
Identification of the Inner Cell Mass and the Trophectoderm Responses after an In Vitro Exposure to Glucose and Insulin during the Preimplantation Period in the Rabbit Embryo

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Embryo In-Vitro Development
2.2. RNA Sequencing
2.3. Quantification of Total Cell Number in Whole Embryos
2.4. Quantification of Apoptotic and Proliferating Cells in ICM and TE
2.5. Statistical Analysis
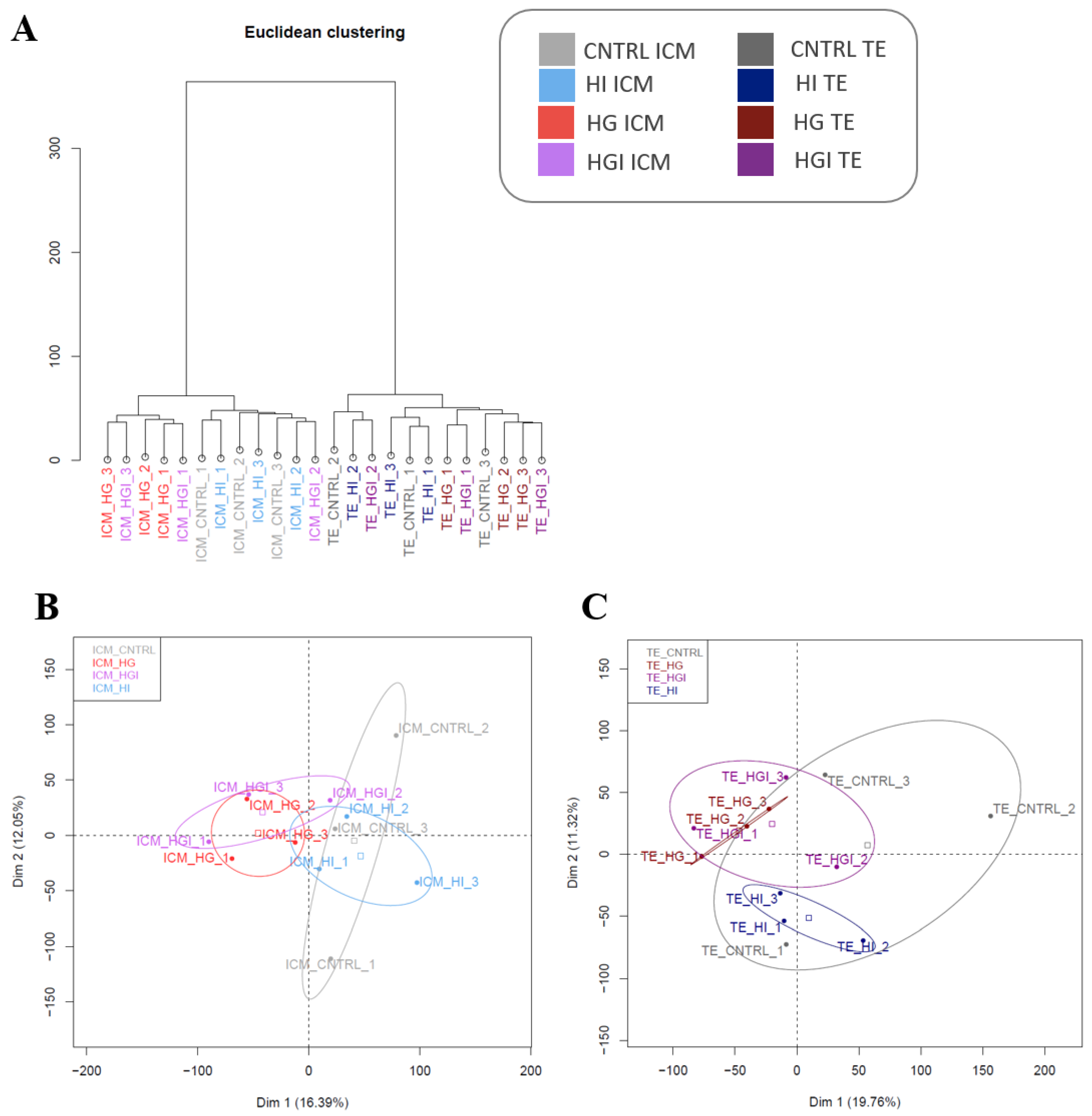
3. Results
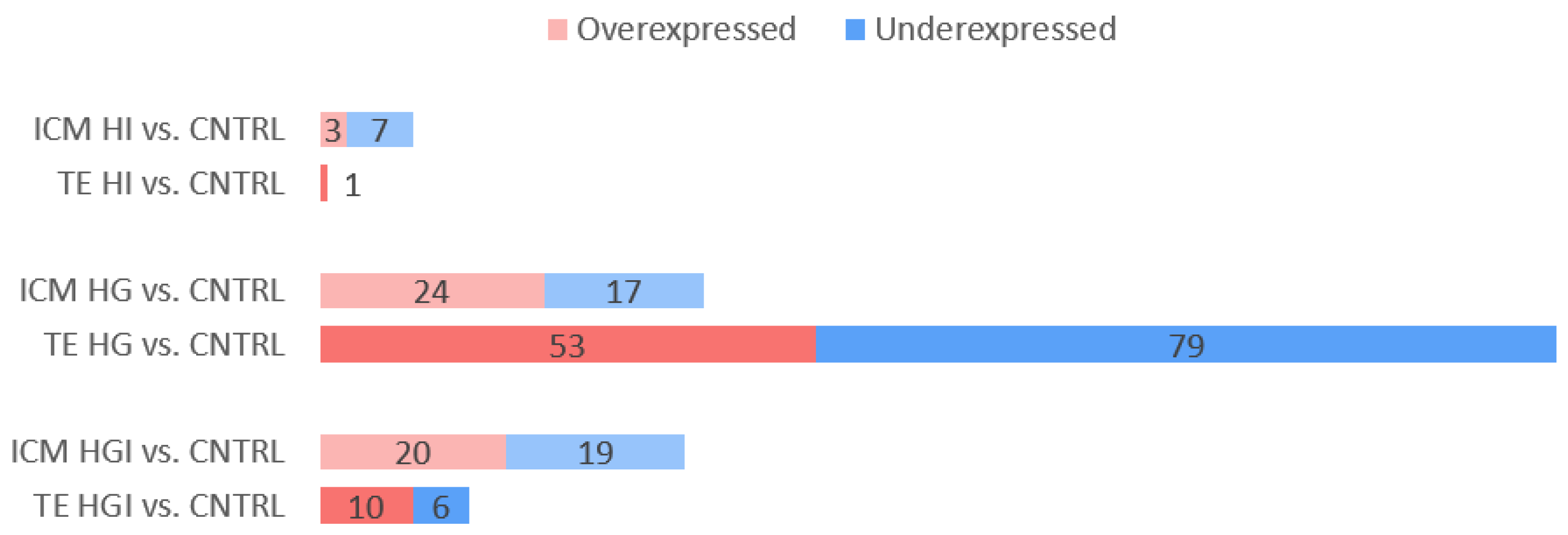
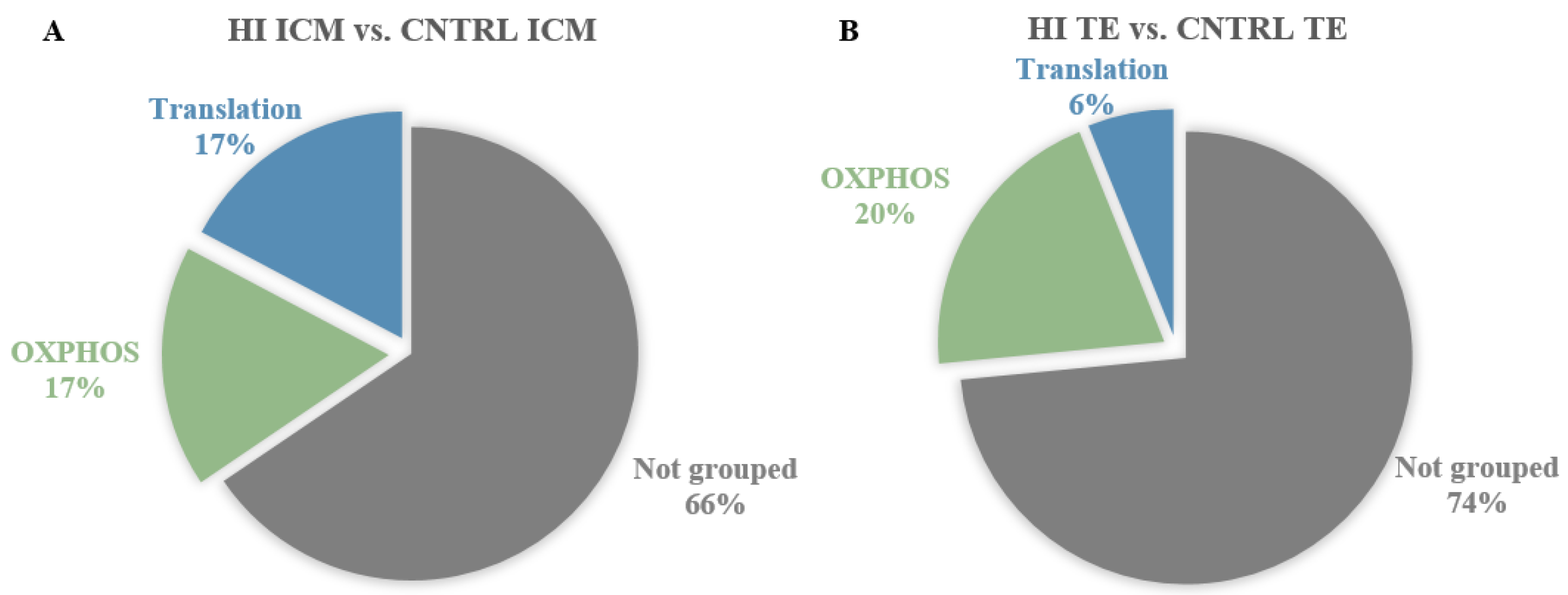
3.1. Impact of High Insulin In Vitro Exposure
3.1.1. In ICM, High Insulin Induced Changes in Cellular Energy Metabolic Pathways
3.1.2. In TE, High Insulin Impacted Cellular Energy Metabolism and Oxidative Stress Pathways
3.1.3. High Insulin Induced Common Responses in ICM and TE
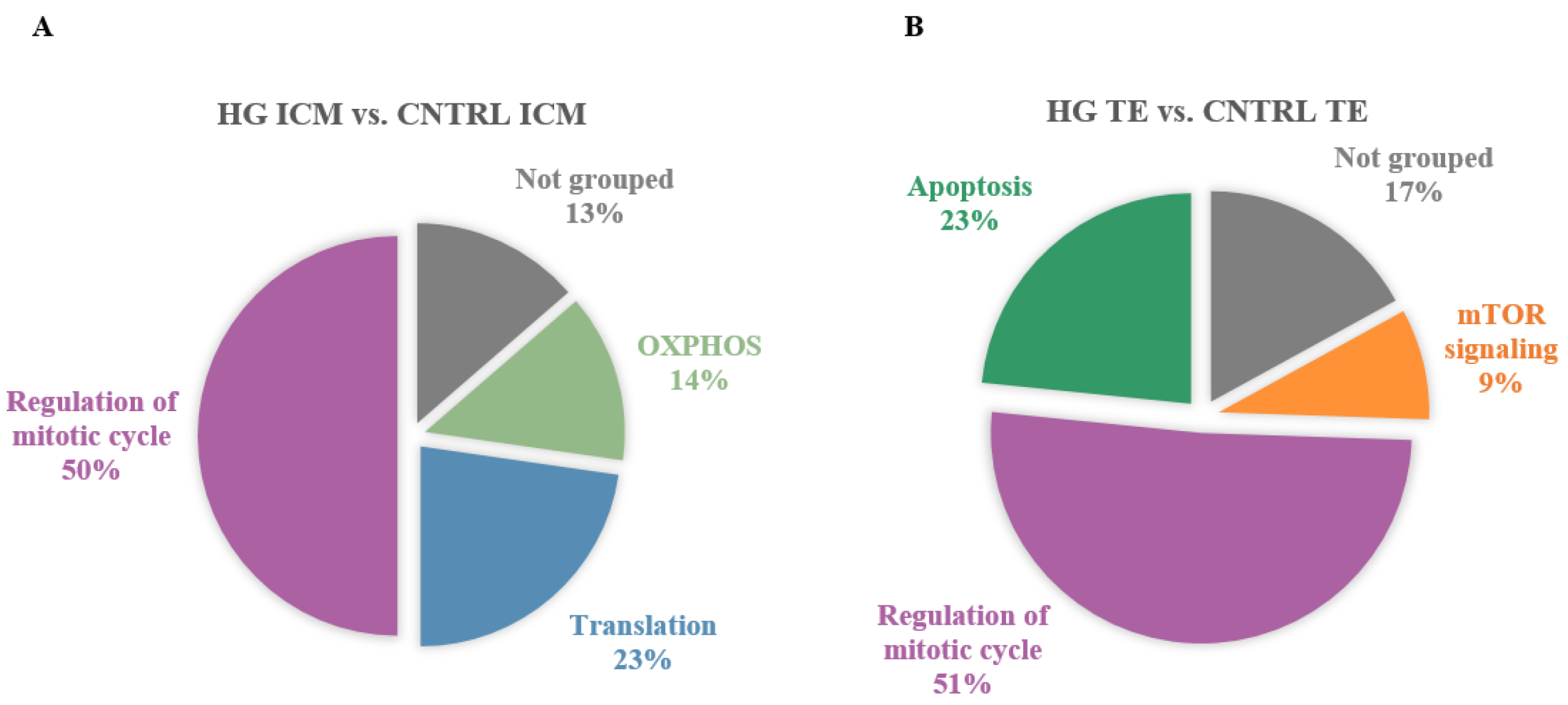
3.2. Impact of High Glucose In Vitro Exposure
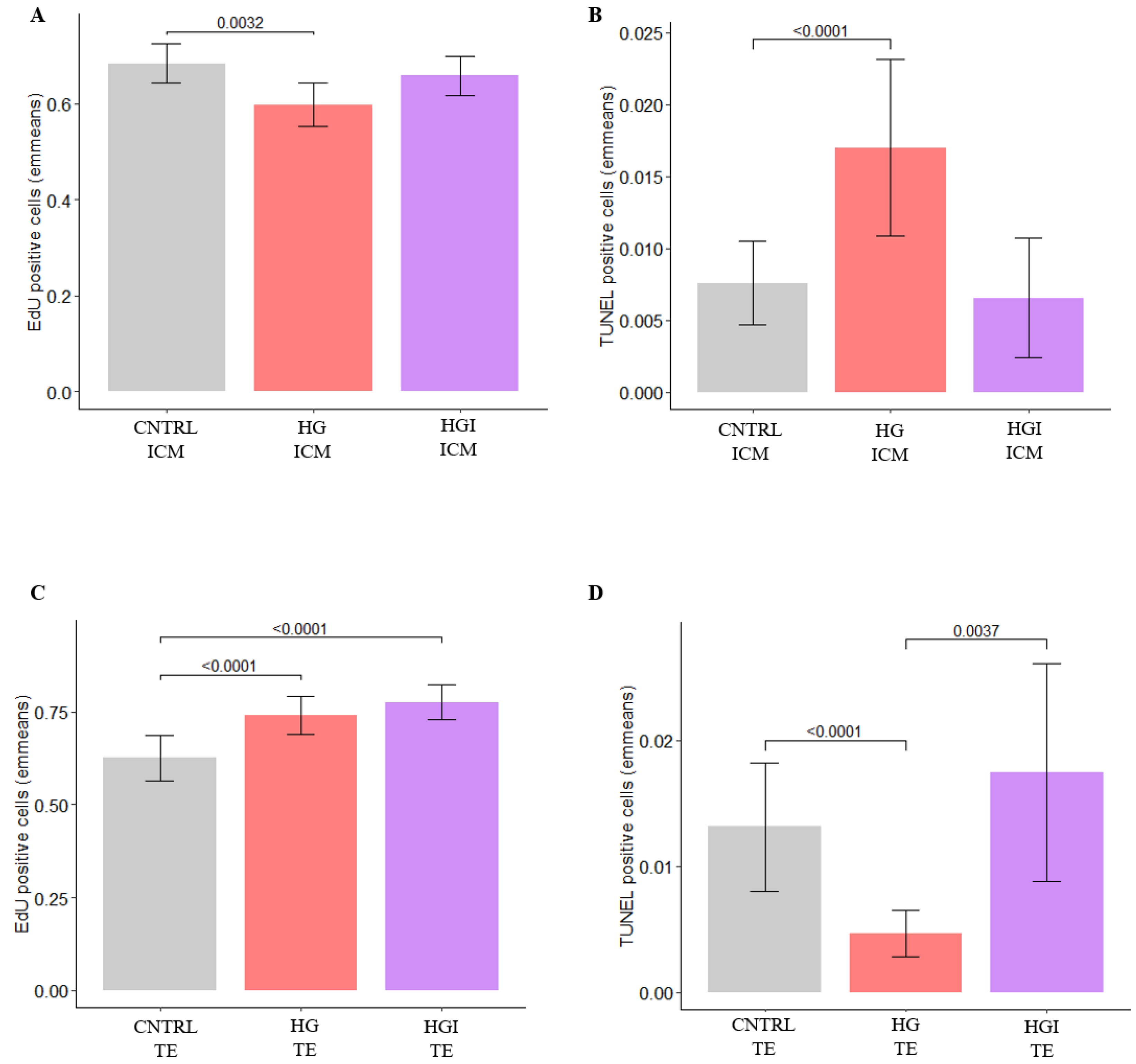
3.2.1. In ICM, High Glucose Altered OXPHOS, Decreased Proliferation, Increased Apoptosis
3.2.2. In TE, High Glucose Impacted Metabolic Pathways, Increased Proliferation, and Decreased Apoptosis
3.2.3. High Glucose Induced Common and Specific Responses in ICM and TE
3.3. Impact of High Glucose and High Insulin In Vitro Exposure
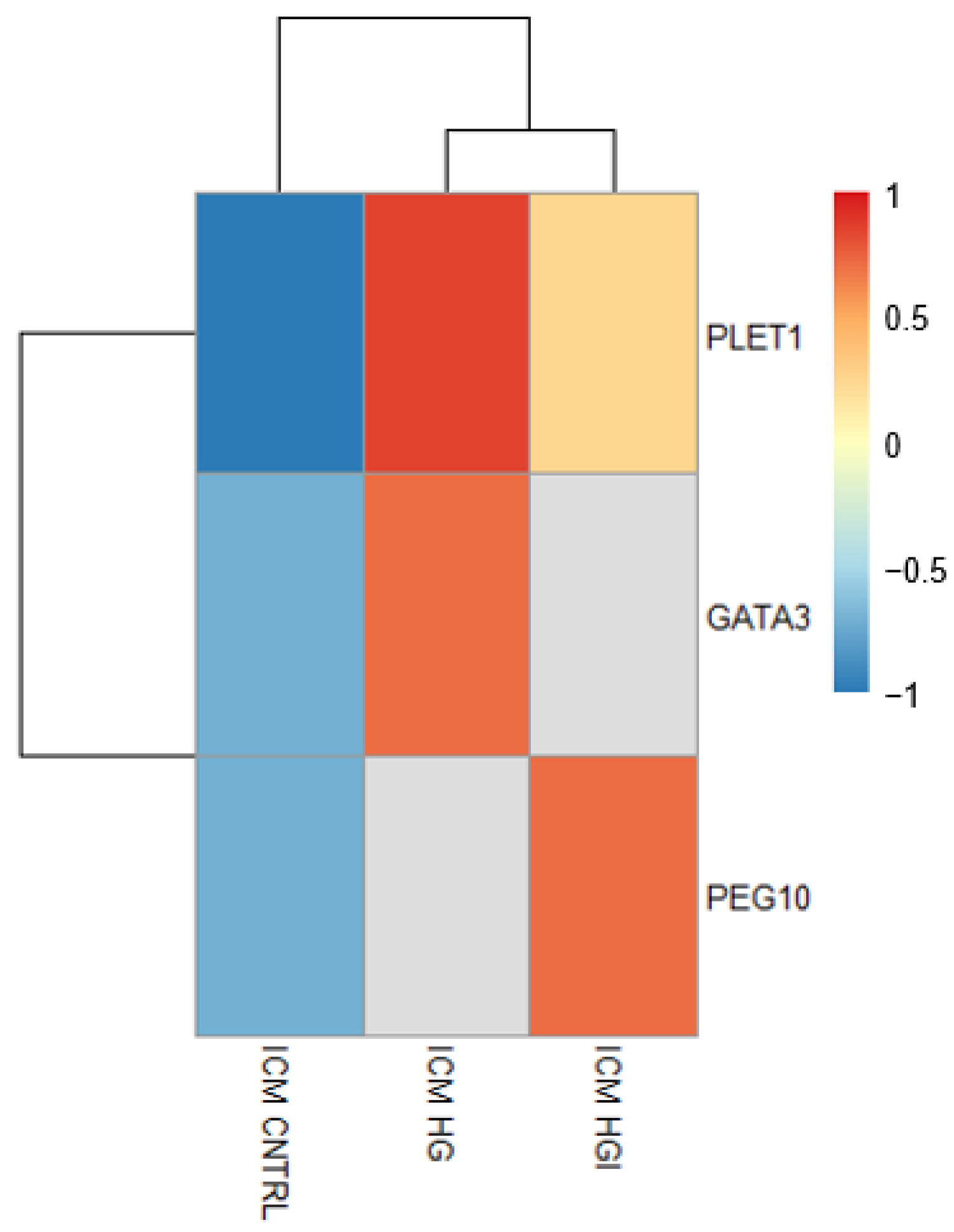
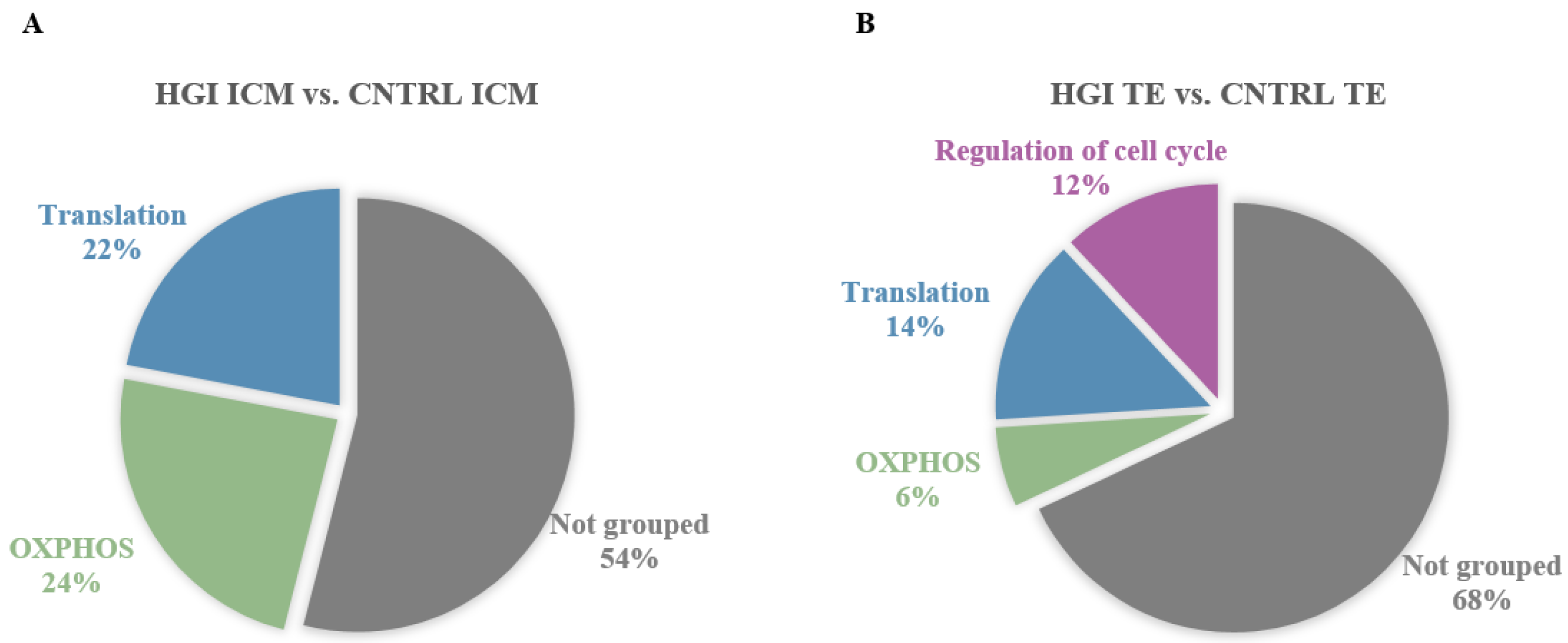
3.3.1. In ICM, Alteration of OXPHOS and ROS by High Glucose and High Insulin
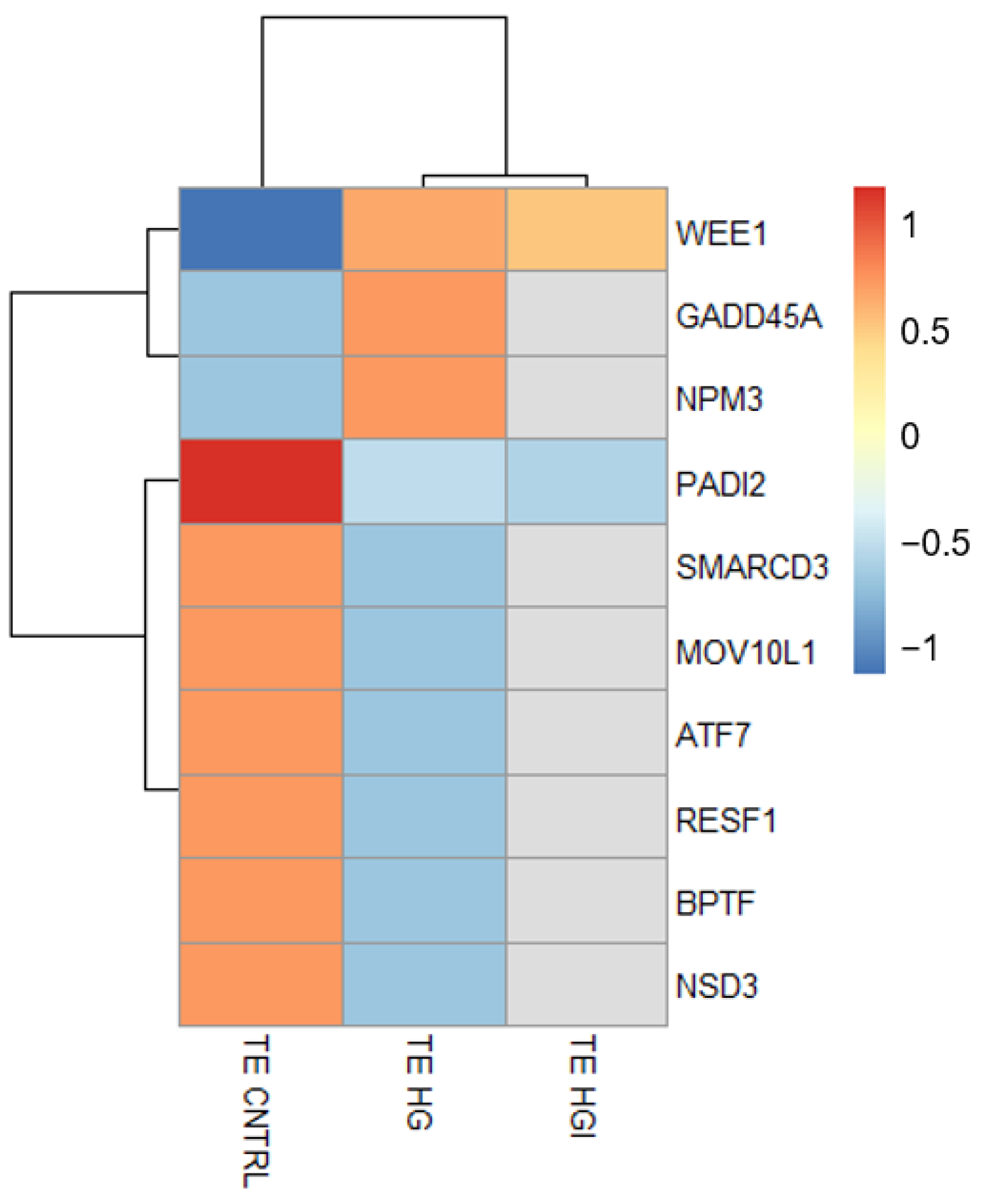
3.3.2. In TE, Alteration of OXPHOS, ROS, and Proliferation by High Glucose and High Insulin
3.3.3. High Glucose and High Insulin Induced Common and Specific Responses in ICM and TE
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- InteInternational Diabetes Federation. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, Belgium, 2021; ISBN 978-2-930229-98-0. [Google Scholar]
- Thomas, D.D.; Corkey, B.E.; Istfan, N.W.; Apovian, C.M. Hyperinsulinemia: An Early Indicator of Metabolic Dysfunction. J. Endocr. Soc. 2019, 3, 1727–1747. [Google Scholar] [CrossRef]
