
Functional Distinctions of Endometrial Cancer-Associated Mutations in the Fibroblast Growth Factor Receptor 2 Gene

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Growth Factors and Inhibitors
2.3. Expression Vectors
2.4. Generation of Stable Clones of COS7 Cells Expressing WT and Mutant FGFR2
2.5. Ectodomain Shedding Assays
2.6. Immunoblot Analysis
2.7. Anchorage-Independent Growth Assays
2.8. Statistical Analysis
3. Results
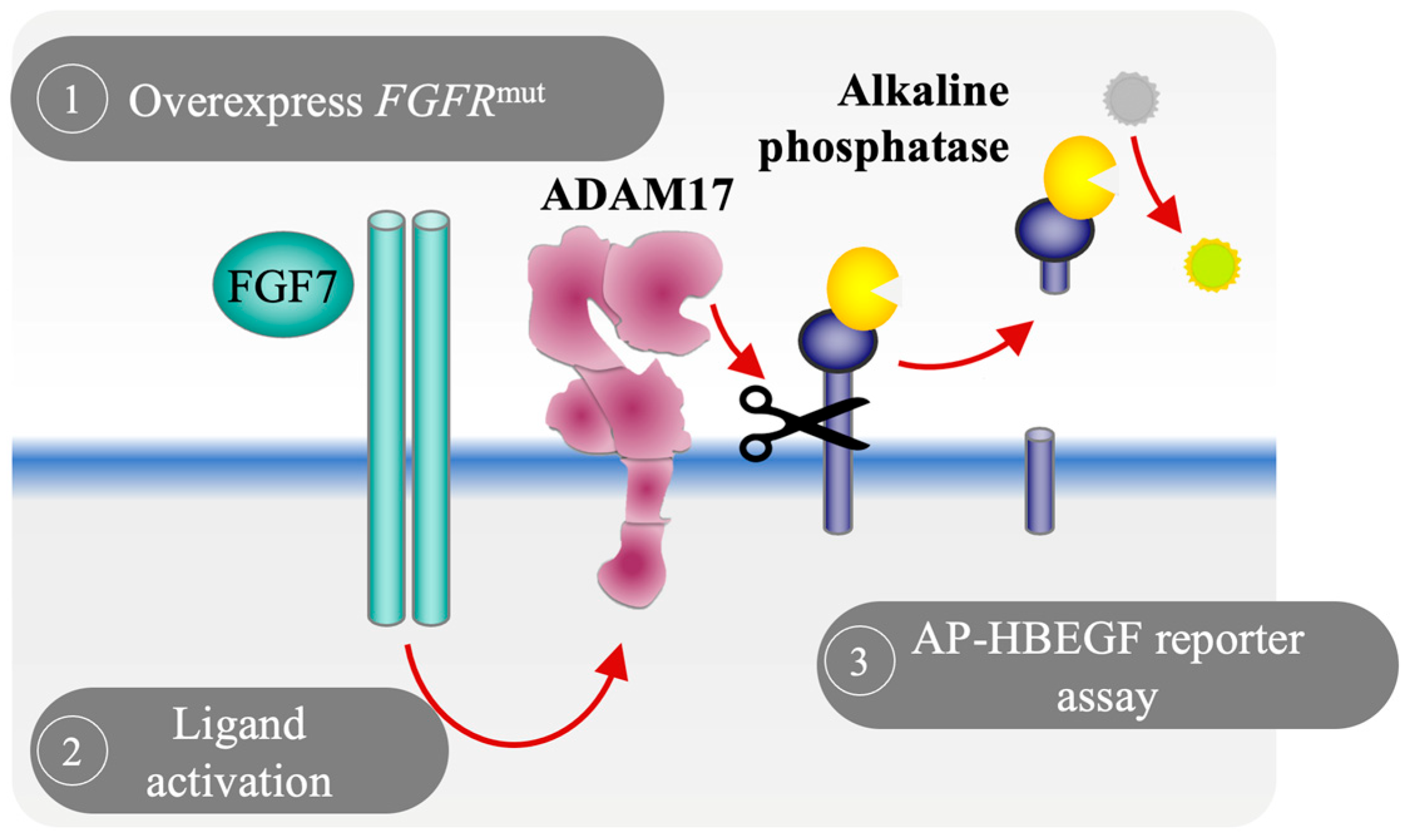
3.1. Development of a Cell-Based Reporter Assay to Measure FGFR2 Activation In Vitro
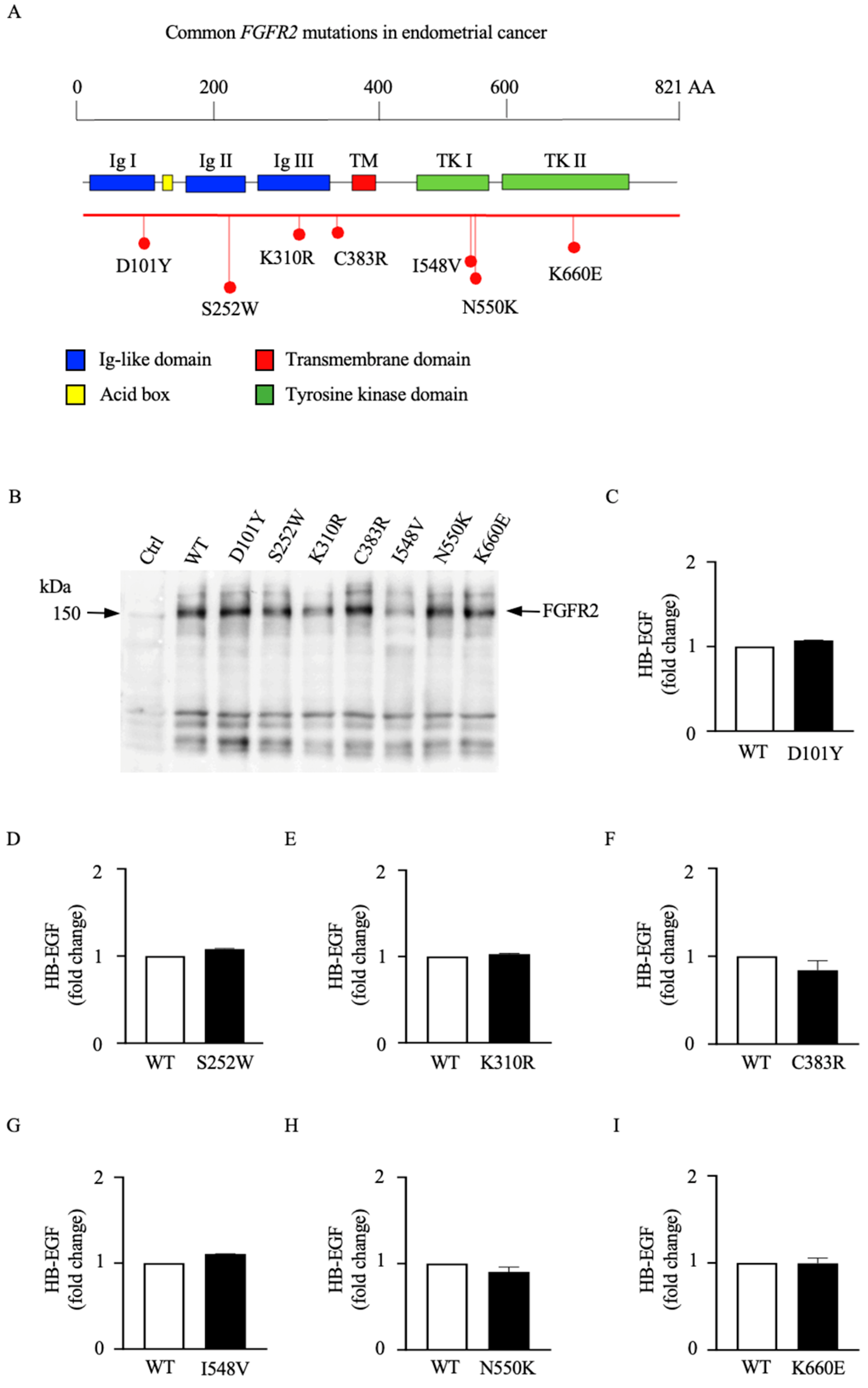
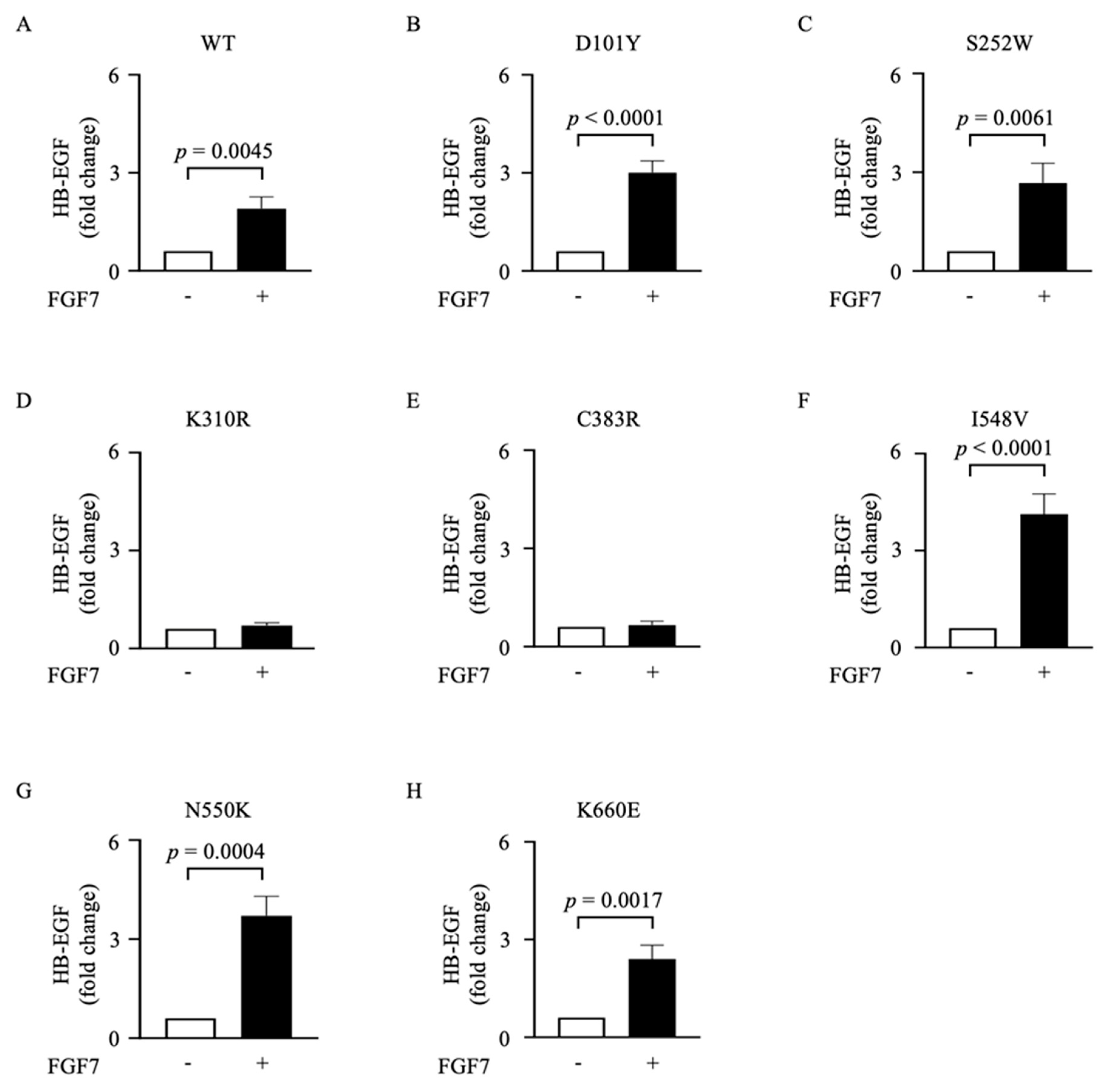
3.2. Different Mutations in the FGFR2 Gene Are Functionally Distinct in Their Abilities to Activate ADAM17
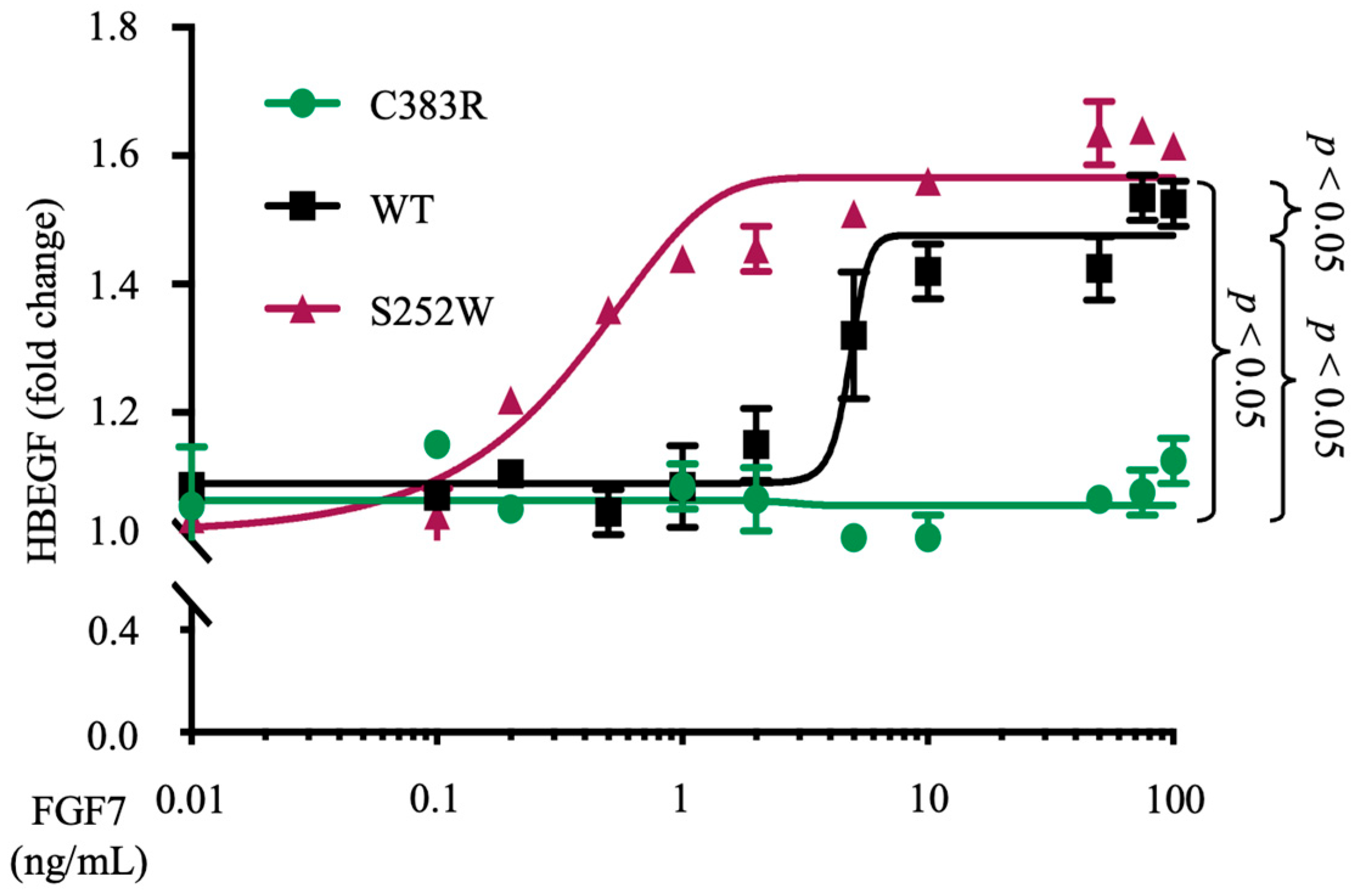
3.3. FGFR2 (S252W) Increases the Sensitivity of FGF7 Ligand-Induced ADAM17 Activity
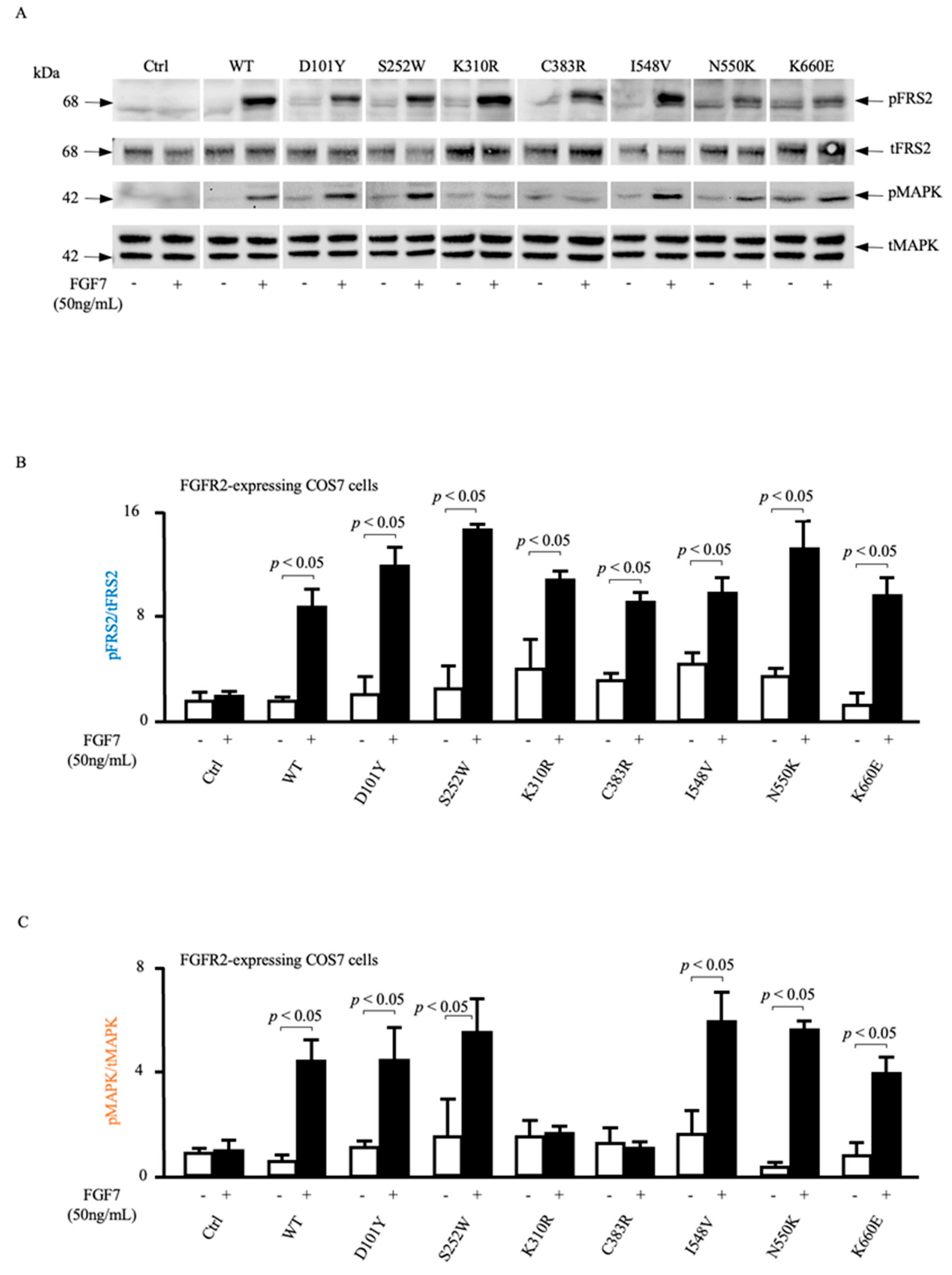
3.4. ADAM17-Dependent MAPK Phosphorylation Is Impaired in Loss-of-Function FGFR2 Mutants
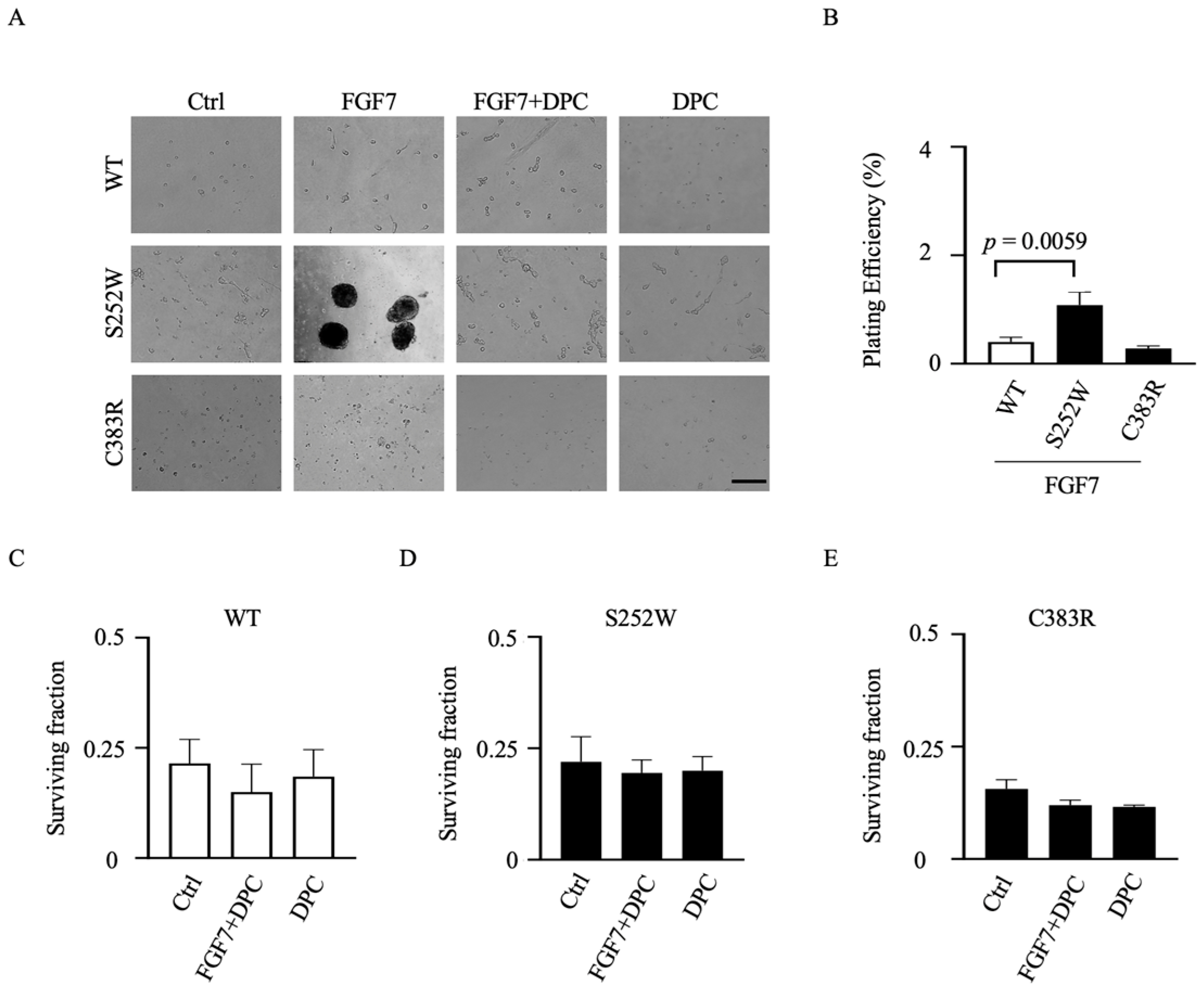
3.5. Transformed Colony Formation in FGFR2-Expressing NIH 3T3 Cell Lines Depends on ADAM17 Activation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM17 | A disintegrin and metalloprotease 17 |
| AP | Alkaline phosphatase |
| EC | Endometrial cancer |
| EGFR | Epidermal growth factor receptor |
| FGF | Fibroblast growth factor |
| FGFR2 | Fibroblast growth factor receptor 2 |
| FRS2 | Fibroblast growth factor receptor substrate 2 |
| HB-EGF | Heparin-binding EGF-like growth factor |
| iRhom2 | Inactive rhomboid protein 2 |
| MAPK | Mitogen-activated protein kinase |
References
