
Patient Selection Approaches in FGFR Inhibitor Trials—Many Paths to the Same End?


 ,
,  and
and 
Abstract
:1. Introduction
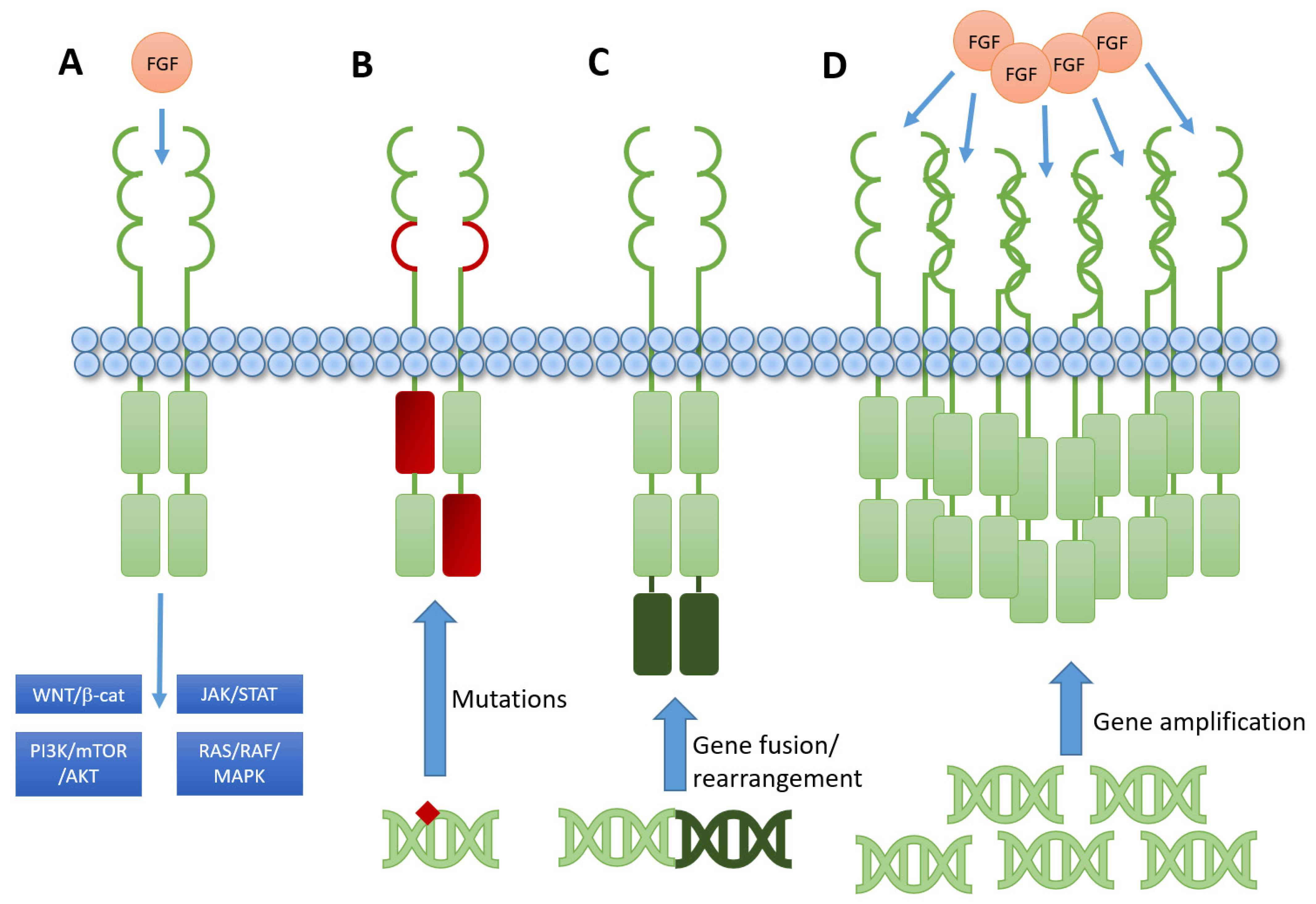
2. Brief Introduction to FGFR Biology and Signaling
3. FGFR Inhibitors in Clinical Trials
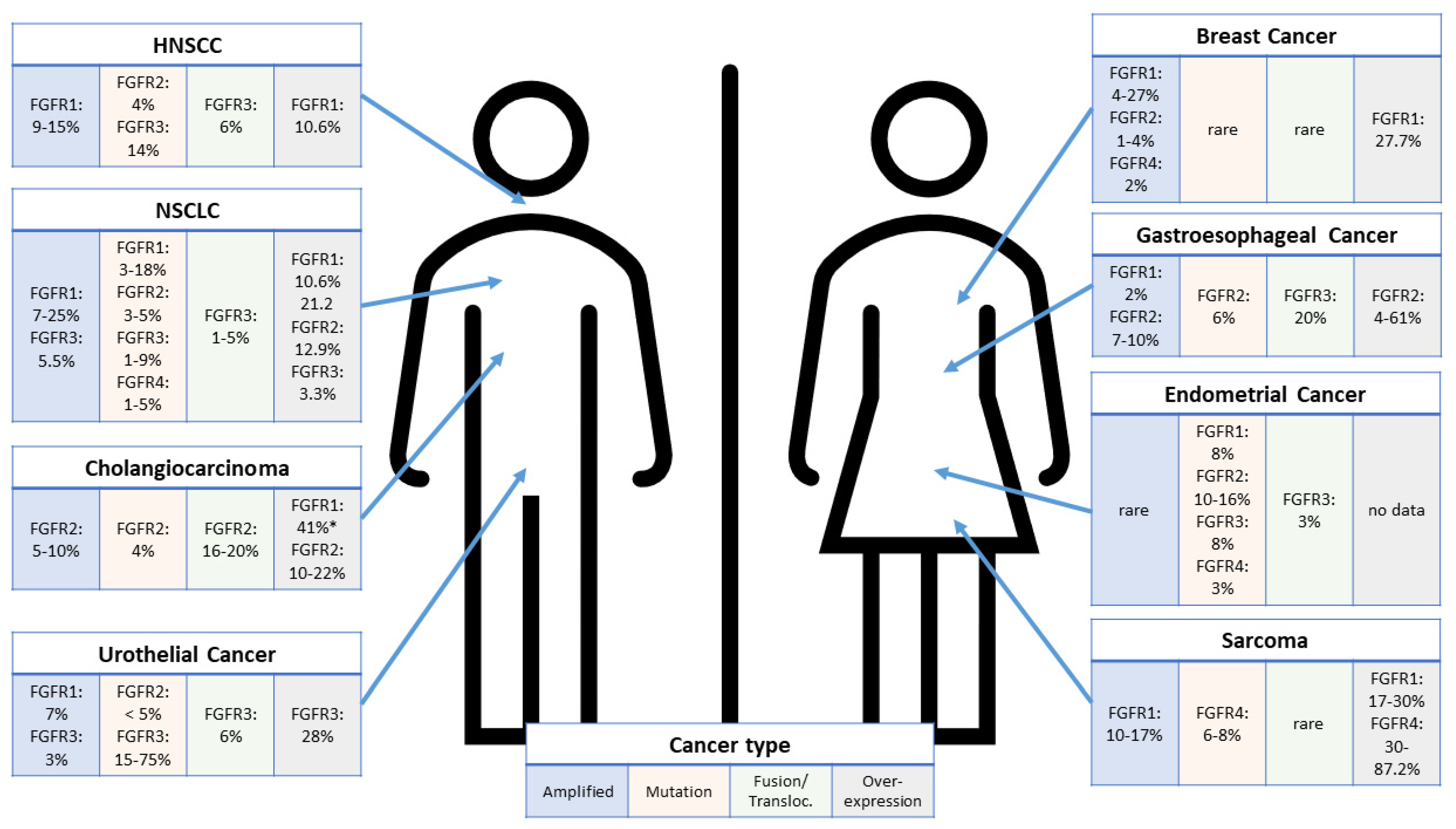
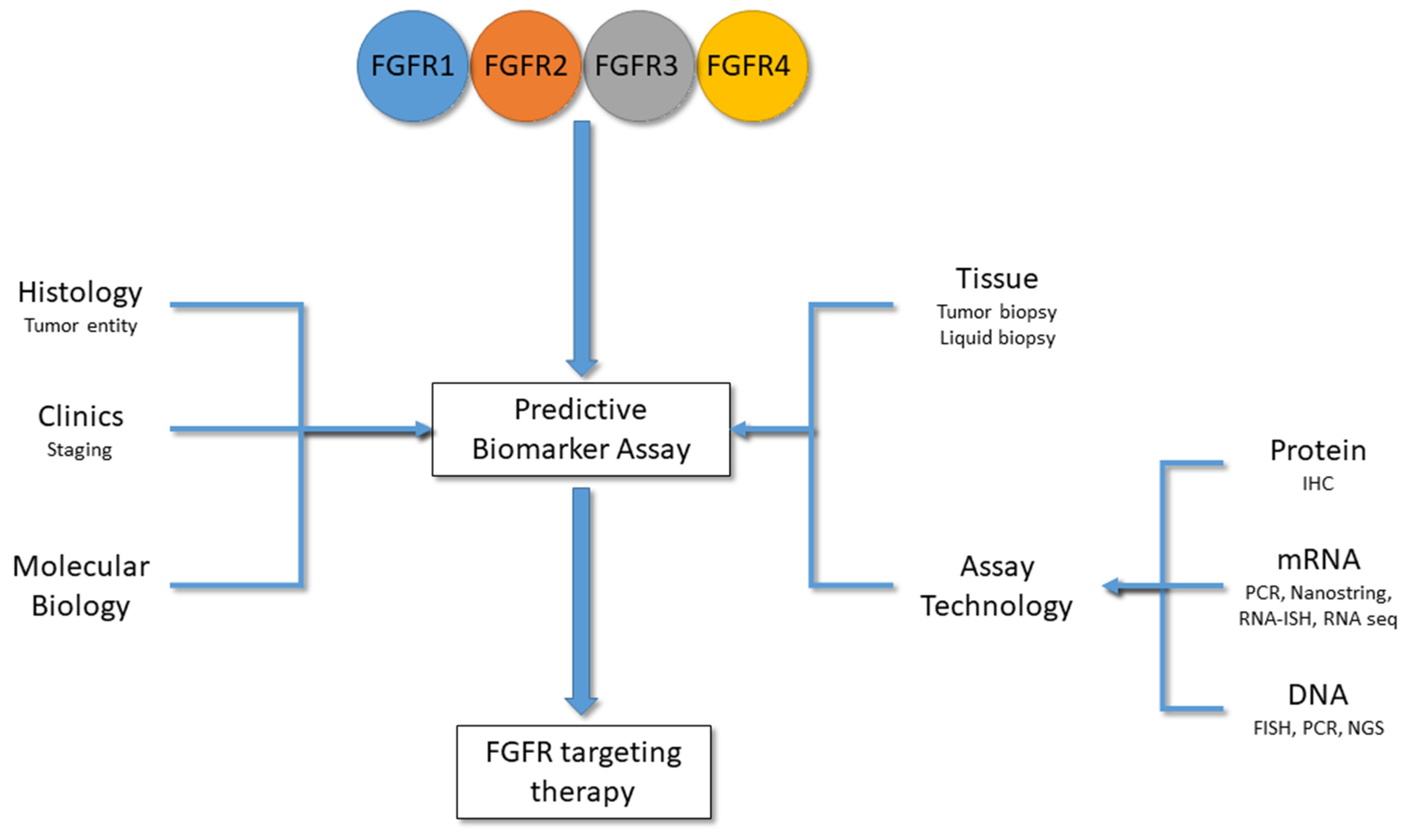
4. Predictive Biomarkers for FGFR Inhibitors
4.1. Immunohistochemistry
4.2. FISH/CISH to Detect Gene Copy Number Variations
4.3. mRNA Expression Technologies
4.4. Next-Generation DNA Sequencing (NGS)
4.5. NGS of ctDNA (Liquid Biopsies)
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Weiss, F.; Lauffenburger, D.; Friedl, P. Towards Targeting of Shared Mechanisms of Cancer Metastasis and Therapy Resistance. Nat. Rev. Cancer 2022, 22, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, C.; Li, S.; Bai, H.; Wang, J. Tumor Immune Microenvironment in Epidermal Growth Factor Receptor-Mutated Non-Small Cell Lung Cancer before and after Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Treatment: A Narrative Review. Transl. Lung Cancer Res. 2021, 10, 3823–3839. [Google Scholar] [CrossRef]
- National Guideline Alliance (UK). Use of Molecular Biomarkers to Guide Systemic Therapy: Colorectal Cancer (Update): Evidence Review B1; NICE Evidence Reviews Collection; National Institute for Health and Care Excellence (NICE): London, UK, 2020; ISBN 978-1-4731-3657-1. [Google Scholar]
- Jørgensen, J.T.; Winther, H.; Askaa, J.; Andresen, L.; Olsen, D.; Mollerup, J. A Companion Diagnostic With Significant Clinical Impact in Treatment of Breast and Gastric Cancer. Front. Oncol. 2021, 11, 676939. [Google Scholar] [CrossRef] [PubMed]
- Papachristos, A.; Sivolapenko, G.B. Pharmacogenomics, Pharmacokinetics and Circulating Proteins as Biomarkers for Bevacizumab Treatment Optimization in Patients with Cancer: A Review. J. Pers. Med. 2020, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Kang, C. Infigratinib: First Approval. Drugs 2021, 81, 1355–1360. [Google Scholar] [CrossRef]
- Schuler, M.; Cho, B.C.; Sayehli, C.M.; Navarro, A.; Soo, R.A.; Richly, H.; Cassier, P.A.; Tai, D.; Penel, N.; Nogova, L.; et al. Rogaratinib in Patients with Advanced Cancers Selected by FGFR MRNA Expression: A Phase 1 Dose-Escalation and Dose-Expansion Study. Lancet Oncol. 2019, 20, 1454–1466. [Google Scholar] [CrossRef]
- Subbiah, V.; Iannotti, N.O.; Gutierrez, M.; Smith, D.C.; Féliz, L.; Lihou, C.F.; Tian, C.; Silverman, I.M.; Ji, T.; Saleh, M. FIGHT-101, a First-in-Human Study of Potent and Selective FGFR 1-3 Inhibitor Pemigatinib in Pan-Cancer Patients with FGF/FGFR Alterations and Advanced Malignancies. Ann. Oncol. 2022, 33, 522–533. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Bang, Y.-J.; Mansoor, W.; Petty, R.D.; Chao, Y.; Cunningham, D.; Ferry, D.R.; Smith, N.R.; Frewer, P.; Ratnayake, J.; et al. A Randomized, Open-Label Study of the Efficacy and Safety of AZD4547 Monotherapy versus Paclitaxel for the Treatment of Advanced Gastric Adenocarcinoma with FGFR2 Polysomy or Gene Amplification. Ann. Oncol. 2017, 28, 1316–1324. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast Growth Factor Signalling: From Development to Cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, F.C.; O’Sullivan, H.; Smyth, E.; McDermott, R.; Viterbo, A. Fibroblast Growth Factor Receptors, Developmental Corruption and Malignant Disease. Carcinogenesis 2013, 34, 2198–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular Signaling by Fibroblast Growth Factor Receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ibrahimi, O.A.; Olsen, S.K.; Umemori, H.; Mohammadi, M.; Ornitz, D.M. Receptor Specificity of the Fibroblast Growth Factor Family. The Complete Mammalian FGF Family. J. Biol. Chem. 2006, 281, 15694–15700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heroult, M.; Ellinghaus, P.; Ince, S.; Ocker, M. Fibroblast Growth Factor Receptor Signaling in Cancer Biology and Treatment. Curr. Signal Transduct. Ther. 2014, 9, 15–25. [Google Scholar] [CrossRef]
- Ong, S.H.; Guy, G.R.; Hadari, Y.R.; Laks, S.; Gotoh, N.; Schlessinger, J.; Lax, I. FRS2 Proteins Recruit Intracellular Signaling Pathways by Binding to Diverse Targets on Fibroblast Growth Factor and Nerve Growth Factor Receptors. Mol. Cell. Biol. 2000, 20, 979–989. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.H.; Hadari, Y.R.; Gotoh, N.; Guy, G.R.; Schlessinger, J.; Lax, I. Stimulation of Phosphatidylinositol 3-Kinase by Fibroblast Growth Factor Receptors Is Mediated by Coordinated Recruitment of Multiple Docking Proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 6074–6079. [Google Scholar] [CrossRef] [Green Version]
- Kouhara, H.; Hadari, Y.R.; Spivak-Kroizman, T.; Schilling, J.; Bar-Sagi, D.; Lax, I.; Schlessinger, J. A Lipid-Anchored Grb2-Binding Protein That Links FGF-Receptor Activation to the Ras/MAPK Signaling Pathway. Cell 1997, 89, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Hart, K.C.; Robertson, S.C.; Kanemitsu, M.Y.; Meyer, A.N.; Tynan, J.A.; Donoghue, D.J. Transformation and Stat Activation by Derivatives of FGFR1, FGFR3, and FGFR4. Oncogene 2000, 19, 3309–3320. [Google Scholar] [CrossRef] [Green Version]
- Quintanal-Villalonga, Á.; Ferrer, I.; Guruceaga, E.; Cirauqui, C.; Marrugal, Á.; Ojeda, L.; García, S.; Zugazagoitia, J.; Muñoz-Galván, S.; Lopez-Rios, F.; et al. FGFR1 and FGFR4 Oncogenicity Depends on N-Cadherin and Their Co-Expression May Predict FGFR-Targeted Therapy Efficacy. EBioMedicine 2020, 53, 102683. [Google Scholar] [CrossRef]
- Nguyen, T.; Duchesne, L.; Sankara Narayana, G.H.N.; Boggetto, N.; Fernig, D.D.; Uttamrao Murade, C.; Ladoux, B.; Mège, R.-M. Enhanced Cell-Cell Contact Stability and Decreased N-Cadherin-Mediated Migration upon Fibroblast Growth Factor Receptor-N-Cadherin Cross Talk. Oncogene 2019, 38, 6283–6300. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Mège, R.M. N-Cadherin and Fibroblast Growth Factor Receptors Crosstalk in the Control of Developmental and Cancer Cell Migrations. Eur. J. Cell Biol. 2016, 95, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Buchtova, M.; Oralova, V.; Aklian, A.; Masek, J.; Vesela, I.; Ouyang, Z.; Obadalova, T.; Konecna, Z.; Spoustova, T.; Pospisilova, T.; et al. Fibroblast Growth Factor and Canonical WNT/β-Catenin Signaling Cooperate in Suppression of Chondrocyte Differentiation in Experimental Models of FGFR Signaling in Cartilage. Biochim. Biophys. Acta 2015, 1852, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Mavila, N.; James, D.; Utley, S.; Cu, N.; Coblens, O.; Mak, K.; Rountree, C.B.; Kahn, M.; Wang, K.S. Fibroblast Growth Factor Receptor-Mediated Activation of AKT-β-Catenin-CBP Pathway Regulates Survival and Proliferation of Murine Hepatoblasts and Hepatic Tumor Initiating Stem Cells. PLoS ONE 2012, 7, e50401. [Google Scholar] [CrossRef] [Green Version]
