclitaxel-Resistant Non-Small Cell Lung Cancer. Cells. 2022; 11(19):3094. https://doi.org/10.3390/cells11193094
Chicago/Turabian StyleNiu, Xiaxia, Ting Wu, Qishuang Yin, Xinsheng Gu, Gege Li, Changlong Zhou, Mei Ma, Li Su, Shu Tang, Yanan Tian, and et al. 2022. "Combination of Paclitaxel and PXR Antagonist SPA70 Reverses Paclitaxel-Resistant Non-Small Cell Lung Cancer" Cells 11, no. 19: 3094. https://doi.org/10.3390/cells11193094
APA StyleNiu, X., Wu, T., Yin, Q., Gu, X., Li, G., Zhou, C., Ma, M., Su, L., Tang, S., Tian, Y., Yang, M., & Cui, H. (2022). Combination of Paclitaxel and PXR Antagonist SPA70 Reverses Paclitaxel-Resistant Non-Small Cell Lung Cancer. Cells, 11(19), 3094. https://doi.org/10.3390/cells11193094