- Hjort, L.; Novakovic, B.; Grunnet, L.G.; Maple-Brown, L.; Damm, P.; Desoye, G.; Saffery, R. Diabetes in Pregnancy and Epigenetic Mechanisms—How the First 9 Months from Conception Might Affect the Child’s Epigenome and Later Risk of Disease. Lancet Diabetes Endocrinol. 2019, 7, 796–806. [Google Scholar] [CrossRef] [PubMed]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 Diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Francis, E.C.; Dabelea, D.; Ringham, B.M.; Sauder, K.A.; Perng, W. Maternal Blood Glucose Level and Offspring Glucose–Insulin Homeostasis: What Is the Role of Offspring Adiposity? Diabetologia 2020, 64, 83–94. [Google Scholar] [CrossRef]
- Langley-Evans, S.C. Nutrition in Early Life and the Programming of Adult Disease: A Review. J. Hum. Nutr. Diet. 2015, 28, 1–14. [Google Scholar] [CrossRef]
- Watkins, A.J.; Ursell, E.; Panton, R.; Papenbrock, T.; Hollis, L.; Cunningham, C.; Wilkins, A.; Perry, V.H.; Sheth, B.; Kwong, W.Y.; et al. Adaptive Responses by Mouse Early Embryos to Maternal Diet Protect Fetal Growth but Predispose to Adult Onset Disease1. Biol. Reprod. 2007, 78, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, M.A. Impact of Maternal Malnutrition during the Periconceptional Period on Mammalian Preimplantation Embryo Development. Domest. Anim. Endocrinol. 2015, 51, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Kaye, P.L.; Gardner, H.G. Preimplantation Access to Maternal Insulin and Albumin Increases Fetal Growth Rate in Mice. Hum. Reprod. 1999, 14, 3052–3059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acevedo, J.J.; Mendoza-Lujambio, I.; de la Vega-Beltrán, J.L.; Treviño, C.L.; Felix, R.; Darszon, A. KATP Channels in Mouse Spermatogenic Cells and Sperm, and Their Role in Capacitation. Dev. Biol. 2006, 289, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Fraser, R.B.; Waite, S.L.; Wood, K.A.; Martin, K.L. Impact of Hyperglycemia on Early Embryo Development and Embryopathy: In Vitro Experiments Using a Mouse Model. Hum. Reprod. 2007, 22, 3059–3068. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.P.; Sun, C.; Denisenko, O.; Caetano, L.; Aljahdali, A.; Gould, J.M.; Khurana, P. Environmental Exposures around Conception: Developmental Pathways Leading to Lifetime Disease Risk. Int. J. Environ. Res. Public Health 2021, 18, 9380. [Google Scholar] [CrossRef] [PubMed]
- Jungheim, E.S.; Moley, K.H. The Impact of Type 1 and Type 2 Diabetes Mellitus on the Oocyte and the Preimplantation Embryo. Semin. Reprod. Med. 2008, 26, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Ramin, N.; Thieme, R.; Fischer, S.; Schindler, M.; Schmidt, T.; Fischer, B.; Santos, A.N. Maternal Diabetes Impairs Gastrulation and Insulin and IGF-I Receptor Expression in Rabbit Blastocysts. Endocrinology 2010, 151, 4158–4167. [Google Scholar] [CrossRef] [Green Version]