- Carstens, J.L.; Shahi, P.; Van Tsang, S.; Smith, B.; Creighton, C.J.; Zhang, Y.; Seamans, A.; Seethammagari, M.; Vedula, I.; Levitt, J.M.; et al. FGFR1-WNT-TGF-β Signaling in Prostate Cancer Mouse Models Recapitulates Human Reactive Stroma. Cancer Res. 2014, 74, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaidee, R.; Kukongviriyapan, V.; Senggunprai, L.; Prawan, A.; Jusakul, A.; Laphanuwat, P.; Kongpetch, S. Inhibition of FGFR2 Enhances Chemosensitivity to Gemcitabine in Cholangiocarcinoma through the AKT/MTOR and EMT Signaling Pathways. Life Sci. 2022, 296, 120427. [Google Scholar] [CrossRef]
- Roy Burman, D.; Das, S.; Das, C.; Bhattacharya, R. Alternative Splicing Modulates Cancer Aggressiveness: Role in EMT/Metastasis and Chemoresistance. Mol. Biol. Rep. 2021, 48, 897–914. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Bottaro, D.P.; Fleming, T.P.; Smith, C.L.; Burgess, W.H.; Chan, A.M.; Aaronson, S.A. Determination of Ligand-Binding Specificity by Alternative Splicing: Two Distinct Growth Factor Receptors Encoded by a Single Gene. Proc. Natl. Acad. Sci. USA 1992, 89, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast Growth Factors and Their Receptors in Cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Niu, D.; Tham Sjin, R.T.; Dubrovskiy, A.; Zhu, Z.; McDonald, J.J.; Fahnoe, K.; Wang, Z.; Munson, M.; Scholte, A.; et al. Discovery of Selective, Covalent FGFR4 Inhibitors with Antitumor Activity in Models of Hepatocellular Carcinoma. ACS Med. Chem. Lett. 2020, 11, 1899–1904. [Google Scholar] [CrossRef]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.-W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.-Y.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1696–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.-Z.; Roychowdhury, S. Fibroblast Growth Factor Receptors in Cancer: Genetic Alterations, Diagnostics, Therapeutic Targets and Mechanisms of Resistance. Br. J. Cancer 2021, 124, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, C.; Lu, G.; Hu, Z.; Chen, Q.; Du, X. FGF/FGFR Signaling Pathway Involved Resistance in Various Cancer Types. J. Cancer 2020, 11, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Liu, X.; Wang, S.; Zhang, Z.; Wu, Z.; Zhang, X.; Li, J. Prognostic Value of FGFR Gene Amplification in Patients with Different Types of Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e105524. [Google Scholar] [CrossRef]
- Ahmad, I.; Iwata, T.; Leung, H.Y. Mechanisms of FGFR-Mediated Carcinogenesis. Biochim. Biophys. Acta 2012, 1823, 850–860. [Google Scholar] [CrossRef] [Green Version]
- Dutt, A.; Salvesen, H.B.; Chen, T.-H.; Ramos, A.H.; Onofrio, R.C.; Hatton, C.; Nicoletti, R.; Winckler, W.; Grewal, R.; Hanna, M.; et al. Drug-Sensitive FGFR2 Mutations in Endometrial Carcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 8713–8717. [Google Scholar] [CrossRef] [Green Version]
- Gust, K.M.; McConkey, D.J.; Awrey, S.; Hegarty, P.K.; Qing, J.; Bondaruk, J.; Ashkenazi, A.; Czerniak, B.; Dinney, C.P.; Black, P.C. Fibroblast Growth Factor Receptor 3 Is a Rational Therapeutic Target in Bladder Cancer. Mol. Cancer Ther. 2013, 12, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Repetto, M.; Crimini, E.; Giugliano, F.; Morganti, S.; Belli, C.; Curigliano, G. Selective FGFR/FGF Pathway Inhibitors: Inhibition Strategies, Clinical Activities, Resistance Mutations, and Future Directions. Expert Rev. Clin. Pharmacol. 2021, 14, 1233–1252. [Google Scholar] [CrossRef]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and Focal FGFR1 Amplification Associates with Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Yao, Y.-W.; Zeng, J.-L.; Liang, W.-J.; Wang, L.; Bai, C.-Q.; Liu, C.-H.; Song, Y. Prognostic Value of FGFR1 Gene Copy Number in Patients with Non-Small Cell Lung Cancer: A Meta-Analysis. J. Thorac. Dis. 2014, 6, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Cohen, O.; Kowalski, K.J.; Kusiel, J.G.; Buendia-Buendia, J.E.; Cuoco, M.S.; Exman, P.; Wander, S.A.; Waks, A.G.; Nayar, U.; et al. Acquired FGFR and FGF Alterations Confer Resistance to Estrogen Receptor (ER) Targeted Therapy in ER+ Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 5974–5989. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Simpson, P.T.; Turner, N.C.; Lambros, M.B.; Jones, C.; Mackay, A.; Grigoriadis, A.; Sarrio, D.; Savage, K.; Dexter, T.; et al. FGFR1 Emerges as a Potential Therapeutic Target for Lobular Breast Carcinomas. Clin. Cancer Res. 2006, 12, 6652–6662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.; Arao, T.; Hamaguchi, T.; Shimada, Y.; Kato, K.; Oda, I.; Taniguchi, H.; Koizumi, F.; Yanagihara, K.; Sasaki, H.; et al. FGFR2 Gene Amplification and Clinicopathological Features in Gastric Cancer. Br. J. Cancer 2012, 106, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.-J.; Jung, E.-J.; Min, S.Y.; Kim, M.A.; Kim, W.H. Fibroblast Growth Factor Receptor 2 Gene Amplification Status and Its Clinicopathologic Significance in Gastric Carcinoma. Hum. Pathol. 2012, 43, 1559–1566. [Google Scholar] [CrossRef]
- Williams, S.V.; Hurst, C.D.; Knowles, M.A. Oncogenic FGFR3 Gene Fusions in Bladder Cancer. Hum. Mol. Genet. 2013, 22, 795–803. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Urothelial Bladder Carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.-F.; et al. Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [Green Version]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast Growth Factor Receptor 2 Tyrosine Kinase Fusions Define a Unique Molecular Subtype of Cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Bao, Y.; Gabrielpillai, J.; Dietrich, J.; Zarbl, R.; Strieth, S.; Schröck, F.; Dietrich, D. Fibroblast Growth Factor (FGF), FGF Receptor (FGFR), and Cyclin D1 (CCND1) DNA Methylation in Head and Neck Squamous Cell Carcinomas Is Associated with Transcriptional Activity, Gene Amplification, Human Papillomavirus (HPV) Status, and Sensitivity to Tyrosine Kinase Inhibitors. Clin. Epigenetics 2021, 13, 228. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, X.; Wang, X.; Liu, H.; Geck, R.C.; Tewari, A.K.; Xiao, T.; Font-Tello, A.; Lim, K.; Jones, K.L.; et al. FGFR-Inhibitor-Mediated Dismissal of SWI/SNF Complexes from YAP-Dependent Enhancers Induces Adaptive Therapeutic Resistance. Nat. Cell Biol. 2021, 23, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Bogatyrova, O.; Mattsson, J.S.M.; Ross, E.M.; Sanderson, M.P.; Backman, M.; Botling, J.; Brunnström, H.; Kurppa, P.; La Fleur, L.; Strell, C.; et al. FGFR1 Overexpression in Non-Small Cell Lung Cancer Is Mediated by Genetic and Epigenetic Mechanisms and Is a Determinant of FGFR1 Inhibitor Response. Eur. J. Cancer 2021, 151, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Huang, T.; Zou, Q.; Liu, D.; Wang, Y.; Tan, X.; Wei, Y.; Qiu, H. FGFR2 Promotes Expression of PD-L1 in Colorectal Cancer via the JAK/STAT3 Signaling Pathway. J. Immunol. 2019, 202, 3065–3075. [Google Scholar] [CrossRef] [PubMed]
