
A Distinct Hibiscus sabdariffa Extract Prevents Iron Neurotoxicity, a Driver of Multiple Sclerosis Pathology

 , ,
, ,
Abstract
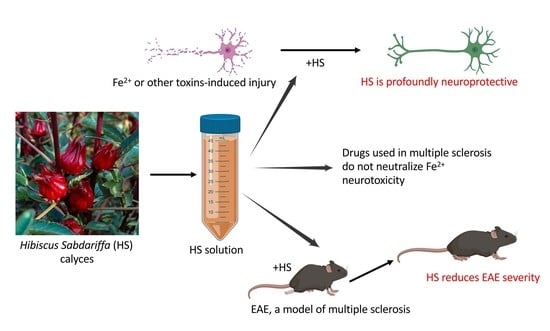
:
1. Introduction
2. Materials and Methods
2.1. Preparation of HS
2.2. Culture of Human and Mouse Neurons, and Evaluation of Neurotoxicity
2.3. Life Imaging of Neurons
2.4. Iron Chelation Analysis
2.5. HORAC and ORAC Assays
2.6. Assessment of HS against Other Mediators of Neuronal Death
2.7. Proliferation of Splenocytes
2.8. EAE and Spinal Cord Histology
2.9. Statistical Analysis
3. Results
3.1. The Killing of Human Neurons in Culture by Iron Is Attenuated by HS, and This Is Not Due to Direct Iron Chelation
3.2. Mouse Neurons Are Also Protected by HS against Iron Neurotoxicity in Culture
3.3. Life Imaging of Neurons
3.4. HS Is Superior to MS Medications for Neuroprotection against Iron
3.5. Neuroprotection against Iron Is Not Related to Anthocyanin Content
3.6. Neuroprotection against Iron Is Not Proportional to Anti-Oxidant Activity
3.7. HS Also Protects Neurons against Other Stressors
3.8. HS Does Not Affect the Proliferation of T Cells in Culture
3.9. HS Reduces the Severity of Murine EAE and Confers Neuroprotection In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Klineova, S.; Lublin, F.D. Clinical Course of Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- George, I.C.; El Mendili, M.M.; Inglese, M.; Azevedo, C.J.; Kantarci, O.; Lebrun, C.; Siva, A.; Okuda, D.T.; Pelletier, D. Cerebellar volume loss in radiologically isolated syndrome. Mult. Scler. 2019, 1352458519887346. [Google Scholar] [CrossRef]
- Lebrun, C.; Kantarci, O.H.; Siva, A.; Pelletier, D.; Okuda, D.T.; Risconsortium. Anomalies Characteristic of Central Nervous System Demyelination: Radiologically Isolated Syndrome. Neurol. Clin. 2018, 36, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Li, R. Cellular immunology of relapsing multiple sclerosis: Interactions, checks, and balances. Lancet Neurol. 2021, 20, 470–483. [Google Scholar] [CrossRef]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef]
- Stephenson, E.; Nathoo, N.; Mahjoub, Y.; Dunn, J.F.; Yong, V.W. Iron in multiple sclerosis: Roles in neurodegeneration and repair. Nat. Rev. Neurol. 2014, 10, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Yong, H.Y.F.; Yong, V.W. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat. Rev. Neurol. 2022, 18, 40–55. [Google Scholar] [CrossRef]
- Cronin, S.J.F.; Woolf, C.J.; Weiss, G.; Penninger, J.M. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front. Mol. Biosci. 2019, 6, 116. [Google Scholar] [CrossRef] [Green Version]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; Gonzalez-Billault, C.; Nunez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef]
- Stankiewicz, J.M.; Brass, S.D. Role of iron in neurotoxicity: A cause for concern in the elderly? Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.J.; Borquez, D.A.; Nunez, M.T. Inflaming the Brain with Iron. Antioxidants 2021, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Haider, L. Inflammation, Iron, Energy Failure, and Oxidative Stress in the Pathogenesis of Multiple Sclerosis. Oxid. Med. Cell Longev. 2015, 2015, 725370. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Huang, X.; Kropat, J.; Henras, A.; Merchant, S.S.; Dickson, R.C.; Chanfreau, G.F. Sphingolipid signaling mediates iron toxicity. Cell Metab. 2012, 16, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Joppe, K.; Roser, A.E.; Maass, F.; Lingor, P. The Contribution of Iron to Protein Aggregation Disorders in the Central Nervous System. Front. Neurosci. 2019, 13, 15. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, H.; Choong, C.J.; Baba, K. Parkinson’s disease and iron. J. Neural Transm. 2020, 127, 181–187. [Google Scholar] [CrossRef]
- Uranga, R.M.; Salvador, G.A. Unraveling the Burden of Iron in Neurodegeneration: Intersections with Amyloid Beta Peptide Pathology. Oxid. Med. Cell Longev. 2018, 2018, 2850341. [Google Scholar] [CrossRef]
- Spotorno, N.; Acosta-Cabronero, J.; Stomrud, E.; Lampinen, B.; Strandberg, O.T.; van Westen, D.; Hansson, O. Relationship between cortical iron and tau aggregation in Alzheimer’s disease. Brain 2020, 143, 1341–1349. [Google Scholar] [CrossRef]
- Faissner, S.; Mishra, M.; Kaushik, D.K.; Wang, J.; Fan, Y.; Silva, C.; Rauw, G.; Metz, L.; Koch, M.; Yong, V.W. Systematic screening of generic drugs for progressive multiple sclerosis identifies clomipramine as a promising therapeutic. Nat. Commun. 2017, 8, 1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippi, M.; Bruck, W.; Chard, D.; Fazekas, F.; Geurts, J.J.G.; Enzinger, C.; Hametner, S.; Kuhlmann, T.; Preziosa, P.; Rovira, A.; et al. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol. 2019, 18, 198–210. [Google Scholar] [CrossRef]
- Hagemeier, J.; Ramanathan, M.; Schweser, F.; Dwyer, M.G.; Lin, F.; Bergsland, N.; Weinstock-Guttman, B.; Zivadinov, R. Iron-related gene variants and brain iron in multiple sclerosis and healthy individuals. Neuroimage Clin. 2018, 17, 530–540. [Google Scholar] [CrossRef] [PubMed]
- LeVine, S.M.; Bilgen, M.; Lynch, S.G. Iron accumulation in multiple sclerosis: An early pathogenic event. Expert Rev. Neurother. 2013, 13, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Elkady, A.M.; Cobzas, D.; Sun, H.; Blevins, G.; Wilman, A.H. Progressive iron accumulation across multiple sclerosis phenotypes revealed by sparse classification of deep gray matter. J. Magn. Reson. Imaging 2017, 46, 1464–1473. [Google Scholar] [CrossRef] [PubMed]
- Zivadinov, R.; Tavazzi, E.; Bergsland, N.; Hagemeier, J.; Lin, F.; Dwyer, M.G.; Carl, E.; Kolb, C.; Hojnacki, D.; Ramasamy, D.; et al. Brain Iron at Quantitative MRI Is Associated with Disability in Multiple Sclerosis. Radiology 2018, 289, 487–496. [Google Scholar] [CrossRef]
- Haider, L.; Simeonidou, C.; Steinberger, G.; Hametner, S.; Grigoriadis, N.; Deretzi, G.; Kovacs, G.G.; Kutzelnigg, A.; Lassmann, H.; Frischer, J.M. Multiple sclerosis deep grey matter: The relation between demyelination, neurodegeneration, inflammation and iron. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- Hametner, S.; Wimmer, I.; Haider, L.; Pfeifenbring, S.; Bruck, W.; Lassmann, H. Iron and neurodegeneration in the multiple sclerosis brain. Ann. Neurol. 2013, 74, 848–861. [Google Scholar] [CrossRef]
- Haider, L.; Zrzavy, T.; Hametner, S.; Hoftberger, R.; Bagnato, F.; Grabner, G.; Trattnig, S.; Pfeifenbring, S.; Bruck, W.; Lassmann, H. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 2016, 139, 807–815. [Google Scholar] [CrossRef] [Green Version]