- Rousseau-Ralliard, D.; Couturier-Tarrade, A.; Thieme, R.; Brat, R.; Rolland, A.; Boileau, P.; Aubrière, M.-C.; Daniel, N.; Dahirel, M.; Derisoud, E.; et al. A Short Periconceptional Exposure to Maternal Type-1 Diabetes Is Sufficient to Disrupt the Feto-Placental Phenotype in a Rabbit Model. Mol. Cell. Endocrinol. 2019, 480, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Moley, K.H.; Chi, M.M.Y.M.-Y.; Mueckler, M.M. Maternal Hyperglycemia Alters Glucose Transport and Utilization in Mouse Preimplantation Embryos. Am. J. Physiol. Metab. 1998, 275, E38–E47. [Google Scholar] [CrossRef]
- Leunda-Casi, A.; de Hertogh, R.; Pampfer, S. Decreased Expression of Fibroblast Growth Factor-4 and Associated Dysregulation of Trophoblast Differentiation in Mouse Blastocysts Exposed to High D-Glucose in Vitro. Diabetologia 2001, 44, 1318–1325. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.H.; Moley, K.H. Glucose Transporters in Gametes and Preimplantation Embryos. Trends Endocrinol. Metab. 2009, 20, 483–489. [Google Scholar] [CrossRef]
- Gardner, D.K.; Harvey, A.J. Blastocyst Metabolism. Reprod. Fertil. Dev. 2015, 27, 638–654. [Google Scholar] [CrossRef]
- Navarrete Santos, A.; Ramin, N.; Tonack, S.; Fischer, B. Cell Lineage-Specific Signaling of Insulin and Insulin-Like Growth Factor I in Rabbit Blastocysts. Endocrinology 2008, 149, 515–524. [Google Scholar] [CrossRef]
- Canon, E.; Jouneau, L.; Blachère, T.; Peynot, N.; Daniel, N.; Boulanger, L.; Maulny, L.; Archilla, C.; Voisin, S.; Jouneau, A.; et al. Progressive Methylation of POU5F1 Regulatory Regions during Blastocyst Development. Reproduction 2018, 156, 145–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchereau, W.; Jouneau, L.; Archilla, C.; Aksoy, I.; Moulin, A.; Daniel, N.; Peynot, N.; Calderari, S.; Joly, T.; Godet, M.; et al. Major Transcriptomic, Epigenetic and Metabolic Changes Underlie the Pluripotency Continuum in Rabbit Preimplantation Embryos. Development 2022, 149, dev200538. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.P.; Kwong, W.Y.; Porter, R.; Ursell, E.; Fesenko, I.; Wilkins, A.; Miller, D.J.; Watkins, A.J.; Eckert, J.J. The Embryo and Its Future. Biol. Reprod. 2004, 71, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staud, F.; Karahoda, R. Trophoblast: The Central Unit of Fetal Growth, Protection and Programming. Int. J. Biochem. Cell Biol. 2018, 105, 35–40. [Google Scholar] [CrossRef]