- Sweis, R.F.; Spranger, S.; Bao, R.; Paner, G.P.; Stadler, W.M.; Steinberg, G.; Gajewski, T.F. Molecular Drivers of the Non-T-Cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol. Res. 2016, 4, 563–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, T.L.; Weir, W.H.; Mayhew, G.M.; Shibata, Y.; Eulitt, P.; Uronis, J.M.; Zhou, M.; Nielsen, M.; Smith, A.B.; Woods, M.; et al. Fibroblast Growth Factor Receptor 3 Alterations and Response to Immune Checkpoint Inhibition in Metastatic Urothelial Cancer: A Real World Experience. Br. J. Cancer 2021, 125, 1251–1260. [Google Scholar] [CrossRef]
- Grantzau, T.; Toft, B.G.; Melchior, L.C.; Elversang, J.; Stormoen, D.R.; Omland, L.H.; Pappot, H. PD-L1 Expression and FGFR-Mutations among Danish Patients Diagnosed with Metastatic Urothelial Carcinoma: A Retrospective and Descriptive Study. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2022, 130, 498–506. [Google Scholar] [CrossRef]
- Akhand, S.S.; Liu, Z.; Purdy, S.C.; Abdullah, A.; Lin, H.; Cresswell, G.M.; Ratliff, T.L.; Wendt, M. Pharmacologic Inhibition of FGFR Modulates the Metastatic Immune Microenvironment and Promotes Response to Immune Checkpoint Blockade. Cancer Immunol. Res. 2020, 8, 1542–1553. [Google Scholar] [CrossRef]
- Deng, H.; Kan, A.; Lyu, N.; Mu, L.; Han, Y.; Liu, L.; Zhang, Y.; Duan, Y.; Liao, S.; Li, S.; et al. Dual Vascular Endothelial Growth Factor Receptor and Fibroblast Growth Factor Receptor Inhibition Elicits Antitumor Immunity and Enhances Programmed Cell Death-1 Checkpoint Blockade in Hepatocellular Carcinoma. Liver Cancer 2020, 9, 338–357. [Google Scholar] [CrossRef]
- Palakurthi, S.; Kuraguchi, M.; Zacharek, S.J.; Zudaire, E.; Huang, W.; Bonal, D.M.; Liu, J.; Dhaneshwar, A.; DePeaux, K.; Gowaski, M.R.; et al. The Combined Effect of FGFR Inhibition and PD-1 Blockade Promotes Tumor-Intrinsic Induction of Antitumor Immunity. Cancer Immunol. Res. 2019, 7, 1457–1471. [Google Scholar] [CrossRef]
- Wu, Y.; Yi, Z.; Li, J.; Wei, Y.; Feng, R.; Liu, J.; Huang, J.; Chen, Y.; Wang, X.; Sun, J.; et al. FGFR Blockade Boosts T Cell Infiltration into Triple-Negative Breast Cancer by Regulating Cancer-Associated Fibroblasts. Theranostics 2022, 12, 4564–4580. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Komatsuda, H.; Yamaki, H.; Kumai, T.; Hayashi, R.; Wakisaka, R.; Nagato, T.; Ohkuri, T.; Kosaka, A.; Ohara, K.; et al. Immunomodulation via FGFR Inhibition Augments FGFR1 Targeting T-Cell Based Antitumor Immunotherapy for Head and Neck Squamous Cell Carcinoma. Oncoimmunology 2022, 11, 2021619. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M. Fibroblast Growth Factor Signaling in Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Paving the Way to Hepatocellular Carcinoma. World J. Gastroenterol. 2020, 26, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Seitz, T.; Hellerbrand, C. Role of Fibroblast Growth Factor Signalling in Hepatic Fibrosis. Liver Int. 2021, 41, 1201–1215. [Google Scholar] [CrossRef]
- Tsimafeyeu, I.; Ludes-Meyers, J.; Stepanova, E.; Daeyaert, F.; Khochenkov, D.; Joose, J.-B.; Solomko, E.; Van Akene, K.; Peretolchina, N.; Yin, W.; et al. Targeting FGFR2 with Alofanib (RPT835) Shows Potent Activity in Tumour Models. Eur. J. Cancer 2016, 61, 20–28. [Google Scholar] [CrossRef]
- Herbert, C.; Schieborr, U.; Saxena, K.; Juraszek, J.; De Smet, F.; Alcouffe, C.; Bianciotto, M.; Saladino, G.; Sibrac, D.; Kudlinzki, D.; et al. Molecular Mechanism of SSR128129E, an Extracellularly Acting, Small-Molecule, Allosteric Inhibitor of FGF Receptor Signaling. Cancer Cell 2016, 30, 176–178. [Google Scholar] [CrossRef]
- Loriot, Y.; Schuler, M.; Iyer, G.; Witt, O.; Doi, T.; Qin, S.; Tabernero, J.; Reardon, D.A.; Massard, C.; Palmer, D.H.; et al. Tumor Agnostic Efficacy and Safety of Erdafitinib in Patients (Pts) with Advanced Solid Tumors with Prespecified Fibroblast Growth Factor Receptor Alterations (FGFRalt) in RAGNAR: Interim Analysis (IA) Results. J. Clin. Oncol. 2022, 40, 3007. [Google Scholar] [CrossRef]
- Kuriwaki, I.; Kameda, M.; Iikubo, K.; Hisamichi, H.; Kawamoto, Y.; Kikuchi, S.; Moritomo, H.; Terasaka, T.; Iwai, Y.; Noda, A.; et al. Discovery of ASP5878: Synthesis and Structure-Activity Relationships of Pyrimidine Derivatives as Pan-FGFRs Inhibitors with Improved Metabolic Stability and Suppressed HERG Channel Inhibitory Activity. Bioorg. Med. Chem. 2022, 59, 116657. [Google Scholar] [CrossRef]
- Yamamoto, N.; Ryoo, B.-Y.; Keam, B.; Kudo, M.; Lin, C.-C.; Kunieda, F.; Ball, H.A.; Moran, D.; Komatsu, K.; Takeda, K.; et al. A Phase 1 Study of Oral ASP5878, a Selective Small-Molecule Inhibitor of Fibroblast Growth Factor Receptors 1–4, as a Single Dose and Multiple Doses in Patients with Solid Malignancies. Investig. New Drugs 2020, 38, 445–456. [Google Scholar] [CrossRef]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J.; et al. AZD4547: An Orally Bioavailable, Potent, and Selective Inhibitor of the Fibroblast Growth Factor Receptor Tyrosine Kinase Family. Cancer Res. 2012, 72, 2045–2056. [Google Scholar] [CrossRef]
- Coombes, R.C.; Badman, P.D.; Lozano-Kuehne, J.P.; Liu, X.; Macpherson, I.R.; Zubairi, I.; Baird, R.D.; Rosenfeld, N.; Garcia-Corbacho, J.; Cresti, N.; et al. Results of the Phase IIa RADICAL Trial of the FGFR Inhibitor AZD4547 in Endocrine Resistant Breast Cancer. Nat. Commun. 2022, 13, 3246. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Shen, R.; Berger, M.F.; Ferry, D.; Soria, J.-C.; Mathewson, A.; Rooney, C.; Smith, N.R.; Cullberg, M.; Kilgour, E.; et al. A Phase Ib Open-Label Multicenter Study of AZD4547 in Patients with Advanced Squamous Cell Lung Cancers. Clin. Cancer Res. 2017, 23, 5366–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.K.; Hong, F.; Vaklavas, C.; Cheng, H.H.; Hammerman, P.; Mitchell, E.P.; Zwiebel, J.A.; Ivy, S.P.; Gray, R.J.; Li, S.; et al. Phase II Study of AZD4547 in Patients With Tumors Harboring Aberrations in the FGFR Pathway: Results From the NCI-MATCH Trial (EAY131) Subprotocol W. J. Clin. Oncol. 2020, 38, 2407–2417. [Google Scholar] [CrossRef]
