
Retinoid Agonists in the Targeting of Heterotopic Ossification


{kind=link}
{kind=link}
Abstract
:1. Introduction
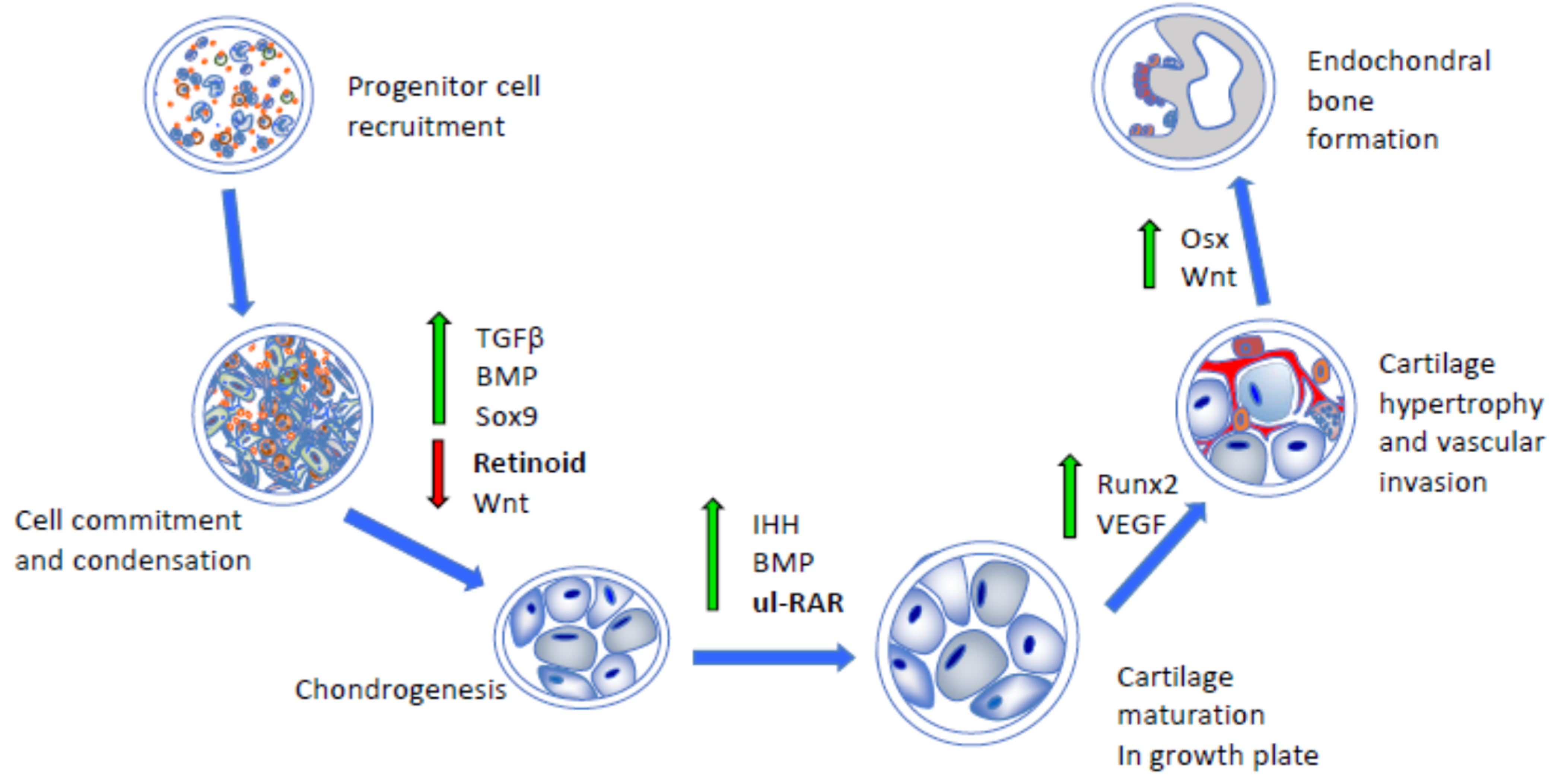
2. RARs, Retinoids, and Skeletal Development and Growth
3. Antagonistic Action of Retinoid Signaling on Chondrogenesis and Bone Morphogenetic Protein (BMP) Signaling
4. Heterotopic Ossification and Pre-Clinical Studies on RARγ Agonists
5. Target and Off-Target Effects
6. Clinical Trials on the RARγ Agonist Palovarotene
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glyselinck, N.B.; Duester, G. Retinoic acid signaling pathways. Development 2019, 146, dev167502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudas, L.J. Emerging roles for retinoids in regeneration and differentiation in normal and disease states. Biochim. Biophys. Acta 2012, 1821, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.A.; McCaffery, P.; Dräger, U.C.; De Luca, L.M. Retinoids in Embryonal Development. Physiol. Rev. 2000, 80, 1021–1054. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, E.M.; Dadon, S.B.-E.; Reifen, R. The vicious cycle of vitamin a deficiency: A review. Crit. Rev. Food Sci. Nutr. 2017, 57, 3703–3714. [Google Scholar] [CrossRef]
- Wolbach, S.B. Vitamin-A deficiency and excess in relation to skeletal growth. J. Bone Jt. Surg.-Am. Vol. 1947, 29, 171–192. [Google Scholar]
- Wolbach, S.B.; Hegsted, D.M. Hypervitaminosis A in young ducks; the epiphyseal cartilages. AMA. Arch. Pathol. 1953, 55, 47–54. [Google Scholar] [PubMed]
- Conaway, H.H.; Henning, P.; Lerner, U.H. Vitamin A Metabolism, Action, and Role in Skeletal Homeostasis. Endocr. Rev. 2013, 34, 766–797. [Google Scholar] [CrossRef] [Green Version]
- Blaner, W.S.; Olson, J.A. Retinol and retinoic acid metabolism. In The Retinoids: Biology, Chemistry, and Medicine, 2nd ed.; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Raven Press: New York, NY, USA, 1994; pp. 229–255. [Google Scholar]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science 1995, 268, 1039–1041. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A Membrane Receptor for Retinol Binding Protein Mediates Cellular Uptake of Vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef]
- Kane, M.A. Analysis, occurrence, and function of 9-cis-retinoic acid. Biochim. Biophys. Acta. 2012, 1821, 10–20. [Google Scholar] [CrossRef]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [CrossRef]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sucov, H.M.; Evans, R.M. Retinoic acid and retinoic acid receptors in development. Mol. Neurobiol. 1995, 10, 169–184. [Google Scholar] [CrossRef]
- Perimann, T.; Rangarajian, P.N.; Umesono, K.; Evans, R.M. Determinants of selective RAR and TR recognition of direct re-peat HREs. Genes Dev. 1993, 7, 1411–1422. [Google Scholar] [CrossRef] [Green Version]
- Umesono, K.; Murakami, K.K.; Thompson, C.C.; Evans, R. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell 1991, 65, 1255–1266. [Google Scholar] [CrossRef]
- Kedishvili, N.Y. Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res. 2013, 54, 1744–1760. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.A.; Kane, M.; Okabe, T.; Enomoto-Iwamoto, M.; Napoli, J.L.; Pacifici, M.; Iwamoto, M. Endogenous Retinoids in Mammalian Growth Plate Cartilage: Analysis and roles in matrix homeostasis and turnover. J. Biol. Chem. 2010, 285, 36674–36681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campo-Paysaa, F.; Marlétaz, F.; Laudet, V.; Schubert, M. Retinoic acid signaling in development: Tissue-specific functions and evolutionary origins. Genes 2008, 46, 640–656. [Google Scholar] [CrossRef]