- Packer, L.; Witt, E.H.; Tritschler, H.J. alpha-Lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef]
- Whiteman, M.; Tritschler, H.; Halliwell, B. Protection against peroxynitrite-dependent tyrosine nitration and alpha 1-antiproteinase inactivation by oxidized and reduced lipoic acid. FEBS Lett. 1996, 379, 74–76. [Google Scholar] [CrossRef] [Green Version]
- Spain, R.; Powers, K.; Murchison, C.; Heriza, E.; Winges, K.; Yadav, V.; Cameron, M.; Kim, E.; Horak, F.; Simon, J.; et al. Lipoic acid in secondary progressive MS: A randomized controlled pilot trial. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serban, C.; Sahebkar, A.; Ursoniu, S.; Andrica, F.; Banach, M. Effect of sour tea (Hibiscus sabdariffa L.) on arterial hypertension: A systematic review and meta-analysis of randomized controlled trials. J. Hypertens. 2015, 33, 1119–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafpour Boushehri, S.; Karimbeiki, R.; Ghasempour, S.; Ghalishourani, S.S.; Pourmasoumi, M.; Hadi, A.; Mbabazi, M.; Pour, Z.K.; Assarroudi, M.; Mahmoodi, M.; et al. The efficacy of sour tea (Hibiscus sabdariffa L.) on selected cardiovascular disease risk factors: A systematic review and meta-analysis of randomized clinical trials. Phytother. Res. 2020, 34, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Grajeda-Iglesias, C.; Figueroa-Espinoza, M.C.; Barouh, N.; Barea, B.; Fernandes, A.; de Freitas, V.; Salas, E. Isolation and Characterization of Anthocyanins from Hibiscus sabdariffa Flowers. J. Nat. Prod. 2016, 79, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Maciel, L.G.; do Carmo, M.A.V.; Azevedo, L.; Daguer, H.; Molognoni, L.; de Almeida, M.M.; Granato, D.; Rosso, N.D. Hibiscus sabdariffa anthocyanins-rich extract: Chemical stability, in vitro antioxidant and antiproliferative activities. Food Chem. Toxicol. 2018, 113, 187–197. [Google Scholar] [CrossRef]
- Zhang, R.R.; Hu, R.D.; Lu, X.Y.; Ding, X.Y.; Huang, G.Y.; Duan, L.X.; Zhang, S.J. Polyphenols from the flower of Hibiscus syriacus Linn ameliorate neuroinflammation in LPS-treated SH-SY5Y cell. Biomed. Pharmacother. 2020, 130, 110517. [Google Scholar] [CrossRef]
- Sogo, T.; Terahara, N.; Hisanaga, A.; Kumamoto, T.; Yamashiro, T.; Wu, S.; Sakao, K.; Hou, D.X. Anti-inflammatory activity and molecular mechanism of delphinidin 3-sambubioside, a Hibiscus anthocyanin. Biofactors 2015, 41, 58–65. [Google Scholar] [CrossRef]
- Bakhtiari, E.; Hosseini, A.; Mousavi, S.H. Protective effect of Hibiscus sabdariffa against serum/glucose deprivation-induced PC12 cells injury. Avicenna J. Phytomed. 2015, 5, 231–237. [Google Scholar]
- Chiaino, E.; Micucci, M.; Cosconati, S.; Novellino, E.; Budriesi, R.; Chiarini, A.; Frosini, M. Olive Leaves and Hibiscus Flowers Extracts-Based Preparation Protect Brain from Oxidative Stress-Induced Injury. Antioxidants 2020, 9, 806. [Google Scholar] [CrossRef]