- Ebiike, H.; Taka, N.; Matsushita, M.; Ohmori, M.; Takami, K.; Hyohdoh, I.; Kohchi, M.; Hayase, T.; Nishii, H.; Morikami, K.; et al. Discovery of [5-Amino-1-(2-Methyl-3H-Benzimidazol-5-Yl)Pyrazol-4-Yl]-(1H-Indol-2-Yl)Methanone (CH5183284/Debio 1347), An Orally Available and Selective Fibroblast Growth Factor Receptor (FGFR) Inhibitor. J. Med. Chem. 2016, 59, 10586–10600. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.H.; Hierro, C.; Heist, R.S.; Cleary, J.M.; Meric-Bernstam, F.; Tabernero, J.; Janku, F.; Gandhi, L.; Iafrate, A.J.; Borger, D.R.; et al. A Phase I, Open-Label, Multicenter, Dose-Escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Patients with Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin. Cancer Res. 2019, 25, 2699–2707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.G.; Yu, Y.; Eathiraj, S.; Wang, Y.; Savage, R.E.; Lapierre, J.-M.; Schwartz, B.; Abbadessa, G. Preclinical Activity of ARQ 087, a Novel Inhibitor Targeting FGFR Dysregulation. PLoS ONE 2016, 11, e0162594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzaferro, V.; El-Rayes, B.F.; Droz Dit Busset, M.; Cotsoglou, C.; Harris, W.P.; Damjanov, N.; Masi, G.; Rimassa, L.; Personeni, N.; Braiteh, F.; et al. Derazantinib (ARQ 087) in Advanced or Inoperable FGFR2 Gene Fusion-Positive Intrahepatic Cholangiocarcinoma. Br. J. Cancer 2019, 120, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trudel, S.; Li, Z.H.; Wei, E.; Wiesmann, M.; Chang, H.; Chen, C.; Reece, D.; Heise, C.; Stewart, A.K. CHIR-258, a Novel, Multitargeted Tyrosine Kinase Inhibitor for the Potential Treatment of t(4;14) Multiple Myeloma. Blood 2005, 105, 2941–2948. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Grünwald, V.; Ravaud, A.; Ou, Y.-C.; Castellano, D.; Lin, C.-C.; Gschwend, J.E.; Harzstark, A.; Beall, S.; Pirotta, N.; et al. Phase II Results of Dovitinib (TKI258) in Patients with Metastatic Renal Cell Cancer. Clin. Cancer Res. 2014, 20, 3012–3022. [Google Scholar] [CrossRef] [Green Version]
- Watanabe Miyano, S.; Yamamoto, Y.; Kodama, K.; Miyajima, Y.; Mikamoto, M.; Nakagawa, T.; Kuramochi, H.; Funasaka, S.; Nagao, S.; Sugi, N.H.; et al. E7090, a Novel Selective Inhibitor of Fibroblast Growth Factor Receptors, Displays Potent Antitumor Activity and Prolongs Survival in Preclinical Models. Mol. Cancer Ther. 2016, 15, 2630–2639. [Google Scholar] [CrossRef]
- Koyama, T.; Shimizu, T.; Iwasa, S.; Fujiwara, Y.; Kondo, S.; Kitano, S.; Yonemori, K.; Shimomura, A.; Iizumi, S.; Sasaki, T.; et al. First-in-Human Phase I Study of E7090, a Novel Selective Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced Solid Tumors. Cancer Sci. 2020, 111, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, Y.; Sudo, K.; Kojima, Y.; Okuma, H.; Kohsaka, S.; Machida, R.; Ichimura, M.; Anjo, K.; Kurishita, K.; Okita, N.; et al. A Multicenter Investigator-Initiated Phase 2 Trial of E7090 in Patients with Advanced or Recurrent Solid Tumor with Fibroblast Growth Factor Receptor (FGFR) Gene Alteration: FORTUNE Trial. BMC Cancer 2022, 22, 869. [Google Scholar] [CrossRef] [PubMed]
- Perera, T.P.S.; Jovcheva, E.; Mevellec, L.; Vialard, J.; De Lange, D.; Verhulst, T.; Paulussen, C.; Van De Ven, K.; King, P.; Freyne, E.; et al. Discovery and Pharmacological Characterization of JNJ-42756493 (Erdafitinib), a Functionally Selective Small-Molecule FGFR Family Inhibitor. Mol. Cancer Ther. 2017, 16, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Montazeri, K.; Bellmunt, J. Erdafitinib for the Treatment of Metastatic Bladder Cancer. Expert Rev. Clin. Pharmacol. 2020, 13, 1–6. [Google Scholar] [CrossRef]
- Sootome, H.; Fujita, H.; Ito, K.; Ochiiwa, H.; Fujioka, Y.; Ito, K.; Miura, A.; Sagara, T.; Ito, S.; Ohsawa, H.; et al. Futibatinib Is a Novel Irreversible FGFR 1-4 Inhibitor That Shows Selective Antitumor Activity against FGFR-Deregulated Tumors. Cancer Res. 2020, 80, 4986–4997. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, A.; Zecchetto, C.; Casalino, S.; Gaule, M.; Pesoni, C.; Merz, V.; Contarelli, S.; Pietrobono, S.; Benhadji, K.A.; Melisi, D. Clinical Response to Futibatinib in Patients with High-Level FGFR2-Amplified Advanced Gastric Cancer: Two Case Reports. Clin. Drug Investig. 2022, 42, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Shitara, K.; Kojima, T.; Kuboki, Y.; Matsubara, N.; Bando, H.; Yoh, K.; Naito, Y.; Hirai, H.; Kurokawa, Y.; et al. Phase I Study of the Irreversible FGFR1-4 Inhibitor Futibatinib in Japanese Patients with Advanced Solid Tumors. Cancer Sci. 2022. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Bahleda, R.; Hierro, C.; Sanson, M.; Bridgewater, J.; Arkenau, H.-T.; Tran, B.; Kelley, R.K.; Park, J.O.; Javle, M.; et al. Futibatinib, an Irreversible FGFR1-4 Inhibitor, in Patients with Advanced Solid Tumors Harboring FGF/FGFR Aberrations: A Phase I Dose-Expansion Study. Cancer Discov. 2022, 12, 402–415. [Google Scholar] [CrossRef]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-Dichloro-3,5-Dimethoxy-Phenyl)-1-{6-[4-(4-Ethyl-Piperazin-1-Yl)-Phenylamino]-Pyrimidin-4-Yl}-1-Methyl-Urea (NVP-BGJ398), a Potent and Selective Inhibitor of the Fibroblast Growth Factor Receptor Family of Receptor Tyrosine Kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef]
- Lassman, A.B.; Sepúlveda-Sánchez, J.M.; Cloughesy, T.F.; Gil-Gil, M.J.; Puduvalli, V.K.; Raizer, J.J.; De Vos, F.Y.F.; Wen, P.Y.; Butowski, N.A.; Clement, P.M.J.; et al. Infigratinib in Patients with Recurrent Gliomas and FGFR Alterations: A Multicenter Phase II Study. Clin. Cancer Res. 2022, 28, 2270–2277. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Li, W.-Y.; Chen, D.; Henry, J.R.; Li, H.-Y.; Chen, Z.; Zia-Ebrahimi, M.; Bloem, L.; Zhai, Y.; Huss, K.; et al. A Novel, Selective Inhibitor of Fibroblast Growth Factor Receptors That Shows a Potent Broad Spectrum of Antitumor Activity in Several Tumor Xenograft Models. Mol. Cancer Ther. 2011, 10, 2200–2210. [Google Scholar] [CrossRef] [Green Version]
- Michael, M.; Bang, Y.-J.; Park, Y.S.; Kang, Y.-K.; Kim, T.M.; Hamid, O.; Thornton, D.; Tate, S.C.; Raddad, E.; Tie, J. A Phase 1 Study of LY2874455, an Oral Selective Pan-FGFR Inhibitor, in Patients with Advanced Cancer. Target. Oncol. 2017, 12, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Holmström, T.H.; Moilanen, A.-M.; Ikonen, T.; Björkman, M.L.; Linnanen, T.; Wohlfahrt, G.; Karlsson, S.; Oksala, R.; Korjamo, T.; Samajdar, S.; et al. ODM-203, a Selective Inhibitor of FGFR and VEGFR, Shows Strong Antitumor Activity, and Induces Antitumor Immunity. Mol. Cancer Ther. 2019, 18, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bono, P.; Massard, C.; Peltola, K.J.; Azaro, A.; Italiano, A.; Kristeleit, R.S.; Curigliano, G.; Lassen, U.; Arkenau, H.-T.; Hakulinen, P.; et al. Phase I/IIa, Open-Label, Multicentre Study to Evaluate the Optimal Dosing and Safety of ODM-203 in Patients with Advanced or Metastatic Solid Tumours. ESMO Open 2020, 5, e001081. [Google Scholar] [CrossRef]