- Cunningham, T.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederreither, K.; Dollé, P. Retinoic acid in development: Towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Subbarayan, V.; Dollé, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef]
- White, R.J.; Nie, Q.; Lander, A.D.; Schilling, T.F. Complex Regulation of cyp26a1 Creates a Robust Retinoic Acid Gradient in the Zebrafish Embryo. PLoS Biol. 2007, 5, e304. [Google Scholar] [CrossRef] [PubMed]
- Duester, G. Retinoic Acid Synthesis and Signaling during Early Organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.D.; Evans, R. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nat. Cell Biol. 1995, 377, 454–457. [Google Scholar] [CrossRef]
- Everett, L.; Hansen, M.; Hannenhalli, S. Regulating the Regulators: Modulators of Transcription Factor Activity. Adv. Struct. Saf. Stud. 2010, 674, 297–312. [Google Scholar] [CrossRef]
- Ferreira, R.; Napoli, J.; Enver, T.; Bernardino, L.; Ferreira, L. Advances and challenges in retinoid delivery systems in regen-erative and therapeutic medicine. Nat. Comm. 2020, 11, 4265. [Google Scholar] [CrossRef]
- Pacifici, M. Retinoid roles and action in skeletal development and growth provide the rationale for an ongoing heterotopic ossification prevention trial. Bone 2018, 109, 267–275. [Google Scholar] [CrossRef]
- Gutierrez-Mazeriegos, J.; Schubert, M.; Laudet, V. Evolution of retinoic acid receptors and retinoic acid signaling. In The Bio-Chemistry of Retinoic Acid Receptors I: Structure, Activation, and Function at the Molecular Level; Asson-Batres, M.A., Rochette-Egly, C., Eds.; Springer: New York, NY, USA, 2014; pp. 55–76. [Google Scholar]
- Dollé, P.; Fraulob, V.; Kastner, P.; Chambon, P. Developmental expression of murine retinoid X receptor (RXR) genes. Mech. Dev. 1994, 45, 91–104. [Google Scholar] [CrossRef]
- Dolle’, P.; Ruberte, E.; Kastner, P.; Petkovich, M.; Stoner, C.M.; Gudas, L.J.; Chambon, P. Differential expression of genes en-coding α, β and γ retinoic acid receptors and CRABP in the developing limbs of the mouse. Nature 1989, 342, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, e002. [Google Scholar] [CrossRef] [Green Version]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dolle, P.; Dierich, A.; Gorry, P.; Gansmuller, A.; Chambon, P. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 1994, 120, 2723–2748. [Google Scholar] [CrossRef] [PubMed]
- Wellik, D.M.; Capecchi, M.R. Hox10 and Hox11 Genes Are Required to Globally Pattern the Mammalian Skeleton. Science 2003, 301, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Balmer, J.E.; Blomhoff, R. Gene expression regulation by retinoic acid. J. Lipid Res. 2002, 43, 1773–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, E.; Golden, E.B.; Kirsch, T.; Adams, S.L.; Chandraratna, R.A.S.; Michaille, J.-J.; Pacifici, M. Retinoid signaling is re-quired for chondrocyte maturation and endochondral bone formation during limb skeletogenesis. Dev. Biol. 1999, 208, 375–391. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.A.; Kondo, N.; Okabe, T.; Takeshita, N.; Pilchak, D.M.; Koyama, E.; Ochiai, T.; Jensen, D.; Chu, M.-L.; Kane, M.A.; et al. Retinoic acid receptors are required for skeletal growth, matrix homeostasis and growth plate function in postnatal mouse. Dev. Biol. 2009, 328, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Kozhemyakina, E.; Lassar, A.B.; Zelzer, E. A pathway to bone: Signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 2015, 142, 817–831. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, V.; Bhattaram, P. Vertebrate Skeletogenesis. Curr. Top. Dev. Biol. 2010, 90, 291–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovchinnikov, D.A.; Deng, J.M.; Ogunrinu, G.; Behringer, R.R. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 2000, 26, 145–146. [Google Scholar] [CrossRef]
- Kane, M.A.; Chen, N.; Sparks, S.; Napoli, J.L. Quantification of endogenous retinoic acid in limited biological samples by LC/MS/MS. Biochem. J. 2005, 388, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Rossant, J.; Zirngibi, R.; Cado, D.; Shago, M.; Giguere, V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991, 5, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Minegishi, Y.; Sakai, Y.; Yahara, Y.; Akiyama, H.; Yoshikawa, H.; Hosokawa, K.; Tsumaki, N. Cyp26b1 within the growth plate regulates bone growth in juvenile mice. Biochem. Biophys. Res. Commun. 2014, 454, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Pignolo, R.J.; Foley, K.L. Nonhereditary Heterotopic Ossification: Implications for Injury, Arthropathy, and Aging. Clin. Rev. Bone Miner. Metab. 2005, 3, 261–266. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia Ossificans Progressiva: Clinical and Genetic Aspects. Orphanet J. Rare Dis. 2011, 6, 80. [Google Scholar] [CrossRef] [Green Version]
- Shimono, K.; Morrison, T.N.; Tung, W.-E.; Chandraratna, R.A.S.; Williams, J.A.; Iwamoto, M.; Pacifici, M. Inhibition of ectopic bone formation by a selective retinoic acid receptor α-agonist: A new therapy for heterotopic ossification? J. Orthop. Res. 2010, 28, 271–277. [Google Scholar] [CrossRef]
- Hinchliffe, J.R.; Johnson, D.R. The Development of the Vertebrate Limb; Oxford University Press: New York, NY, USA, 1980; pp. 72–83. [Google Scholar]
- Salazar, V.S.; Gamer, L.W.; Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 2016, 12, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Chaboissier, M.-C.; Martin, J.F.; Schedl, A.; De Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002, 16, 2813–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Lyons, J.P.; Mori-Akiyama, Y.; Yand, X.; Zhang, R.; Zhang, Z.; Deng, J.M.; Takato, M.M.; Nakamura, T.; Beh-ringer, R.R.; et al. Interaction between Sox9 and β-catenin control chondrocyte differentiation. Genes Dev. 2004, 18, 1072–1087. [Google Scholar] [CrossRef] [Green Version]
- Day, T.; Guo, X.; Garrett-Beal, L.; Yang, Y. Wnt/β-Catenin Signaling in Mesenchymal Progenitors Controls Osteoblast and Chondrocyte Differentiation during Vertebrate Skeletogenesis. Dev. Cell 2005, 8, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Weston, A.D.; Hoffman, L.M.; Underhill, T.M. Revisiting the role of retinoid signaling in skeletal development. Birth Defects Res. Part C Embryo Today Rev. 2003, 69, 156–173. [Google Scholar] [CrossRef]