- Hritcu, L.; Foyet, H.S.; Stefan, M.; Mihasan, M.; Asongalem, A.E.; Kamtchouing, P. Neuroprotective effect of the methanolic extract of Hibiscus asper leaves in 6-hydroxydopamine-lesioned rat model of Parkinson’s disease. J. Ethnopharmacol. 2011, 137, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Foyet, H.S.; Hritcu, L.; Ciobica, A.; Stefan, M.; Kamtchouing, P.; Cojocaru, D. Methanolic extract of Hibiscus asper leaves improves spatial memory deficits in the 6-hydroxydopamine-lesion rodent model of Parkinson’s disease. J. Ethnopharmacol. 2011, 133, 773–779. [Google Scholar] [CrossRef] [PubMed]
- El-Shiekh, R.A.; Ashour, R.M.; Abd El-Haleim, E.A.; Ahmed, K.A.; Abdel-Sattar, E. Hibiscus sabdariffa L.: A potent natural neuroprotective agent for the prevention of streptozotocin-induced Alzheimer’s disease in mice. Biomed. Pharmacother. 2020, 128, 110303. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.X.; Tong, X.; Terahara, N.; Luo, D.; Fujii, M. Delphinidin 3-sambubioside, a Hibiscus anthocyanin, induces apoptosis in human leukemia cells through reactive oxygen species-mediated mitochondrial pathway. Arch. Biochem. Biophys. 2005, 440, 101–109. [Google Scholar] [CrossRef]
- Lee, J.; Durst, R.W.; Wrolstad, R.E. Determination of total monomeric anthocyanin pigment content of fruit juices, beverages, natural colorants, and wines by the pH differential method: Collaborative study. J. AOAC Int. 2005, 88, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecil, G.G.; Larsen, P.H.; Corley, S.M.; Herx, L.M.; Besson, A.; Goodyer, C.G.; Yong, V.W. Interleukin-1 is a key regulator of matrix metalloproteinase-9 expression in human neurons in culture and following mouse brain trauma in vivo. J. Neurosci. Res. 2000, 61, 212–224. [Google Scholar] [CrossRef]
- Faissner, S.; Mahjoub, Y.; Mishra, M.; Haupeltshofer, S.; Hahn, J.N.; Gold, R.; Koch, M.; Metz, L.M.; Ben-Hur, T.; Yong, V.W. Unexpected additive effects of minocycline and hydroxychloroquine in models of multiple sclerosis: Prospective combination treatment for progressive disease? Mult. Scler. 2018, 24, 1543–1556. [Google Scholar] [CrossRef]
- Cua, R.C.; Lau, L.W.; Keough, M.B.; Midha, R.; Apte, S.S.; Yong, V.W. Overcoming neurite-inhibitory chondroitin sulfate proteoglycans in the astrocyte matrix. Glia 2013, 61, 972–984. [Google Scholar] [CrossRef]
- Dong, Y.; D’Mello, C.; Pinsky, W.; Lozinski, B.M.; Kaushik, D.K.; Ghorbani, S.; Moezzi, D.; Brown, D.; Melo, F.C.; Zandee, S.; et al. Oxidized phosphatidylcholines found in multiple sclerosis lesions mediate neurodegeneration and are neutralized by microglia. Nat. Neurosci. 2021, 24, 489–503. [Google Scholar] [CrossRef]
- Mishra, M.K.; Wang, J.; Keough, M.B.; Fan, Y.; Silva, C.; Sloka, S.; Hayardeny, L.; Bruck, W.; Yong, V.W. Laquinimod reduces neuroaxonal injury through inhibiting microglial activation. Ann. Clin. Transl. Neurol. 2014, 1, 409–422. [Google Scholar] [CrossRef]
- Sloka, S.; Zhornitsky, S.; Silva, C.; Metz, L.M.; Yong, V.W. 1,25-Dihydroxyvitamin D3 Protects against Immune-Mediated Killing of Neurons in Culture and in Experimental Autoimmune Encephalomyelitis. PLoS ONE 2015, 10, e0144084. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Moezzi, D.; Dong, Y.; Koch, M.; Yong, V.W. Combination of Hydroxychloroquine and Indapamide Attenuates Neurodegeneration in Models Relevant to Multiple Sclerosis. Neurotherapeutics 2021, 18, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Grenier, D.; Huot, M.P.; Mayrand, D. Iron-chelating activity of tetracyclines and its impact on the susceptibility of Actinobacillus actinomycetemcomitans to these antibiotics. Antimicrob. Agents Chemother. 2000, 44, 763–766. [Google Scholar] [CrossRef] [Green Version]