- Fischer, B.; Chavatte-Palmer, P.; Viebahn, C.; Navarrete Santos, A.; Duranthon, V. Rabbit as a Reproductive Model for Human Health. Reproduction 2012, 144, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, D.; Sjunnesson, Y.; Humblot, P.; Sirard, M.A.; Andersson, G.; Gustafsson, H.; Båge, R. Insulin Exposure during in Vitro Bovine Oocyte Maturation Changes Blastocyst Gene Expression and Developmental Potential. Reprod. Fertil. Dev. 2017, 29, 876–889. [Google Scholar] [CrossRef] [Green Version]
- Tarrade, A.; Rousseau-Ralliard, D.; Aubrière, M.C.; Peynot, N.; Dahirel, M.; Bertrand-Michel, J.; Aguirre-Lavin, T.; Morel, O.; Beaujean, N.; Duranthon, V.; et al. Sexual Dimorphism of the Feto-Placental Phenotype in Response to a High Fat and Control Maternal Diets in a Rabbit Model. PLoS ONE 2013, 8, e83458. [Google Scholar] [CrossRef] [Green Version]
- Sanz, G.; Daniel, N.; Aubrière, M.C.; Archilla, C.; Jouneau, L.; Jaszczyszyn, Y.; Duranthon, V.; Chavatte-Palmer, P.; Jouneau, A. Differentiation of Derived Rabbit Trophoblast Stem Cells under Fluid Shear Stress to Mimic the Trophoblastic Barrier. Biochim. Biophys. Acta-Gen. Subj. 2019, 1863, 1608–1618. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinform. 2015, 16, 1–7. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2019, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbon, S.; Douglass, E.; Good, B.M.; Unni, D.R.; Harris, N.L.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; Hartline, E.; et al. The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Savage, S.R.; Shi, Z.; Liao, Y.; Zhang, B. Graph Algorithms for Condensing and Consolidating Gene Set Analysis Results. Mol. Cell. Proteomics 2019, 18, S141–S152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.H.; Apeltsin, L.; Newman, A.M.; Baumbach, J.; Wittkop, T.; Su, G.; Bader, G.D.; Ferrin, T.E. ClusterMaker: A Multi-Algorithm Clustering Plugin for Cytoscape. BMC Bioinform. 2011, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Gürke, J.; Schindler, M.; Pendzialek, S.M.; Thieme, R.; Grybel, K.J.; Heller, R.; Spengler, K.; Fleming, T.P.; Fischer, B.; Navarrete Santos, A. Maternal Diabetes Promotes MTORC1 Downstream Signalling in Rabbit Preimplantation Embryos. Reproduction 2016, 151, 465–476. [Google Scholar] [CrossRef]