- Liu, P.C.C.; Koblish, H.; Wu, L.; Bowman, K.; Diamond, S.; DiMatteo, D.; Zhang, Y.; Hansbury, M.; Rupar, M.; Wen, X.; et al. INCB054828 (Pemigatinib), a Potent and Selective Inhibitor of Fibroblast Growth Factor Receptors 1, 2, and 3, Displays Activity against Genetically Defined Tumor Models. PLoS ONE 2020, 15, e0231877. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for Previously Treated, Locally Advanced or Metastatic Cholangiocarcinoma: A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Wong, M.J.; Moran, L.; Wardwell, S.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Clackson, T.; Rivera, V.M. Ponatinib (AP24534), a Multitargeted Pan-FGFR Inhibitor with Activity in Multiple FGFR-Amplified or Mutated Cancer Models. Mol. Cancer Ther. 2012, 11, 690–699. [Google Scholar] [CrossRef] [Green Version]
- Ahn, D.H.; Uson Junior, P.L.S.; Masci, P.; Kosiorek, H.; Halfdanarson, T.R.; Mody, K.; Babiker, H.; DeLeon, T.; Sonbol, M.B.; Gores, G.; et al. A Pilot Study of Pan-FGFR Inhibitor Ponatinib in Patients with FGFR-Altered Advanced Cholangiocarcinoma. Investig. New Drugs 2022, 40, 134–141. [Google Scholar] [CrossRef]
- Fairhurst, R.A.; Knoepfel, T.; Buschmann, N.; Leblanc, C.; Mah, R.; Todorov, M.; Nimsgern, P.; Ripoche, S.; Niklaus, M.; Warin, N.; et al. Discovery of Roblitinib (FGF401) as a Reversible-Covalent Inhibitor of the Kinase Activity of Fibroblast Growth Factor Receptor 4. J. Med. Chem. 2020, 63, 12542–12573. [Google Scholar] [CrossRef]
- Collin, M.-P.; Lobell, M.; Hübsch, W.; Brohm, D.; Schirok, H.; Jautelat, R.; Lustig, K.; Bömer, U.; Vöhringer, V.; Héroult, M.; et al. Discovery of Rogaratinib (BAY 1163877): A Pan-FGFR Inhibitor. ChemMedChem 2018, 13, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, S.; Politz, O.; Bender, S.; Héroult, M.; Lustig, K.; Thuss, U.; Kneip, C.; Kopitz, C.; Zopf, D.; Collin, M.-P.; et al. Rogaratinib: A Potent and Selective Pan-FGFR Inhibitor with Broad Antitumor Activity in FGFR-Overexpressing Preclinical Cancer Models. Int. J. Cancer 2019, 145, 1346–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynes, M.W.; Hinz, T.K.; Gao, D.; Martini, M.; Marek, L.A.; Ware, K.E.; Edwards, M.G.; Böhm, D.; Perner, S.; Helfrich, B.A.; et al. FGFR1 MRNA and Protein Expression, Not Gene Copy Number, Predict FGFR TKI Sensitivity across All Lung Cancer Histologies. Clin. Cancer Res. 2014, 20, 3299–3309. [Google Scholar] [CrossRef] [Green Version]
- Smyth, E.C.; Turner, N.; Pearson, A.; Peckitt, C.; Chau, I.; Watkins, D.; Kilgour, E.; Smith, N.R.; Gillbanks, A.; Chua, S.; et al. Phase II Study of AZD4547 in FGFR Amplified Tumours: Gastroesophageal Cancer (GC) Cohort Clinical and Translational Results. Ann. Oncol. 2015, 26, ix42. [Google Scholar] [CrossRef] [Green Version]
- Göke, F.; Franzen, A.; Hinz, T.K.; Marek, L.A.; Yoon, P.; Sharma, R.; Bode, M.; von Maessenhausen, A.; Lankat-Buttgereit, B.; Göke, A.; et al. FGFR1 Expression Levels Predict BGJ398 Sensitivity of FGFR1-Dependent Head and Neck Squamous Cell Cancers. Clin. Cancer Res. 2015, 21, 4356–4364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard-Pierrot, I.; Brams, A.; Dunois-Lardé, C.; Caillault, A.; Diez de Medina, S.G.; Cappellen, D.; Graff, G.; Thiery, J.P.; Chopin, D.; Ricol, D.; et al. Oncogenic Properties of the Mutated Forms of Fibroblast Growth Factor Receptor 3b. Carcinogenesis 2006, 27, 740–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.W.; Kim, Y.-H.; Jeong, P.; Park, C.; Kim, W.T.; Ryu, D.H.; Cha, E.-J.; Ha, Y.-S.; Kim, T.-H.; Kwon, T.G.; et al. Expression Levels of FGFR3 as a Prognostic Marker for the Progression of Primary PT1 Bladder Cancer and Its Association with Mutation Status. Oncol. Lett. 2017, 14, 3817–3824. [Google Scholar] [CrossRef] [Green Version]
- Guancial, E.A.; Werner, L.; Bellmunt, J.; Bamias, A.; Choueiri, T.K.; Ross, R.; Schutz, F.A.; Park, R.S.; O’Brien, R.J.; Hirsch, M.S.; et al. FGFR3 Expression in Primary and Metastatic Urothelial Carcinoma of the Bladder. Cancer Med. 2014, 3, 835–844. [Google Scholar] [CrossRef]
- Jogo, T.; Nakamura, Y.; Shitara, K.; Bando, H.; Yasui, H.; Esaki, T.; Terazawa, T.; Satoh, T.; Shinozaki, E.; Nishina, T.; et al. Circulating Tumor DNA Analysis Detects FGFR2 Amplification and Concurrent Genomic Alterations Associated with FGFR Inhibitor Efficacy in Advanced Gastric Cancer. Clin. Cancer Res. 2021, 27, 5619–5627. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Enzinger, P.C.; Kang, Y.-K.; Yamaguchi, K.; Qin, S.; Lee, K.-W.; Oh, S.C.; Li, J.; Turk, H.M.; Teixeira, A.C.; et al. Randomized Double-Blind Placebo-Controlled Phase 2 Study of Bemarituzumab Combined with Modified FOLFOX6 (MFOLFOX6) in First-Line (1L) Treatment of Advanced Gastric/Gastroesophageal Junction Adenocarcinoma (FIGHT). J. Clin. Oncol. 2021, 39, 160. [Google Scholar] [CrossRef]
- Schuler, M.; Nogova, L.; Heidenreich, A.; Tai, D.; Cassier, P.; Richly, H.; Cho, B.C.; Sayehli, C.M.; Bender, S.; Ocker, M.; et al. Anti-Tumor Activity of the Pan-FGFR Inhibitor Rogaratinib in Patients with Advanced Urothelial Carcinomas Selected Based on Tumor FGFR MRNA Expression Levels. Ann. Oncol. 2017, 28, v295–v329. [Google Scholar] [CrossRef]
- Kim, E.K.; Cho, Y.A.; Koh, Y.W.; Shin, H.A.; Cho, B.C.; Yoon, S.O. Prognostic Implications of Fibroblast Growth Factor Receptor 1 (FGFR1) Gene Amplification and Protein Overexpression in Hypopharyngeal and Laryngeal Squamous Cell Carcinoma. BMC Cancer 2020, 20, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theelen, W.S.; Mittempergher, L.; Willems, S.M.; Bosma, A.J.; Peters, D.D.; van der Noort, V.; Japenga, E.J.; Peeters, T.; Koole, K.; Šuštić, T.; et al. FGFR1, 2 and 3 Protein Overexpression and Molecular Aberrations of FGFR3 in Early Stage Non-Small Cell Lung Cancer. J. Pathol. Clin. Res. 2016, 2, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Tomiguchi, M.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Goto-Yamaguchi, L.; Fujiki, Y.; Fujiwara, S.; Sueta, A.; Hayashi, M.; Takeshita, T.; Inao, T.; et al. Fibroblast Growth Factor Receptor-1 Protein Expression Is Associated with Prognosis in Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor-2-Negative Primary Breast Cancer. Cancer Sci. 2016, 107, 491–498. [Google Scholar] [CrossRef]