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Lander, E.S. Lessons from the Cancer Genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef]
- Stratton, M.R. Exploring the Genomes of Cancer Cells: Progress and Promise. Science 2011, 331, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Bianco, B.; Barbosa, C.P.; Trevisan, C.M.; Laganà, A.S.; Montagna, E. Endometrial cancer: A genetic point of view. Transl. Cancer Res. 2020, 9, 7706–7715. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Gartside, M.G.; Dejeza, L.C.; Powell, M.A.; Mallon, M.A.; Davies, H.; Mohammadi, M.; Futreal, P.A.; Stratton, M.R.; Trent, J.M.; et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 2007, 26, 7158–7162. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.-L.; Tuo, X.-M.; Rong, Y.; Zhang, K.; Guo, Y. Fibroblast growth factor receptor signaling as therapeutic targets in female reproductive system cancers. J. Cancer 2020, 11, 7264–7275. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Pollock, P.M. FGFR2 as a molecular target in endometrial cancer. Futur. Oncol. 2009, 5, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Dutt, A.; Salvesen, H.B.; Chen, T.-H.; Ramos, A.H.; Onofrio, R.C.; Hatton, C.; Nicoletti, R.; Winckler, W.; Grewal, R.; Hanna, M.; et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 8713–8717. [Google Scholar] [CrossRef] [PubMed]
- Winterhoff, B.; Konecny, G.E. Targeting fibroblast growth factor pathways in endometrial cancer. Curr. Probl. Cancer 2017, 41, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.S.; Secord, A.A. Targeting molecular pathways in endometrial cancer: A focus on the FGFR pathway. Cancer