- Jhamandas, J.H.; Mactavish, D. beta-Amyloid protein (Abeta) and human amylin regulation of apoptotic genes occurs through the amylin receptor. Apoptosis 2012, 17, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Rekha, K.R.; Inmozhi Sivakamasundari, R. Geraniol Protects Against the Protein and Oxidative Stress Induced by Rotenone in an In Vitro Model of Parkinson’s Disease. Neurochem. Res. 2018, 43, 1947–1962. [Google Scholar] [CrossRef]
- Wiesner, D.A.; Dawson, G. Staurosporine induces programmed cell death in embryonic neurons and activation of the ceramide pathway. J. Neurochem. 1996, 66, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Wuerch, E.; Mishra, M.; Melo, H.; Ebacher, V.; Yong, V.W. Quantitative analysis of spinal cord neuropathology in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2022, 362, 577777. [Google Scholar] [CrossRef]
- Rommer, P.S.; Milo, R.; Han, M.H.; Satyanarayan, S.; Sellner, J.; Hauer, L.; Illes, Z.; Warnke, C.; Laurent, S.; Weber, M.S.; et al. Immunological Aspects of Approved MS Therapeutics. Front. Immunol. 2019, 10, 1564. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Campolo, M.; Casili, G.; Lanza, M.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Multiple mechanisms of dimethyl fumarate in amyloid beta-induced neurotoxicity in human neuronal cells. J. Cell Mol. Med. 2018, 22, 1081–1094. [Google Scholar] [CrossRef]
- Di Menna, L.; Molinaro, G.; Di Nuzzo, L.; Riozzi, B.; Zappulla, C.; Pozzilli, C.; Turrini, R.; Caraci, F.; Copani, A.; Battaglia, G.; et al. Fingolimod protects cultured cortical neurons against excitotoxic death. Pharmacol. Res. 2013, 67, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Di Dario, M.; Capitolo, E.; Chaabane, L.; Newcombe, J.; Martino, G.; Farina, C. Fingolimod may support neuroprotection via blockade of astrocyte nitric oxide. Ann. Neurol. 2014, 76, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Jeffery, D.; Kappos, L.; Montalban, X.; Boyko, A.; Rocca, M.A.; Filippi, M.; Group, A.S. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N. Engl. J. Med. 2012, 366, 1000–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippi, M.; Rocca, M.A.; Pagani, E.; De Stefano, N.; Jeffery, D.; Kappos, L.; Montalban, X.; Boyko, A.N.; Comi, G.; Group, A.S. Placebo-controlled trial of oral laquinimod in multiple sclerosis: MRI evidence of an effect on brain tissue damage. J. Neurol. Neurosurg. Psychiatry 2014, 85, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Gentile, A.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Rizzo, F.R.; Centonze, D.; Mandolesi, G. Laquinimod ameliorates excitotoxic damage by regulating glutamate re-uptake. J. Neuroinflammation 2018, 15, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Bassani, C.; De Angelis, A.; Ruffini, F.; Ottoboni, L.; Comi, G.; Martino, G.; Farina, C. Siponimod (BAF312) Activates Nrf2 While Hampering NFkappaB in Human Astrocytes, and Protects From Astrocyte-Induced Neurodegeneration. Front. Immunol. 2020, 11, 635. [Google Scholar] [CrossRef]