- Ahn, S.; Lee, J.; Hong, M.; Kim, S.T.; Park, S.H.; Choi, M.G.; Lee, J.-H.; Sohn, T.S.; Bae, J.M.; Kim, S.; et al. FGFR2 in Gastric Cancer: Protein Overexpression Predicts Gene Amplification and High H-Index Predicts Poor Survival. Mod. Pathol. 2016, 29, 1095–1103. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, R.; Imamura, Y.; Nakamura, K.; Ishimoto, T.; Nakagawa, S.; Miyake, K.; Nakaji, Y.; Tsuda, Y.; Iwatsuki, M.; Baba, Y.; et al. Fibroblast Growth Factor Receptor 2 Expression, but Not Its Genetic Amplification, Is Associated with Tumor Growth and Worse Survival in Esophagogastric Junction Adenocarcinoma. Oncotarget 2016, 7, 19748–19761. [Google Scholar] [CrossRef]
- Uson Junior, P.L.S.; DeLeon, T.T.; Bogenberger, J.M.; Pai, R.K.; Kosiorek, H.E.; Yin, J.; Ahn, D.H.; Sonbol, M.B.; Bekaii-Saab, T.; Mansfield, A.S.; et al. FGFR2-IIIb Expression by Immunohistochemistry Has High Specificity in Cholangiocarcinoma with FGFR2 Genomic Alterations. Dig. Dis. Sci. 2022, 67, 3797–3805. [Google Scholar] [CrossRef]
- van Rhijn, B.W.G.; Mertens, L.S.; Mayr, R.; Bostrom, P.J.; Real, F.X.; Zwarthoff, E.C.; Boormans, J.L.; Abas, C.; van Leenders, G.J.L.H.; Götz, S.; et al. FGFR3 Mutation Status and FGFR3 Expression in a Large Bladder Cancer Cohort Treated by Radical Cystectomy: Implications for Anti-FGFR3 Treatment? Eur. Urol. 2020, 78, 682–687. [Google Scholar] [CrossRef]
- Moes-Sosnowska, J.; Skupinska, M.; Lechowicz, U.; Szczepulska-Wojcik, E.; Skronska, P.; Rozy, A.; Stepniewska, A.; Langfort, R.; Rudzinski, P.; Orlowski, T.; et al. FGFR1-4 RNA-Based Gene Alteration and Expression Analysis in Squamous Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2022, 23, 10506. [Google Scholar] [CrossRef]
- Dietrich, D. FGFR-targeted therapy in head and neck carcinomas. HNO 2021, 69, 172–184. [Google Scholar] [CrossRef]
- Santolla, M.F.; Maggiolini, M. The FGF/FGFR System in Breast Cancer: Oncogenic Features and Therapeutic Perspectives. Cancers 2020, 12, 3029. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Frezzetti, D.; Gallo, M.; Normanno, N. FGFR-Targeted Therapeutics for the Treatment of Breast Cancer. Expert Opin. Investig. Drugs 2017, 26, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Hong, J.Y.; Kim, K.; Kim, K.-M.; Kang, S.Y.; Lee, T.; Kim, S.T.; Park, S.H.; Park, Y.S.; Lim, H.Y.; et al. Detection of Fusion Genes Using a Targeted RNA Sequencing Panel in Gastrointestinal and Rare Cancers. J. Oncol. 2020, 2020, 4659062. [Google Scholar] [CrossRef]
- Gu, W.; Yang, J.; Wang, Y.; Xu, J.; Wang, X.; Du, F.; Hu, X.; Guo, H.; Song, C.; Tao, R.; et al. Comprehensive Identification of FGFR1-4 Alterations in 5557 Chinese Patients with Solid Tumors by next-Generation Sequencing. Am. J. Cancer Res. 2021, 11, 3893–3906. [Google Scholar] [PubMed]
- Napolitano, A.; Ostler, A.E.; Jones, R.L.; Huang, P.H. Fibroblast Growth Factor Receptor (FGFR) Signaling in GIST and Soft Tissue Sarcomas. Cells 2021, 10, 1533. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Schuler, M.; Kang, Y.-K.; Yen, C.-J.; Edeline, J.; Choo, S.P.; Lin, C.-C.; Okusaka, T.; Weiss, K.-H.; Macarulla, T.; et al. A First-in-Human Phase 1/2 Study of FGF401 and Combination of FGF401 with Spartalizumab in Patients with Hepatocellular Carcinoma or Biomarker-Selected Solid Tumors. J. Exp. Clin. Cancer Res. CR 2022, 41, 189. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Zhan, P.; Gavine, P.R.; Morgan, S.; Womack, C.; Ni, X.; Shen, D.; Bang, Y.-J.; Im, S.-A.; Ho Kim, W.; et al. FGFR2 Amplification Has Prognostic Significance in Gastric Cancer: Results from a Large International Multicentre Study. Br. J. Cancer 2014, 110, 967–975. [Google Scholar] [CrossRef]
- Seo, A.N.; Jin, Y.; Lee, H.J.; Sun, P.-L.; Kim, H.; Jheon, S.; Kim, K.; Lee, C.-T.; Chung, J.-H. FGFR1 Amplification Is Associated with Poor Prognosis and Smoking in Non-Small-Cell Lung Cancer. Virchows Arch. Int. J. Pathol. 2014, 465, 547–558. [Google Scholar] [CrossRef]
- Hur, J.Y.; Chao, J.; Kim, K.; Kim, S.T.; Kim, K.-M.; Klempner, S.J.; Lee, J. High-Level FGFR2 Amplification Is Associated with Poor Prognosis and Lower Response to Chemotherapy in Gastric Cancers. Pathol. Res. Pract. 2020, 216, 152878. [Google Scholar] [CrossRef]
- Kuboki, Y.; Schatz, C.A.; Koechert, K.; Schubert, S.; Feng, J.; Wittemer-Rump, S.; Ziegelbauer, K.; Krahn, T.; Nagatsuma, A.K.; Ochiai, A. In Situ Analysis of FGFR2 MRNA and Comparison with FGFR2 Gene Copy Number by Dual-Color in Situ Hybridization in a Large Cohort of Gastric Cancer Patients. Gastric Cancer 2018, 21, 401–412. [Google Scholar] [CrossRef]
- Pearson, A.; Smyth, E.; Babina, I.S.; Herrera-Abreu, M.T.; Tarazona, N.; Peckitt, C.; Kilgour, E.; Smith, N.R.; Geh, C.; Rooney, C.; et al. High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov. 2016, 6, 838–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schildhaus, H.-U.; Heukamp, L.C.; Merkelbach-Bruse, S.; Riesner, K.; Schmitz, K.; Binot, E.; Paggen, E.; Albus, K.; Schulte, W.; Ko, Y.-D.; et al. Definition of a Fluorescence In-Situ Hybridization Score Identifies High- and Low-Level FGFR1 Amplification Types in Squamous Cell Lung Cancer. Mod. Pathol. 2012, 25, 1473–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaibori, M.; Sakai, K.; Ishizaki, M.; Matsushima, H.; De Velasco, M.A.; Matsui, K.; Iida, H.; Kitade, H.; Kwon, A.-H.; Nagano, H.; et al. Increased FGF19 Copy Number Is Frequently Detected in Hepatocellular Carcinoma with a Complete Response after Sorafenib Treatment. Oncotarget 2016, 7, 49091–49098. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Flanagan, J.; Su, N.; Wang, L.-C.; Bui, S.; Nielson, A.; Wu, X.; Vo, H.-T.; Ma, X.-J.; Luo, Y. RNAscope: A Novel in Situ RNA Analysis Platform for Formalin-Fixed, Paraffin-Embedded Tissues. J. Mol. Diagn. JMD 2012, 14, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiss, G.K.; Bumgarner, R.E.; Birditt, B.; Dahl, T.; Dowidar, N.; Dunaway, D.L.; Fell, H.P.; Ferree, S.; George, R.D.; Grogan, T.; et al. Direct Multiplexed Measurement of Gene Expression with Color-Coded Probe Pairs. Nat. Biotechnol. 2008, 26, 317–325. [Google Scholar] [CrossRef]
- Sánchez-Guixé, M.; Hierro, C.; Jiménez, J.; Viaplana, C.; Villacampa, G.; Monelli, E.; Brasó-Maristany, F.; Ogbah, Z.; Parés, M.; Guzmán, M.; et al. High FGFR1-4 MRNA Expression Levels Correlate with Response to Selective FGFR Inhibitors in Breast Cancer. Clin. Cancer Res. 2022, 28, 137–149. [Google Scholar] [CrossRef]
- Silverman, I.M.; Li, M.; Murugesan, K.; Krook, M.A.; Javle, M.M.; Kelley, R.K.; Borad, M.J.; Roychowdhury, S.; Meng, W.; Yilmazel, B.; et al. Validation and Characterization of FGFR2 Rearrangements in Cholangiocarcinoma with Comprehensive Genomic Profiling. J. Mol. Diagn. JMD 2022, 24, 351–364. [Google Scholar] [CrossRef]