Treat. Rev. 2014, 40, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Gartside, M.; Powell, M.A.; Wellens, C.L.; Gao, F.; Mutch, D.G.; Goodfellow, P.J.; Pollock, P.M. FGFR2 point mutations in 466 endometrioid endometrial tumors: Relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS ONE 2012, 7, e30801. [Google Scholar] [CrossRef]
- Krakstad, C.; Birkeland, E.; Seidel, D.; Kusonmano, K.; Petersen, K.; Mjøs, S.; Hoivik, E.A.; Wik, E.; Halle, M.K.; Øyan, A.M.; et al. High-Throughput Mutation Profiling of Primary and Metastatic Endometrial Cancers Identifies KRAS, FGFR2 and PIK3CA to Be Frequently Mutated. PLoS ONE 2012, 7, e52795. [Google Scholar] [CrossRef] [PubMed]
- Konecny, G.E.; Finkler, N.; Garcia, A.A.; Lorusso, D.; Lee, P.S.; Rocconi, R.P.; Fong, P.C.; Squires, M.; Mishra, K.; Upalawanna, A.; et al. Second-line dovitinib (TKI258) in patients with FGFR2-mutated or FGFR2-non-mutated advanced or metastatic endometrial cancer: A non-randomised, open-label, two-group, two-stage, phase 2 study. Lancet Oncol. 2015, 16, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Sarabipour, S.; Hristova, K. Mechanism of FGF receptor dimerization and activation. Nat. Commun. 2016, 7, 10262. [Google Scholar] [CrossRef] [PubMed]
- Karp, J.M.; Sparks, S.; Cowburn, D. Effects of FGFR2 kinase activation loop dynamics on catalytic activity. PLOS Comput. Biol. 2017, 13, e1005360. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Rose, B.A.; Force, T.; Wang, Y.; van der Velden, J.; Stienen, G.J.M.; You, J.; Wu, J.; Zhang, Q.; Ye, Y.; Wang, S.; et al. Mitogen-Activated Protein Kinase Signaling in the Heart: Angels Versus Demons