- Gentile, A.; Musella, A.; Bullitta, S.; Fresegna, D.; De Vito, F.; Fantozzi, R.; Piras, E.; Gargano, F.; Borsellino, G.; Battistini, L.; et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J. Neuroinflammation 2016, 13, 207. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Sharifi-Rad, J.; Cappellini, F.; Reiner, Z.; Zorzan, D.; Imran, M.; Sener, B.; Kilic, M.; El-Shazly, M.; Fahmy, N.M.; et al. The Therapeutic Potential of Anthocyanins: Current Approaches Based on Their Molecular Mechanism of Action. Front. Pharmacol. 2020, 11, 1300. [Google Scholar] [CrossRef]
- Zdunska, K.; Dana, A.; Kolodziejczak, A.; Rotsztejn, H. Antioxidant Properties of Ferulic Acid and Its Possible Application. Skin Pharmacol. Physiol. 2018, 31, 332–336. [Google Scholar] [CrossRef]
- Packer, L.; Tritschler, H.J.; Wessel, K. Neuroprotection by the metabolic antioxidant alpha-lipoic acid. Free Radic. Biol. Med. 1997, 22, 359–378. [Google Scholar] [CrossRef]
- Scott, B.C.; Aruoma, O.I.; Evans, P.J.; O’Neill, C.; Van der Vliet, A.; Cross, C.E.; Tritschler, H.; Halliwell, B. Lipoic and dihydrolipoic acids as antioxidants. A critical evaluation. Free Radic. Res. 1994, 20, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Marracci, G.; Lovera, J.; Woodward, W.; Bogardus, K.; Marquardt, W.; Shinto, L.; Morris, C.; Bourdette, D. Lipoic acid in multiple sclerosis: A pilot study. Mult. Scler. 2005, 11, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Takeuchi, H.; Horiuchi, H.; Hanyu, T.; Kawanokuchi, J.; Jin, S.; Parajuli, B.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Fingolimod phosphate attenuates oligomeric amyloid beta-induced neurotoxicity via increased brain-derived neurotrophic factor expression in neurons. PLoS ONE 2013, 8, e61988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkl, M.; Frascolla, S.; Amormino, C.; Volpe, E.; Tuosto, L. T Helper Cells: The Modulators of Inflammation in Multiple Sclerosis. Cells 2020, 9, 482. [Google Scholar] [CrossRef] [Green Version]
- Kaskow, B.J.; Baecher-Allan, C. Effector T Cells in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- De Stefano, N.; Matthews, P.M.; Fu, L.; Narayanan, S.; Stanley, J.; Francis, G.S.; Antel, J.P.; Arnold, D.L. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain 1998, 121, 1469–1477. [Google Scholar] [CrossRef]
- Eshaghi, A.; Prados, F.; Brownlee, W.J.; Altmann, D.R.; Tur, C.; Cardoso, M.J.; De Angelis, F.; van de Pavert, S.H.; Cawley, N.; De Stefano, N.; et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann. Neurol. 2018, 83, 210–222. [Google Scholar] [CrossRef] [Green Version]
- Zivadinov, R.; Sepcic, J.; Nasuelli, D.; De Masi, R.; Bragadin, L.M.; Tommasi, M.A.; Zambito-Marsala, S.; Moretti, R.; Bratina, A.; Ukmar, M.; et al. A longitudinal study of brain atrophy and cognitive disturbances in the early phase of relapsing-remitting multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2001, 70, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Alcaide-Leon, P.; Cybulsky, K.; Sankar, S.; Casserly, C.; Leung, G.; Hohol, M.; Selchen, D.; Montalban, X.; Bharatha, A.; Oh, J. Quantitative spinal cord MRI in radiologically isolated syndrome. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e436. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, C.J.; Overton, E.; Khadka, S.; Buckley, J.; Liu, S.; Sampat, M.; Kantarci, O.; Lebrun Frenay, C.; Siva, A.; Okuda, D.T.; et al. Early CNS neurodegeneration in radiologically isolated syndrome. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e102. [Google Scholar] [CrossRef] [Green Version]