- Shao, W.-J.; Tao, L.-Y.; Xie, J.-Y.; Gao, C.; Hu, J.-H.; Zhao, R.-Q. Exposure of Preimplantation Embryos to Insulin Alters Expression of Imprinted Genes. Comp. Med. 2007, 57, 482–486. [Google Scholar] [PubMed]
- Schindler, M.; Pendzialek, S.M.; Grybel, K.; Seeling, T.; Santos, A.N. Metabolic Profiling in Blastocoel Fluid and Blood Plasma of Diabetic Rabbits. Int. J. Mol. Sci. 2020, 21, 919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez, A.; Madrid-Bury, N.; Fernández, R.; Pérez-Garnelo, S.; Moreira, P.; Pintado, B.; de la Fuente, J.; Gutiérrez-Adán, A. Hyperglycemia-Induced Apoptosis Affects Sex Ratio of Bovine and Murine Preimplantation Embryos. Mol. Reprod. Dev. 2003, 65, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Moley, K.H. Diabetes and Preimplantation Events of Embryogenesis. Semin. Reprod. Endocrinol. 1999, 17, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Pampfer, S. Apoptosis in Rodent Peri-Implantation Embryos: Differential Susceptibility of Inner Cell Mass and Trophectoderm Cell Lineages—A Review. Placenta 2000, 21, 3–10. [Google Scholar] [CrossRef]
- Harvey, M.B.; Kaye, P.L. Insulin Increases the Cell Number of the Inner Cell Mass and Stimulates Morphological Development of Mouse Blastocysts in Vitro. Development 1990, 110, 963–967. [Google Scholar] [CrossRef]
- Gardner, H.G.; Kaye, P.L. Insulin Increases Cell Numbers and Morphological Development in Mouse Pre-Implantation Embryos in Vitro. Reprod. Fertil. Dev. 1991, 3, 79–91. [Google Scholar] [CrossRef]
- Augustin, R.; Pocar, P.; Wrenzycki, C.; Niemann, H.; Fischer, B. Mitogenic and Anti-Apoptotic Activity of Insulin on Bovine Embryos Produced in Vitro. Reproduction 2003, 126, 91–99. [Google Scholar] [CrossRef]
- Chi, M.M.-Y.; Schlein, A.L.; Moley, K.H. High Insulin-Like Growth Factor 1 (IGF-1) and Insulin Concentrations Trigger Apoptosis in the Mouse Blastocyst via Down-Regulation of the IGF-1 Receptor. Endocrinology 2000, 141, 4784–4792. [Google Scholar] [CrossRef]
- Kaneko, K.J. Metabolism of Preimplantation Embryo Development: A Bystander or an Active Participant? 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 120, ISBN 9780128014288. [Google Scholar]