- Eichele, G.; Thaller, C. Characterization of concentration gradients of a morphologically active retinoid in the chick limb bud. J. Cell Biol. 1987, 105, 1917–1923. [Google Scholar] [CrossRef] [Green Version]
- Pacifici, M.; Cossu, G.; Molinaro, M.; Tato, F. Vitamin A inhibits chondrogenesis but not myogenesis. Exp. Cell Res. 1980, 129, 469–474. [Google Scholar] [CrossRef]
- Bénazet, J.-D.; Zeller, R. Vertebrate Limb Development: Moving from Classical Morphogen Gradients to an Integrated 4-Dimensional Patterning System. Cold Spring Harb. Perspect. Biol. 2009, 1, a001339. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Weston, A.D.; Underhill, T.M. Molecular mechanisms regulating chondroblast differentiation. J. Bone Jt. Surg. Am. 2003, 85, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Weston, A.D.; Chandraratna, R.A.; Torchia, J.; Underhill, T.M. Requirement for RAR-mediated gene repression in skeletal progenitor differentiation. J. Cell Biol. 2002, 158, 39–51. [Google Scholar] [CrossRef]
- Weston, A.D.; Rosen, V.; Chandraratna, R.A.; Underhill, T.M. Regulation of Skeletal Progenitor Differentiation by the Bmp and Retinoid Signaling Pathways. J. Cell Biol. 2000, 148, 679–690. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Garcha, K.; Karamboulas, K.; Cowan, M.F.; Drysdale, L.M.; Horton, W.A.; Underhill, T.M. BMP action in skeletogenesis involves attenuation of retinoid signaling. J. Cell Biol. 2006, 174, 101–113. [Google Scholar] [CrossRef]
- Meyers, C.; Lisiecki, J.; Miller, S.; Levin, A.; Fayad, L.; Ding, C.; Sono, T.; McCarthy, E.; Levi, B.; James, A.W. Heterotopic Ossification: A Comprehensive Review. JBMR Plus 2019, 3, e10172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, F.S.; A Zasloff, M.; A Kitterman, J.; Shore, E.M.; Hong, C.C.; Rocke, D.M. Early Mortality and Cardiorespiratory Failure in Patients with Fibrodysplasia Ossificans Progressiva. J. Bone Jt. Surg. Am. 2010, 92, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shore, E.M.; Xu, M.; Feldman, G.J.; A Fenstermacher, D.; Cho, T.-J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Gregson, C.L.; Hollingworth, P.; Williams, M.; Petrie, K.A.; Bullock, A.N.; Brown, M.A.; Tobias, J.H.; Triffitt, J. A novel ACVR1 mutation in the glycine/serine-rich domain foud in the most begign case of a fibrodysplasia ossificans progressiva variant reported to date. Bone 2011, 48, 654–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacifici, M.; Shore, E.M. Common mutations in ALK2/ACVR1, a multi-facet receptor, have roles in distinct musculoskeletal and neural orphan disorders. Cytokine Growth Factor Rev. 2016, 27, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Al Mukaddam, M.; Pignolo, R.J. Genetics of Fibrodysplasia Ossificans Progressiva. eLS 2021, 2, 1–8. Available online: https://onlinelibrary.wiley.com/doi/10.1002/9780470015902.a0029154 (accessed on 18 November 2021). [CrossRef]