- Eshaghi, A.; Marinescu, R.V.; Young, A.L.; Firth, N.C.; Prados, F.; Jorge Cardoso, M.; Tur, C.; De Angelis, F.; Cawley, N.; Brownlee, W.J.; et al. Progression of regional grey matter atrophy in multiple sclerosis. Brain 2018, 141, 1665–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stromillo, M.L.; Giorgio, A.; Rossi, F.; Battaglini, M.; Hakiki, B.; Malentacchi, G.; Santangelo, M.; Gasperini, C.; Bartolozzi, M.L.; Portaccio, E.; et al. Brain metabolic changes suggestive of axonal damage in radiologically isolated syndrome. Neurology 2013, 80, 2090–2094. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Munger, K.L.; Cortese, M.; Barro, C.; Healy, B.C.; Niebuhr, D.W.; Scher, A.I.; Kuhle, J.; Ascherio, A. Serum Neurofilament Light Chain Levels in Patients With Presymptomatic Multiple Sclerosis. JAMA Neurol. 2020, 77, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Matute-Blanch, C.; Villar, L.M.; Alvarez-Cermeno, J.C.; Rejdak, K.; Evdoshenko, E.; Makshakov, G.; Nazarov, V.; Lapin, S.; Midaglia, L.; Vidal-Jordana, A.; et al. Neurofilament light chain and oligoclonal bands are prognostic biomarkers in radiologically isolated syndrome. Brain 2018, 141, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, M.; Magliozzi, R.; Ciccarelli, O.; Geurts, J.J.; Reynolds, R.; Martin, R. Exploring the origins of grey matter damage in multiple sclerosis. Nat. Rev. Neurosci. 2015, 16, 147–158. [Google Scholar] [CrossRef]
- Mendiola, A.S.; Ryu, J.K.; Bardehle, S.; Meyer-Franke, A.; Ang, K.K.; Wilson, C.; Baeten, K.M.; Hanspers, K.; Merlini, M.; Thomas, S.; et al. Transcriptional profiling and therapeutic targeting of oxidative stress in neuroinflammation. Nat. Immunol. 2020, 21, 513–524. [Google Scholar] [CrossRef]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Macrez, R.; Stys, P.K.; Vivien, D.; Lipton, S.A.; Docagne, F. Mechanisms of glutamate toxicity in multiple sclerosis: Biomarker and therapeutic opportunities. Lancet Neurol. 2016, 15, 1089–1102. [Google Scholar] [CrossRef]
- Woo, M.S.; Ufer, F.; Rothammer, N.; Di Liberto, G.; Binkle, L.; Haferkamp, U.; Sonner, J.K.; Engler, J.B.; Hornig, S.; Bauer, S.; et al. Neuronal metabotropic glutamate receptor 8 protects against neurodegeneration in CNS inflammation. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta 2016, 1862, 506–510. [Google Scholar] [CrossRef]
- Adamczyk, B.; Adamczyk-Sowa, M. New Insights into the Role of Oxidative Stress Mechanisms in the Pathophysiology and Treatment of Multiple Sclerosis. Oxid. Med. Cell Longev. 2016, 2016, 1973834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Horssen, J.; Schreibelt, G.; Drexhage, J.; Hazes, T.; Dijkstra, C.D.; van der Valk, P.; de Vries, H.E. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic. Biol. Med. 2008, 45, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Nikic, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Bruck, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Kemp, K.; