
Towards a Better Vision of Retinoic Acid Signaling during Eye Development


Abstract
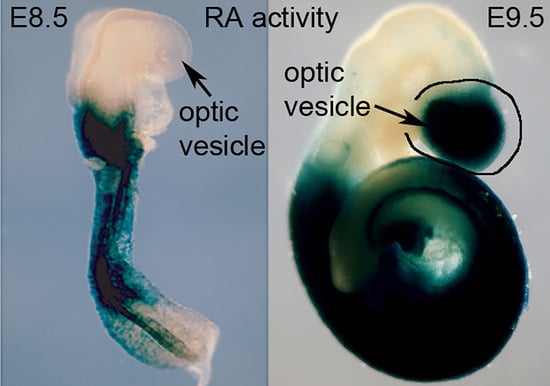
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Requirement of Retinoic Acid for Optic Cup Formation
3. Requirement of Retinoic Acid for Morphogenesis of Anterior Eye Structures
4. Identification of RA Direct Target Genes and Essential RAREs during Eye Formation
Funding
Acknowledgments
Conflicts of Interest
References
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.P.; Ma, J.X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes Dev. 2007, 21, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Molotkov, A.; Manabe, S.-I.; Donmoyer, C.M.; Deltour, L.; Foglio, M.H.; Cuenca, A.E.; Blaner, W.S.; Lipton, S.A.; Duester, G. Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol. Cell. Biol. 2003, 23, 4637–4648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupé, V.; Matt, N.; Garnier, J.-M.; Chambon, P.; Mark, M.; Ghyselinck, N.B. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. USA 2003, 100, 14036–14041. [Google Scholar] [CrossRef] [Green Version]
- Mic, F.A.; Molotkov, A.; Molotkova, N.; Duester, G. Raldh2 expression in optic vesicle generates a retinoic acid signal needed for invagination of retina during optic cup formation. Dev. Dyn. 2004, 231, 270–277. [Google Scholar] [CrossRef]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [Green Version]
- Germain, P.; Iyer, J.; Zechel, C.; Gronemeyer, H. Co-regulator recruitment and the mechanism of retinoic acid receptor synergy. Nature 2002, 415, 187–192. [Google Scholar] [CrossRef]
- Perissi, V.; Rosenfeld, M.G. Controlling nuclear receptors: The circular logic of cofactor cycles. Nat. Rev. Mol. Cell Biol. 2005, 6, 542–554. [Google Scholar] [CrossRef]
- Lonard, D.M.; O’Malley, B.W. Nuclear receptor coregulators: Modulators of pathology and therapeutic targets. Nat. Rev. Endocrinol. 2012, 8, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Berenguer, M.; Meyer, K.F.; Yin, J.; Duester, G. Discovery of genes required for body axis and limb formation by global identification of retinoic acid-regulated epigenetic marks. PLoS Biol. 2020, 18, e3000719. [Google Scholar] [CrossRef]
- Nedelec, B.; Rozet, J.M.; Fares Taie, L. Genetic architecture of retinoic-acid signaling-associated ocular developmental defects. Hum. Genet. 2019, 138, 937–955. [Google Scholar] [CrossRef] [PubMed]
- Slavotinek, A. Genetics of anophthalmia and microphthalmia. Part 2: Syndromes associated with anophthalmia-microphthalmia. Hum. Genet. 2019, 138, 831–846. [Google Scholar] [CrossRef]
- Mory, A.; Ruiz, F.X.; Dagan, E.; Yakovtseva, E.A.; Kurolap, A.; Pares, X.; Farres, J.; Gershoni-Baruch, R. A missense mutation in ALDH1A3 causes isolated microphthalmia/anophthalmia in nine individuals from an inbred Muslim kindred. Eur. J. Hum. Genet. 2014, 22, 419–422. [Google Scholar] [CrossRef] [Green Version]
- Plaisancie, J.; Bremond-Gignac, D.; Demeer, B.; Gaston, V.; Verloes, A.; Fares-Taie, L.; Gerber, S.; Rozet, J.M.; Calvas, P.; Chassaing, N. Incomplete penetrance of biallelic ALDH1A3 mutations. Eur. J. Med. Gen. 2016, 59, 215–218. [Google Scholar] [CrossRef]
- Williams, A.L.; Bohnsack, B.L. What’s retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis 2019, 57, e23308. [Google Scholar] [CrossRef] [PubMed]
- Weisschuh, N.; Dressler, P.; Schuettauf, F.; Wolf, C.; Wissinger, B.; Gramer, E. Novel mutations of FOXC1 and PITX2 in patients with Axenfeld-Rieger malformations. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3846–3852. [Google Scholar] [CrossRef] [Green Version]
- Molotkov, A.; Molotkova, N.; Duester, G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development 2006, 133, 1901–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matt, N.; Dupé, V.; Garnier, J.-M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.L.; Gage, P.J. Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum. Mol. Genet. 2005, 14, 3347–3359. [Google Scholar] [CrossRef]
- Kidson, S.H.; Kume, T.; Deng, K.; Winfrey, V.; Hogan, B.L. The forkhead/winged-helix gene, Mf1, is necessary for the normal development of the cornea and formation of the anterior chamber in the mouse eye. Dev. Biol. 1999, 211, 306–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warkany, J.; Schraffenberger, S. Congenital malformations induced in rats by maternal vitamin A deficiency. I. Defects of the eye. Arch. Ophthalmol. 1946, 35, 150–169. [Google Scholar] [CrossRef]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dollé, P.; Dierich, A.; Gorry, P.; Gansmuller, A.; Chambon, P. Function of the retinoic acid receptors (RARs) during development. (I) Craniofacial and skeletal abnormalities in RAR double mutants. Development 1994, 120, 2723–2748. [Google Scholar] [CrossRef]
- Niederreither, K.; Subbarayan, V.; Dollé, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef]
- Mic, F.A.; Haselbeck, R.J.; Cuenca, A.E.; Duester, G. Novel retinoic acid generating activities in the neural tube and heart identified by conditional rescue of Raldh2 null mutant mice. Development 2002, 129, 2271–2282. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Grondona, J.M.; Mark, M.; Gansmuller, A.; LeMeur, M.; Decimo, D.; Vonesch, J.-L.; Dollé, P.; Chambon, P. Genetic analysis of RXRa developmental function: Convergence of RXR and RAR signaling pathways in heart and eye morphogenesis. Cell 1994, 78, 987–1003. [Google Scholar] [CrossRef]
- Goto, S.; Onishi, A.; Misaki, K.; Yonemura, S.; Sugita, S.; Ito, H.; Ohigashi, Y.; Ema, M.; Sakaguchi, H.; Nishida, K.; et al. Neural retina-specific Aldh1a1 controls dorsal choroidal vascular development via Sox9 expression in retinal pigment epithelial cells. eLife 2018, 7, e32358. [Google Scholar] [CrossRef] [Green Version]
- Chawla, B.; Schley, E.; Williams, A.L.; Bohnsack, B.L. Retinoic Acid and Pitx2 Regulate Early Neural Crest Survival and Migration in Craniofacial and Ocular Development. Birth Defects Res. Part B 2016, 107, 126–135. [Google Scholar] [CrossRef]
- Gage, P.J.; Qian, M.; Wu, D.; Rosenberg, K.I. The canonical Wnt signaling antagonist DKK2 is an essential effector of PITX2 function during normal eye development. Dev. Biol. 2008, 317, 310–324. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Duester, G. Retinoic acid signaling in perioptic mesenchyme represses Wnt signaling via induction of Pitx2 and Dkk2. Dev. Biol. 2010, 340, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghyselinck, N.B.; Duester, G. Retinoic acid signaling pathways. Development 2019, 146, dev167502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutier, E.; Ye, T.; Choukrallah, M.A.; Urban, S.; Osz, J.; Chatagnon, A.; Delacroix, L.; Langer, D.; Rochel, N.; Moras, D.; et al. Retinoic Acid Receptors Recognize the Mouse Genome through Binding Elements with Diverse Spacing and Topology. J. Biol. Chem. 2012, 287, 26328–26341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalevee, S.; Anno, Y.N.; Chatagnon, A.; Samarut, E.; Poch, O.; Laudet, V.; Benoit, G.; Lecompte, O.; Rochette-Egly, C. Genome-wide