- Bocciardi, R.; Bordo, D.; Di Duca, M.; Di Rocco, M.; Ravazzolo, R. Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: Confirmations and advancements. Eur. J. Hum. Genet. 2008, 17, 311–318. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht-Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Pignolo, R.J.; Bedford-Gay, C.; Liljesthröm, M.; Durbin-Johnson, B.P.; Shore, E.M.; Rocke, D.M.; Kaplan, F.S. The Natural History of Flare-Ups in Fibrodysplasia Ossificans Progressiva (FOP): A Comprehensive Global Assessment. J. Bone Miner. Res. 2016, 31, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Kan, L.; Lounev, V.Y.; Pignolo, R.J.; Duan, L.; Liu, Y.; Stock, S.R.; McGuire, T.L.; Lu, B.; Gerard, N.P.; Shore, E.M.; et al. Sub-stance P signaling mediates BMP-dependent heterotopic ossification. J. Cell. Biochem. 2011, 112, 2757–2772. [Google Scholar] [CrossRef] [Green Version]
- Scarlett, R.F.; Rocke, D.; Kantanie, S.; Patel, J.B.; Shore, E.M.; Kaplan, F.S. Influenza-like Viral Illnesses and Flare-ups of Fibrodysplasia Ossificans Progressiva. Clin. Orthop. Relat. Res. 2004, 423, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Shore, E.M.; Kaplan, F.S. Inherited human diseases of heterotopic bone formation. Nat. Rev. Rheumatol. 2010, 6, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Gannon, F.H.; Valentine, B.A.; Shore, E.M.; Zasloff, M.A.; Kaplan, F.S. Acute Lymphocytic Infiltration in an Extremely Early Lesion of Fibrodysplasia Ossificans Progressiva. Clin. Orthop. Relat. Res. 1998, 346, 19–25. [Google Scholar] [CrossRef]
- Kaplan, F.S.; A Tabas, J.; Gannon, F.H.; Finkel, G.; Hahn, G.V.; A Zasloff, M. The histopathology of fibrodysplasia ossificans progressiva. An endochondral process. J. Bone Jt. Surg. Am. 1993, 75, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.; Sawyer, J.; Connors, S.; Keough, K.; Shore, E.; Gannon, F.; Glaser, D.; Rocke, D.M.; Zasloff, M.; Folkman, J. Urinary basic fibroblast growth factor. A biochemical marker for preosseous fibroproliferative lesions in patients with fibrodys-plasia ossificans progressiva. Clin. Orthop. Relat. Res. 1998, 346, 59–65. [Google Scholar]
- Kaplan, F.S.; Strear, C.M.; Zasloff, M.A. Radiographic and scintigraphic features of modeling and remodeling in the hetero-topic skeleton of patients who have fibrodysplasia ossificans progressiva. Clin. Orthop. Relat. Res. 1994, 304, 238–247. [Google Scholar]
- Lutwak, L. Myositis ossificans progressiva: Mineral, metabolic and radioactive calcium studies of the effects of hormones. Am. J. Med. 1964, 37, 269–293. [Google Scholar] [CrossRef]
- Chiu, Y.-Y.; Roth, M.D.; Kolis, S.; Rogovitz, D.; Davies, B. Pharamacokinetics of a novle agent, R667, in patients with emphy-sema. Br. J. Clin. Pharmacol. 2007, 63, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Thacher, S.M.; Vasudevan, J.; Chandraratna, R.A.S. Therapeutic applications for ligands of retinoic receptors. Curr. Pharm. Des. 2000, 6, 25–58. [Google Scholar] [CrossRef] [PubMed]
- Underhill, T.M.; Weston, A.D. Retinoids and their receptors in skeletal development. Microsc. Res. Tech. 1998, 43, 137–155. [Google Scholar] [CrossRef]
- Shimono, K.; Tung, W.-E.; Macolino, C.; Chi, A.H.-T.; Didizian, J.H.; Mundy, C.; A Chandraratna, R.; Mishina, Y.; Enomoto-Iwamoto, M.; Pacifici, M.; et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef] [Green Version]
- Weston, A.D.; Blumberg, B.; Underhill, T.M. Active repression by unligated retinoid receptors in development: Less is some-times more. J. Cell Biol. 2003, 161, 223–228. [Google Scholar] [CrossRef]
- Chakkalakal, S.A.; Uchibe, K.; Convente, M.R.; Zhang, D.; Economides, A.N.; Kaplan, F.S.; Pacifici, M.; Iwamoto, M.; Shore, E.M. Palovarotene inhibits heterotopic ossification and maintains limb mobility and growth in mice with the human ACVR1R206H Fibrodysplasia Ossificans Progressiva (FOP) mutation. J. Bone Miner Res. 2016, 31, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Shalita, A.R. Mucosutaneous and systemic toxicity of retinoids: Monitoring and management. Dermatologica 1987, 175, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Pignolo, R.J.; Al Mukaddam, M.; Baujat, G.; Berglund, S.K.; Cheung, A.M.; De Cunto, C. Palovarotene (PVO) for Fibrodysplasia Ossificans Progressiva (FOP): Data from the phase III MOVE trial. In Proceedings of the 2020 Meeting of the American Society for Bone and Mineral Research, Virtual, 11–15 September 2020. [Google Scholar]
- Pignolo, R.J.; Al Mukaddam, M.; Baujat, G.; Brown, M.A.; Cheung, A.M.; De Cunto, C.L. Palovarotene for the Treatment of Fibrodysplasia Ossificans Progressiva in Females Aged > 8 Years Amd Males > 1- Years: Data from the Phase III MOVE Trial. In Proceedings of the IOF-ESCO World Conference on Osteoporosis, Osteoarthritis and Musculoskeletal Diseases, Virtual Congress, 26–28 August 2021; pp. 66–67. Available online: https://www.wco-iof-esceo.org/sites/wco_22/pdf/WCO21-AbstractBook.pdf (accessed on 18 November 2021).
- Armstrong, R.B.; Ashenfelter, K.O. General and reproductive toxicology of retinoids. In The Retinoids: Biology, Chemistry, and Medicine, 2nd ed.; Sporn, M.B., Roberts, A.B., Goodman, D.S., Eds.; Wiley: New York, NY, USA, 1994; pp. 545–572. [Google Scholar]
- A Natural History Study of Fibrodysplasia Ossificans Progressiva (FOP). Available online: https://clinicaltrials.gov/ct2/show/NCT02322255 (accessed on 18 November 2021).
- An Efficacy and Safety Study of Palovarotene to Treat Preosseous Flare-Ups in FOP Subjects. Available online: https://clinicaltrials.gov/ct2/show/NCT02190747 (accessed on 18 November 2021).
- An Open-Label Extension Study of Palovarotene Treatment in Fibrodysplasia Ossificans Progressiva (FOP). Available online: https://clinicaltrials.gov/ct2/show/NCT02279095 (accessed on 18 November 2021).
- An Efficacy and Safety Study of Palovarotene for the Treatment of Fibrodysplasia Ossificans Progressiva. (MOVE). Available online: https://clinicaltrials.gov/ct2/show/NCT03312634 (accessed on 18 November 2021).
- Pignolo, R.J.; Kaplan, F.S. Druggable targets, clinical trial design and proposed pharmacological management in fibrodyspla-sia ossificans progressiva. Expert Opin. Orphan Drugs 2020, 8, 101–109. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pignolo, R.J.; Pacifici, M. Retinoid Agonists in the Targeting of Heterotopic Ossification. Cells 2021, 10, 3245. https://doi.org/10.3390/cells10113245
Pignolo RJ, Pacifici M. Retinoid Agonists in the Targeting of Heterotopic Ossification. Cells. 2021; 10(11):3245. https://doi.org/10.3390/cells10113245
Chicago/Turabian StylePignolo, Robert J., and Maurizio Pacifici. 2021. "Retinoid Agonists in the Targeting of Heterotopic Ossification" Cells 10, no. 11: 3245. https://doi.org/10.3390/cells10113245
APA StylePignolo, R. J., & Pacifici, M. (2021). Retinoid Agonists in the Targeting of Heterotopic Ossification. Cells, 10(11), 3245. https://doi.org/10.3390/cells10113245