- Freedman, A.N.; Klabunde, C.N.; Wiant, K.; Enewold, L.; Gray, S.W.; Filipski, K.K.; Keating, N.L.; Leonard, D.G.B.; Lively, T.; McNeel, T.S.; et al. Use of Next-Generation Sequencing Tests to Guide Cancer Treatment: Results From a Nationally Representative Survey of Oncologists in the United States. JCO Precis. Oncol. 2018, 2, 1–13. [Google Scholar] [CrossRef]
- Vega, D.M.; Nishimura, K.K.; Zariffa, N.; Thompson, J.C.; Hoering, A.; Cilento, V.; Rosenthal, A.; Anagnostou, V.; Baden, J.; Beaver, J.A.; et al. Changes in Circulating Tumor DNA Reflect Clinical Benefit Across Multiple Studies of Patients With Non–Small-Cell Lung Cancer Treated With Immune Checkpoint Inhibitors. JCO Precis. Oncol. 2022, 6, e2100372. [Google Scholar] [CrossRef]
- Duffy, M.J.; Crown, J. Circulating Tumor DNA as a Biomarker for Monitoring Patients with Solid Cancers: Comparison with Standard Protein Biomarkers. Clin. Chem. 2022, hvac121. [Google Scholar] [CrossRef]
- Mi, J.; Han, X.; Wang, R.; Ma, R.; Zhao, D. Circulation Tumour DNA in Predicting Recurrence and Prognosis in Operable Colorectal Cancer Patients: A Meta-Analysis. Eur. J. Clin. Investig. 2022, e13842. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, A.M.; Patel, J.; Janjigian, Y.Y.; Meng, F.; Selcuklu, S.D.; Iyer, G.; Houck-Loomis, B.; Harding, J.J.; O’Reilly, E.M.; Abou-Alfa, G.K.; et al. Noninvasive Detection of Polyclonal Acquired Resistance to FGFR Inhibition in Patients With Cholangiocarcinoma Harboring FGFR2 Alterations. JCO Precis. Oncol. 2021, 5, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Shi, L.; Liu, L.Y.; Fece de la Cruz, F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.S.; et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR Signaling Mediates Resistance to CDK4/6 Inhibitors in ER+ Breast Cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef] [Green Version]
- Nogova, L.; Sequist, L.V.; Perez Garcia, J.M.; Andre, F.; Delord, J.-P.; Hidalgo, M.; Schellens, J.H.M.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [CrossRef]
- Miao, J.-L.; Zhou, J.-H.; Cai, J.-J.; Liu, R.-J. The Association between Fibroblast Growth Factor Receptor 1 Gene Amplification and Lung Cancer: A Meta-Analysis. Arch. Med. Sci. AMS 2020, 16, 16–26. [Google Scholar] [CrossRef]
- Quinn, D.I.; Petrylak, D.P.; Bellmunt, J.; Necchi, A.; Gurney, H.; Lee, J.-L.; Van der Heijden, M.S.; Rosenbaum, E.; Penel, N.; Pang, S.-T.; et al. FORT-1: Phase II/III Study of Rogaratinib versus Chemotherapy (CT) in Patients (Pts) with Locally Advanced or Metastatic Urothelial Carcinoma (UC) Selected Based on FGFR1/3 MRNA Expression. J. Clin. Oncol. 2020, 38, 489. [Google Scholar] [CrossRef]
- Kazdal, D.; Hofman, V.; Christopoulos, P.; Ilié, M.; Stenzinger, A.; Hofman, P. Fusion-Positive Non-Small Cell Lung Carcinoma: Biological Principles, Clinical Practice, and Diagnostic Implications. Genes. Chromosomes Cancer 2022, 61, 244–260. [Google Scholar] [CrossRef]
- Trombetta, D.; Sparaneo, A.; Fabrizio, F.P.; Di Micco, C.M.; Rossi, A.; Muscarella, L.A. NRG1 and NRG2 Fusions in Non-Small Cell Lung Cancer (NSCLC): Seven Years between Lights and Shadows. Expert Opin. Ther. Targets 2021, 25, 865–875. [Google Scholar] [CrossRef]
- Drilon, A.; Duruisseaux, M.; Han, J.-Y.; Ito, M.; Falcon, C.; Yang, S.-R.; Murciano-Goroff, Y.R.; Chen, H.; Okada, M.; Molina, M.A.; et al. Clinicopathologic Features and Response to Therapy of NRG1 Fusion-Driven Lung Cancers: The ENRGy1 Global Multicenter Registry. J. Clin. Oncol. 2021, 39, 2791–2802. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; DuBois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for Paediatric Solid Tumours Harbouring NTRK Gene Fusions: Phase 1 Results from a Multicentre, Open-Label, Phase 1/2 Study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Camidge, D.R.; Bang, Y.-J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.-H.I.; Kim, D.-W.; et al. Activity and Safety of Crizotinib in Patients with ALK-Positive Non-Small-Cell Lung Cancer: Updated Results from a Phase 1 Study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Wolf, J.; Helland, Å.; Oh, I.-J.; Migliorino, M.R.; Dziadziuszko, R.; Wrona, A.; de Castro, J.; Mazieres, J.; Griesinger, F.; Chlistalla, M.; et al. Final Efficacy and Safety Data, and Exploratory Molecular Profiling from the Phase III ALUR Study of Alectinib versus Chemotherapy in Crizotinib-Pretreated ALK-Positive Non-Small-Cell Lung Cancer. ESMO Open 2022, 7, 100333. [Google Scholar] [CrossRef]
- Prawira, A.; Le, T.B.U.; Ho, R.Z.W.; Huynh, H. Upregulation of the ErbB Family by EZH2 in Hepatocellular Carcinoma Confers Resistance to FGFR Inhibitor. J. Cancer Res. Clin. Oncol. 2021, 147, 2955–2968. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, A.; Molina-Pinelo, S.; Cirauqui, C.; Ojeda-Márquez, L.; Marrugal, Á.; Suarez, R.; Conde, E.; Ponce-Aix, S.; Enguita, A.B.; Carnero, A.; et al. FGFR1 Cooperates with EGFR in Lung Cancer Oncogenesis, and Their Combined Inhibition Shows Improved Efficacy. J. Thorac. Oncol. 2019, 14, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Datta, J.; Damodaran, S.; Parks, H.; Ocrainiciuc, C.; Miya, J.; Yu, L.; Gardner, E.P.; Samorodnitsky, E.; Wing, M.R.; Bhatt, D.; et al. Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol. Cancer Ther. 2017, 16, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Cowell, J.K.; Qin, H.; Hu, T.; Wu, Q.; Bhole, A.; Ren, M. Mutation in the FGFR1 Tyrosine Kinase Domain or Inactivation of PTEN Is Associated with Acquired Resistance to FGFR Inhibitors in FGFR1-Driven Leukemia/Lymphomas. Int. J. Cancer 2017, 141, 1822–1829. [Google Scholar] [CrossRef] [Green Version]
- Lau, D.K.; Luk, I.Y.; Jenkins, L.J.; Martin, A.; Williams, D.S.; Schoffer, K.L.; Chionh, F.; Buchert, M.; Sjoquist, K.; Boussioutas, A.; et al. Rapid Resistance of FGFR-Driven Gastric Cancers to Regorafenib and Targeted FGFR Inhibitors Can Be Overcome by Parallel Inhibition of MEK. Mol. Cancer Ther. 2021, 20, 704–715. [Google Scholar] [CrossRef]
- Siefker-Radtke, A.O.; Necchi, A.; Park, S.H.; García-Donas, J.; Huddart, R.A.; Burgess, E.F.; Fleming, M.T.; Rezazadeh Kalebasty, A.; Mellado, B.; Varlamov, S.; et al. Efficacy and Safety of Erdafitinib in Patients with Locally Advanced or Metastatic Urothelial Carcinoma: Long-Term Follow-up of a Phase 2 Study. Lancet Oncol. 2022, 23, 248–258. [Google Scholar] [CrossRef]
- Vaclova, T.; Grazini, U.; Ward, L.; O’Neill, D.; Markovets, A.; Huang, X.; Chmielecki, J.; Hartmaier, R.; Thress, K.S.; Smith, P.D.; et al. Clinical Impact of Subclonal EGFR T790M Mutations in Advanced-Stage EGFR-Mutant Non-Small-Cell Lung Cancers. Nat. Commun. 2021, 12, 1780. [Google Scholar] [CrossRef] [PubMed]