in a Heart-Breaking Tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Carew, T.J. The Roles of MAPK Cascades in Synaptic Plasticity and Memory in Aplysia: Facilitatory Effects and Inhibitory Constraints. Learn. Mem. 2004, 11, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Gamady, A.; Koren, R.; Ron, D.; Liberman, U.A.; Ravid, A. Vitamin D enhances mitogenesis mediated by keratinocyte growth factor receptor in keratinocytes. J. Cell. Biochem. 2003, 89, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Finch, P.W.; Rubin, J.S. Keratinocyte Growth Factor⧸Fibroblast Growth Factor 7, a Homeostatic Factor with Therapeutic Potential for Epithelial Protection and Repair. Adv. Cancer Res. 2004, 91, 69–136. [Google Scholar] [CrossRef] [PubMed]
- Uzan, B.; Figeac, F.; Portha, B.; Movassat, J. Mechanisms of KGF Mediated Signaling in Pancreatic Duct Cell Proliferation and Differentiation. PLoS ONE 2009, 4, e4734. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; Evers, A.; Zhou, W.; Swendeman, S.L.; Wong, P.-M.; Rafii, S.; Reiss, K.; Blobel, C.P. Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat. Commun. 2011, 2, 229. [Google Scholar] [CrossRef] [PubMed]
- Dixit, G.; Gonzalez-Bosquet, J.; Skurski, J.; Devor, E.J.; Dickerson, E.B.; Nothnick, W.B.; Issuree, P.D.; Leslie, K.K.; Maretzky, T. FGFR2 mutations promote endometrial cancer progression through dual engagement of EGFR and Notch signalling pathways. Clin. Transl. Med. 2023, 13, e1223. [Google Scholar] [CrossRef] [PubMed]
- Jeske, Y.W.; Ali, S.; Byron, S.A.; Gao, F.; Mannel, R.S.; Ghebre, R.G.; DiSilvestro, P.A.; Lele, S.B.; Pearl, M.L.; Schmidt, A.P.; et al. FGFR2 mutations are associated with poor outcomes in endometrioid endometrial cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 145, 366–373. [Google Scholar] [CrossRef]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.-M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef]
- Dixit, G.; Schanz, W.; Pappas, B.A.; Maretzky, T. Members of the Fibroblast Growth Factor Receptor Superfamily Are Proteolytically Cleaved by Two Differently Activated Metalloproteases. Int. J. Mol. Sci. 2021, 22, 3165. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; Le Gall, S.M.; Worpenberg-Pietruk, S.; Eder, J.; Overall, C.M.; Huang, X.-Y.; Poghosyan, Z.; Edwards, D.R.; Blobel, C.P. Src Stimulates Fibroblast Growth Factor Receptor-2 Shedding by an ADAM15 Splice Variant Linked to Breast Cancer. Cancer Res 2009, 69, 4573–4576. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Weskamp, G.; Zheng, Y.; Chesneau, V.; Horiuchi, K.; Blobel, C.P. A Sensitive Method to Monitor Ectodomain Shedding of Ligands of the Epidermal Growth Factor Receptor. 2006, 327, 99–114. In Epidermal Growth Factor. Methods in Molecular Biology; Humana Press; Totowa, NJ, USA, 2006; Volume 327, pp. 99–114. [Google Scholar] [CrossRef]
- Le Gall, S.M.; Maretzky, T.; Issuree, P.D.A.; Niu, X.-D.; Reiss, K.; Saftig, P.; Khokha, R.; Lundell, D.; Blobel, C.P. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci. 2010, 123, 3913–3922. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; Yang, G.; Ouerfelli, O.; Overall, C.M.; Worpenberg, S.; Hassiepen, U.; Eder, J.; Blobel, C.P. Characterization of the catalytic activity of the membrane-anchored metalloproteinase ADAM15 in cell-based assays. Biochem. J. 2009, 420, 105–113. [Google Scholar] [CrossRef]