- May-Panloup, P.; Boguenet, M.; El Hachem, H.; Bouet, P.E.; Reynier, P. Embryo and Its Mitochondria. Antioxidants 2021, 10, 139. [Google Scholar] [CrossRef]
- Leese, H.J.; Conaghan, J.; Martin, K.L.; Hardy, K. Early Human Embryo Metabolism. BioEssays 1993, 15, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 8, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Tseng, Y.; White, M.F. Insulin Signaling Meets Mitochondria in Metabolism. Trends Endocrinol. Metab. 2010, 21, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-ΚB) Doing in and to the Mitochondrion? Front. Cell Dev. Biol. 2019, 7, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, F.; Philippe, C.; Antoine, P.J.; Baud, L.; Groyer, A.; Capeau, J.; Cherqui, G. Insulin Activates Nuclear Factor ΚB in Mammalian Cells through a Raf-1- Mediated Pathway. J. Biol. Chem. 1995, 270, 24435–24441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.J.; Liu, Z. Crosstalk of Reactive Oxygen Species and NF-ΚB Signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Reid, M.A.; Dai, Z.; Locasale, J.W. The Impact of Cellular Metabolism on Chromatin Dynamics and Epigenetics. Nat. Cell Biol. 2017, 19, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Arab, K.; Karaulanov, E.; Musheev, M.; Trnka, P.; Schäfer, A.; Grummt, I.; Niehrs, C. GADD45A Binds R-Loops and Recruits TET1 to CpG Island Promoters. Nat. Genet. 2019, 51, 217–223. [Google Scholar] [CrossRef]
- Qian, X.; Zhang, Y. EZH2 Enhances Proliferation and Migration of Trophoblast Cell Lines by Blocking GADD45A-Mediated P38/MAPK Signaling Pathway. Bioengineered 2022, 13, 12583–12597. [Google Scholar] [CrossRef] [PubMed]
- Goller, T.; Vauti, F.; Ramasamy, S.; Arnold, H.-H. Transcriptional Regulator BPTF/FAC1 Is Essential for Trophoblast Differentiation during Early Mouse Development. Mol. Cell. Biol. 2008, 28, 6819–6827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landry, J.; Sharov, A.A.; Piao, Y.; Sharova, L.V.; Xiao, H.; Southon, E.; Matta, J.; Tessarollo, L.; Zhang, Y.E.; Ko, M.S.H.; et al. Essential Role of Chromatin Remodeling Protein Bptf in Early Mouse Embryos and Embryonic Stem Cells. PLoS Genet. 2008, 4, e1000241. [Google Scholar] [CrossRef]
- Beato, M.; Sharma, P. Peptidyl Arginine Deiminase 2 (PADI2)-Mediated Arginine Citrullination Modulates Transcription in Cancer. Int. J. Mol. Sci. 2020, 21, 1351. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Maekawa, T.; Ly, N.H.; Fujita, S.; Muratani, M.; Ando, M.; Katou, Y.; Araki, H.; Miura, F.; Shirahige, K.; et al. ATF7-Dependent Epigenetic Changes are Required for the Intergenerational Effect of a Paternal Low-Protein Diet. Mol. Cell 2020, 78, 445–458.e6. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Maekawa, T.; Yoshida, K.; Muratani, M.; Chatton, B.; Ishii, S. The Transcription Factor ATF7 Controls Adipocyte Differentiation and Thermogenic Gene Programming. iScience 2019, 13, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralston, A.; Cox, B.J.; Nishioka, N.; Sasaki, H.; Chea, E.; Rugg-Gunn, P.; Guo, G.; Robson, P.; Draper, J.S.; Rossant, J. Gata3 Regulates Trophoblast Development Downstream of Tead4 and in Parallel to Cdx2. Development 2010, 137, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Chi, F.; Sharpley, M.S.; Nagaraj, R.; Roy, S.S.; Banerjee, U. Glycolysis-Independent Glucose Metabolism Distinguishes TE from ICM Fate during Mammalian Embryogenesis. Dev. Cell 2020, 53, 9–26.e4. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.; Sienerth, A.R.; Hemberger, M. Plet1 Is an Epigenetically Regulated Cell Surface Protein That Provides Essential Cues to Direct Trophoblast Stem Cell Differentiation. Sci. Rep. 2016, 6, 25112. [Google Scholar] [CrossRef]
- Ono, R.; Nakamura, K.; Inoue, K.; Naruse, M.; Usami, T.; Wakisaka-Saito, N.; Hino, T.; Suzuki-Migishima, R.; Ogonuki, N.; Miki, H.; et al. Deletion of Peg10, an Imprinted Gene Acquired from a Retrotransposon, Causes Early Embryonic Lethality. Nat. Genet. 2006, 38, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.H.; Lee, Y.L.; Fong, S.W.; Wong, C.C.Y.; Ng, E.H.Y.; Yeung, W.S.B. Hyperglycemia Impedes Definitive Endoderm Differentiation of Human Embryonic Stem Cells by Modulating Histone Methylation Patterns. Cell Tissue Res. 2017, 368, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Armistead, B.; Kadam, L.; Drewlo, S.; Kohan-Ghadr, H.-R.R. The Role of NFκB in Healthy and Preeclamptic Placenta: Trophoblasts in the Spotlight. Int. J. Mol. Sci. 2020, 21, 1775. [Google Scholar] [CrossRef] [Green Version]
- Marchand, M.; Horcajadas, J.A.; Esteban, F.J.; McElroy, S.L.; Fisher, S.J.; Giudice, L.C. Transcriptomic Signature of Trophoblast Differentiation in a Human Embryonic Stem Cell Model. Biol. Reprod. 2011, 84, 1258–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, J.D.; Snow, M.H.L. The Development of Trophoblast in Vitro from Blastocysts Containing Varying Amounts of Inner Cell Mass. J. Embryol. Exp. Morphol. 1975, 33, 177–185. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | N Rabbits | N Embryos | Development Arrest Rate | Compacted Embryos Rate | Blastocyst Rate |
|---|---|---|---|---|---|
| CNTRL | 60 | 1090 | 0.034 (0.009) a | 0.303 (0.061) a | 0.638 (0.057) a |
| HI | 21 | 530 | 0.029 (0.009) a | 0.309 (0.063) a | 0.645 (0.059) a |
| HG | 52 | 751 | 0.027 (0.008) a | 0.228 (0.052) b | 0.726 (0.051) b |
| HGI | 35 | 519 | 0.023 (0.007) a | 0.232 (0.053) b | 0.732 (0.051) b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Via y Rada, R.; Daniel, N.; Archilla, C.; Frambourg, A.; Jouneau, L.; Jaszczyszyn, Y.; Charpigny, G.; Duranthon, V.; Calderari, S. Identification of the Inner Cell Mass and the Trophectoderm Responses after an In Vitro Exposure to Glucose and Insulin during the Preimplantation Period in the Rabbit Embryo. Cells 2022, 11, 3766. https://doi.org/10.3390/cells11233766
Via y Rada R, Daniel N, Archilla C, Frambourg A, Jouneau L, Jaszczyszyn Y, Charpigny G, Duranthon V, Calderari S. Identification of the Inner Cell Mass and the Trophectoderm Responses after an In Vitro Exposure to Glucose and Insulin during the Preimplantation Period in the Rabbit Embryo. Cells. 2022; 11(23):3766. https://doi.org/10.3390/cells11233766
Chicago/Turabian StyleVia y Rada, Romina, Nathalie Daniel, Catherine Archilla, Anne Frambourg, Luc Jouneau, Yan Jaszczyszyn, Gilles Charpigny, Véronique Duranthon, and Sophie Calderari. 2022. "Identification of the Inner Cell Mass and the Trophectoderm Responses after an In Vitro Exposure to Glucose and Insulin during the Preimplantation Period in the Rabbit Embryo" Cells 11, no. 23: 3766. https://doi.org/10.3390/cells11233766
APA StyleVia y Rada, R., Daniel, N., Archilla, C., Frambourg, A., Jouneau, L., Jaszczyszyn, Y., Charpigny, G., Duranthon, V., & Calderari, S. (2022). Identification of the Inner Cell Mass and the Trophectoderm Responses after an In Vitro Exposure to Glucose and Insulin during the Preimplantation Period in the Rabbit Embryo. Cells, 11(23), 3766. https://doi.org/10.3390/cells11233766