- Sevillano Fernández, E.; Madurga de Lacalle, R.; Rodriguez Moreno, J.F.; Barquín García, A.; Yagüe Fernández, M.; Navarro Alcaraz, P.; Barba Llacer, M.; Quiralte Pulido, M.; García-Donás Jiménez, J. Prognostic Value and Clinical Significance of FGFR Genomic Alterations (GAs) in Metastatic Urothelial Cancer Patients. J. Clin. Med. 2022, 11, 4483. [Google Scholar] [CrossRef] [PubMed]
- Necchi, A.; Lo Vullo, S.; Raggi, D.; Gloghini, A.; Giannatempo, P.; Colecchia, M.; Mariani, L. Prognostic Effect of FGFR Mutations or Gene Fusions in Patients with Metastatic Urothelial Carcinoma Receiving First-Line Platinum-Based Chemotherapy: Results from a Large, Single-Institution Cohort. Eur. Urol. Focus 2019, 5, 853–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, W.; Wang, G.; Cui, Z.; Xiong, G.; Jiang, X.; Li, Y.; Li, W.; Han, B.; Chen, S.; Shi, B. FGFR3 Destabilizes PD-L1 via NEDD4 to Control T-Cell-Mediated Bladder Cancer Immune Surveillance. Cancer Res. 2022, 82, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Gajate, P.; Morales-Barrera, R.; Lee, J.-L.; Necchi, A.; Penel, N.; Zagonel, V.; Sierecki, M.R.; Bao, W.; Zhou, Y.; et al. Safety and Efficacy of Rogaratinib in Combination with Atezolizumab in Cisplatin-Ineligible Patients (Pts) with Locally Advanced or Metastatic Urothelial Cancer (UC) and FGFR MRNA Overexpression in the Phase Ib/II FORT-2 Study. J. Clin. Oncol. 2021, 39, 4521. [Google Scholar] [CrossRef]
- Siefker-Radtke, A.O.; Loriot, Y.; Siena, S.; Beato, C.; Climent Duran, M.A.; Varlamov, S.; Duran, I.; Tagawa, S.T.; Geoffrois, L.; Mellado, B.; et al. Updated Data from the NORSE Trial of Erdafitinib (ERDA) plus Cetrelimab (CET) in Patients (Pts) with Metastatic or Locally Advanced Urothelial Carcinoma (MUC) and Specific Fibroblast Growth Factor Receptor (FGFR) Alterations. Ann. Oncol. 2020, 31, S584. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Selectivity | Indications | Phase | Biomarker | Reference |
|---|---|---|---|---|---|
| ASP5878 | FGFR1-4 | UC, HCC, sqLC | 1 | FGFR3 fusion or mutation by FISH or PCR (UC), FGF19 overexp (HCC) or FGFR1 overexp (sqLC) by IHC | [69,70] |
| AZD4547 | FGFR1-4 | BC, GC, sqLC, agnostic | 3 | FGFR copy number in ctDNA (BC), FISH (GC, sqLC), any FGFR alteration by NGS in the indication agnostic setting | [11,71,72,73,74] |
| Debio 1347 | FGFR1-3 | Advanced solid tumors | 1/2 | FISH, NGS | [75,76] |
| Derazantinib (ARQ087) | FGFR1-3 | ihCC | 1/2 | FGFR2 fusion by FISH or NGS | [77,78] |
| Dovitinib (TKI-258) | FGFR1 & 3 | RCC and other solid tumors | 3 | No specific biomarker used | [79,80] |
| E7090 | FGFR1-3 | GC, ihCC, advanced solid tumors | 1/2 | FGFR2 amp (GC), FGFR2 fusion (ihCC), NGS | [81,82,83] |
| Erdafitinib (JNJ-42756493) | FGFR1-4 | UC | approved | FGFR2/3 alterations by qRT-PCR | [84,85] |
| Fisogatinib (BLU-554) | FGFR4 | HCC | 1/2 | FGF19 by IHC | [32] |
| Futibatinib (TAS-120) | FGFR1-4 | ihCC, GC, advanced solid tumors | approved | FGFR2 amp (GC), various FGFR aberrations | [86,87,88,89] |
| Infigratinib (BGJ398) | FGFR1-3 | ihCC, gliomas | approved | Any alteration of FGFR1 or FGFR3 (gliomas) or FGFR2 (ihCC) | [90,91,92] |
| LY2874455 | FGFR1-4 | GC, NSCLC | 1 | FGFR1 amp (NSCLC), FGFR2 amp (GC) | [93,94] |
| ODM-203 | FGFR1-4 | Advanced solid tumors | 1 | Any genetic FGFR aberration | [95,96] |
| Pemigatinib (INCB054828) | FGFR1-3 | ihCC | approved | NGS | [97,98] |
| Ponatinib | FGFR1-4 | ihCC | 3 | FGFR2 fusion/rearrangement by FISH or NGS | [99,100] |
| Roblitinib (FGF401) | FGFR4 | HCC | 1/2 | FGFR4 expression by PCR | [101] |
| Rogaratinib (BAY 1163877) | FGFR1-4 | Advanced solid tumors | 1/2 | mRNA expression (RNA-ISH, Nanostring) | [9,102,103] |
| Technology | Pros | Cons | Patient Population * | Prevalence |
|---|---|---|---|---|
| FGFR protein expression | ||||
| Immunohistochemistry | Broadly available, direct measure of receptor expression, keeps spatial resolution, short TAT | No single antibody, needs multiplexing for pan-FGFR inhibitors, Requires pathologist training or central testing | FGFR2b + gastric cancer | 30% |
| FGFR mRNA expression | ||||
| PCR | Sensitive, cheap, short TAT Easy to establish for each FGFR subtype | No preservation of spatial resolution | FGFR4 + HCC pts (Roblitinib) | Unknown |
| Nanostring | Sensitive, highly multiplex testing | Expensive, tumor content needs to be retrospectively calculated | FGFR1/2/3 + all comers (Rogaratinib) FGFR2 + gastric cancer (AZD4547) | Up to 25% Unknown |
| RNA-ISH | Sensitive, keeps spatial resolution, IHC-like workflow, short TAT, multiplex possible | Requires pathologist training or central testing | FGFR1/2/3 + all comers (Rogaratinib) FGFR1&3 + urothelial cancer patients (Rogaratinib) | 25% |
| RNAseq | Sensitive, highly multiplex testing | Expensive, long TAT (several weeks), no preservation of spatial resolution, | Not applied in any FGFR inhibitor trial to date | Unknown |
| FGFR DNA alterations | ||||
| FISH | Keeps spatial resolution | Requires fluorescence microscopy, multiplex possible | FGFR2 + gastric cancer (AZD4547) | 4–7% [11] |
| PCR | Short TAT (7 days) | No preservation of spatial resolution | FGFR2&3 fusion and FGFR3 mutations in urothelial carcinoma (QIAGEN’s FDA approved CDx therascreen® FGFR kit for Erdafitinib) | 20% [7] |
| NGS | Highly multiplex testing | Expensive, long TAT, no preservation of spatial resolution | FGFR2 fusion-positive iCCA (Foundation One™ as FDA approved CDx for Pemigatinib & Infigratinib) | 10% [91,98] |
| FGF ligand | ||||
| IHC | Broadly available, direct measure of receptor expression, keeps spatial resolution, short TAT | No single antibody, needs multiplexing for pan-FGFR inhibitors, Requires pathologist training or central testing | FGF-19 serum levels in HCC (Fisogatinib) | 27% [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ellinghaus, P.; Neureiter, D.; Nogai, H.; Stintzing, S.; Ocker, M. Patient Selection Approaches in FGFR Inhibitor Trials—Many Paths to the Same End? Cells 2022, 11, 3180. https://doi.org/10.3390/cells11193180
Ellinghaus P, Neureiter D, Nogai H, Stintzing S, Ocker M. Patient Selection Approaches in FGFR Inhibitor Trials—Many Paths to the Same End? Cells. 2022; 11(19):3180. https://doi.org/10.3390/cells11193180
Chicago/Turabian StyleEllinghaus, Peter, Daniel Neureiter, Hendrik Nogai, Sebastian Stintzing, and Matthias Ocker. 2022. "Patient Selection Approaches in FGFR Inhibitor Trials—Many Paths to the Same End?" Cells 11, no. 19: 3180. https://doi.org/10.3390/cells11193180
APA StyleEllinghaus, P., Neureiter, D., Nogai, H., Stintzing, S., & Ocker, M. (2022). Patient Selection Approaches in FGFR Inhibitor Trials—Many Paths to the Same End? Cells, 11(19), 3180. https://doi.org/10.3390/cells11193180