- Lugo, T.G.; Witte, O.N. The BCR-ABL oncogene transforms Rat-1 cells and cooperates with v-myc. Mol. Cell Biol. 1989, 9, 1263–1270. [Google Scholar] [PubMed]
- Wang, C.; Liu, H.; Qiu, Q.; Zhang, Z.; Gu, Y.; He, Z. TCRP1 promotes NIH/3T3 cell transformation by over-activating PDK1 and AKT1. Oncogenesis 2017, 6, e323. [Google Scholar] [CrossRef] [PubMed]
- Nagano, O.; Murakami, D.; Hartmann, D.; de Strooper, B.; Saftig, P.; Iwatsubo, T.; Nakajima, M.; Shinohara, M.; Saya, H. Cell–matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca2+ influx and PKC activation. J. Cell Biol. 2004, 165, 893–902. [Google Scholar] [CrossRef] [PubMed]
- la Rubia, E.C.-D.; Martinez-Garcia, E.; Dittmar, G.; Gil-Moreno, A.; Cabrera, S.; Colas, E. Prognostic Biomarkers in Endometrial Cancer: A Systematic Review and Meta-Analysis. J. Clin. Med. 2020, 9, 1900. [Google Scholar] [CrossRef]
- Tang, B.; Li, X.; Maretzky, T.; Perez-Aguilar, J.M.; McIlwain, D.; Xie, Y.; Zheng, Y.; Mak, T.W.; Weinstein, H.; Blobel, C.P. Substrate-selective protein ectodomain shedding by ADAM17 and iRhom2 depends on their juxtamembrane and transmembrane domains. FASEB J. 2020, 34, 4956–4969. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.A.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 11433–11438, Correction in 2021, 118, e2112652118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Dávila, E.M.; Li, X.; Tang, B.; Rabinowitsch, A.I.; Perez-Aguilar, J.M.; Blobel, C.P. Identification of Molecular Determinants in iRhoms1 and 2 That Contribute to the Substrate Selectivity of Stimulated ADAM17. Int. J. Mol. Sci. 2022, 23, 12796. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.I.; Rose-John, S.; Jenkins, B.J. ADAM17: An Emerging Therapeutic Target for Lung Cancer. Cancers 2019, 11, 1218. [Google Scholar] [CrossRef]
- Xiang, Y.; Liu, L.; Wang, Y.; Li, B.; Peng, J.; Feng, D. ADAM17 promotes the invasion of hepatocellular carcinoma via upregulation MMP21. Cancer Cell Int. 2020, 20, 516. [Google Scholar] [CrossRef] [PubMed]
- Mustafi, R.; Dougherty, U.; Mustafi, D.; Ayaloglu-Butun, F.; Fletcher, M.; Adhikari, S.; Sadiq, F.; Meckel, K.; Haider, H.I.; Khalil, A.; et al. ADAM17 is a Tumor Promoter and Therapeutic Target in Western Diet-associated Colon Cancer. Clin. Cancer Res. 2017, 23, 549–561. [Google Scholar] [CrossRef]
- Rogmans, C.; Kuhlmann, J.D.; Hugendieck, G.; Link, T.; Arnold, N.; Weimer, J.P.; Flörkemeier, I.; Rambow, A.C.; Lieb, W.; Maass, N.; et al. ADAM17-A Potential Blood-Based Biomarker for Detection of Early-Stage Ovarian Cancer. Cancers 2021, 13, 5563. [Google Scholar] [CrossRef] [PubMed]
- Zingg, D.; Bhin, J.; Yemelyanenko, J.; Kas, S.M.; Rolfs, F.; Lutz, C.; Lee, J.K.; Klarenbeek, S.; Silverman, I.M.; Annunziato, S.; et al. Truncated FGFR2 is a clinically actionable oncogene in multiple cancers. Nature 2022, 608, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Bahleda, R.; Dienstmann, R.; Infante, J.R.; Mita, A.; Italiano, A.; Calvo, E.; Moreno, V.; Adamo, B.; Gazzah, A.; et al. Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar] [CrossRef]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.H.; Hierro, C.; Heist, R.S.; Cleary, J.M.; Meric-Bernstam, F.; Tabernero, J.; Janku, F.; Gandhi, L.; Iafrate, A.J.; Borger, D.R.; et al. A Phase I, Open-Label, Multicenter, Dose-escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Patients with Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin. Cancer Res. 2019, 25, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Iannotti, N.; Gutierrez, M.; Smith, D.; Féliz, L.; Lihou, C.; Tian, C.; Silverman, I.; Ji, T.; Saleh, M. FIGHT-101, a first-in-human study of potent and selective FGFR 1-3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann. Oncol. 2022, 33, 522–533. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dixit, G.; Pappas, B.A.; Bhardwaj, G.; Schanz, W.; Maretzky, T. Functional Distinctions of Endometrial Cancer-Associated Mutations in the Fibroblast Growth Factor Receptor 2 Gene. Cells 2023, 12, 2227. https://doi.org/10.3390/cells12182227
Dixit G, Pappas BA, Bhardwaj G, Schanz W, Maretzky T. Functional Distinctions of Endometrial Cancer-Associated Mutations in the Fibroblast Growth Factor Receptor 2 Gene. Cells. 2023; 12(18):2227. https://doi.org/10.3390/cells12182227
Chicago/Turabian StyleDixit, Garima, Benjamin A. Pappas, Gourav Bhardwaj, Willow Schanz, and Thorsten Maretzky. 2023. "Functional Distinctions of Endometrial Cancer-Associated Mutations in the Fibroblast Growth Factor Receptor 2 Gene" Cells 12, no. 18: 2227. https://doi.org/10.3390/cells12182227
APA StyleDixit, G., Pappas, B. A., Bhardwaj, G., Schanz, W., & Maretzky, T. (2023). Functional Distinctions of Endometrial Cancer-Associated Mutations in the Fibroblast Growth Factor Receptor 2 Gene. Cells, 12(18), 2227. https://doi.org/10.3390/cells12182227