in Silico Identification of New Conserved and Functional Retinoic Acid Receptor Response Elements (Direct Repeats Separated by 5 bp). J. Biol. Chem. 2011, 286, 33322–33334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschaki, M.; Schneider, C.; Rhinn, M.; Thibault-Carpentier, C.; Dembele, D.; Niederreither, K.; Dolle, P. Transcriptomic analysis of murine embryos lacking endogenous retinoic Acid signaling. PLoS ONE 2013, 8, e62274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Gudas, L.J. Gene expression profiling elucidates a specific role for RARgamma in the retinoic acid-induced differentiation of F9 teratocarcinoma stem cells. Biochem. Pharmacol. 2008, 75, 1129–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupé, V.; Davenne, M.; Brocard, J.; Dollé, P.; Mark, M.; Dierich, A.; Chambon, P.; Rijli, F.M. In vivo functional analysis of the Hoxa-1 3’ retinoic acid response element (3’RARE). Development 1997, 124, 399–410. [Google Scholar] [CrossRef]
- Houle, M.; Sylvestre, J.R.; Lohnes, D. Retinoic acid regulates a subset of Cdx1 function in vivo. Development 2003, 130, 6555–6567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Cunningham, T.J.; Duester, G. Nuclear receptor corepressors Ncor1 and Ncor2 (Smrt) are required for retinoic acid-dependent repression of Fgf8 during somitogenesis. Dev. Biol. 2016, 418, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, S.; Wilde, S.M.; Wood, S.; Logan, M.P. RA Acts in a Coherent Feed-Forward Mechanism with Tbx5 to Control Limb Bud Induction and Initiation. Cell Rep. 2015, 12, 879–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, T.J.; Lancman, J.J.; Berenguer, M.; Dong, P.D.S.; Duester, G. Genomic knockout of two presumed forelimb Tbx5 enhancers reveals they are nonessential for limb development. Cell Rep. 2018, 23, 3146–3151. [Google Scholar] [CrossRef]
- Adachi, N.; Robinson, M.; Goolsbee, A.; Shubin, N.H. Regulatory evolution of Tbx5 and the origin of paired appendages. Proc. Natl. Acad. Sci. USA 2016, 113, 10115–10120. [Google Scholar] [CrossRef] [Green Version]
- Duester, G. Knocking out enhancers to enhance epigenetic research. Trends Genet. 2019, 35, 89. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Laugesen, A.; Helin, K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell 2014, 14, 735–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolovos, P.; Knoch, T.A.; Grosveld, F.G.; Cook, P.R.; Papantonis, A. Enhancers and silencers: An integrated and simple model for their function. Epigenetics Chromatin 2012, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibn-Salem, J.; Kohler, S.; Love, M.I.; Chung, H.R.; Huang, N.; Hurles, M.E.; Haendel, M.; Washington, N.L.; Smedley, D.; Mungall, C.J.; et al. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 2014, 15, 423. [Google Scholar] [CrossRef] [Green Version]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duester, G. Towards a Better Vision of Retinoic Acid Signaling during Eye Development. Cells 2022, 11, 322. https://doi.org/10.3390/cells11030322
Duester G. Towards a Better Vision of Retinoic Acid Signaling during Eye Development. Cells. 2022; 11(3):322. https://doi.org/10.3390/cells11030322
Chicago/Turabian StyleDuester, Gregg. 2022. "Towards a Better Vision of Retinoic Acid Signaling during Eye Development" Cells 11, no. 3: 322. https://doi.org/10.3390/cells11030322
APA StyleDuester, G. (2022). Towards a Better Vision of Retinoic Acid Signaling during Eye Development. Cells, 11(3), 322. https://doi.org/10.3390/cells11030322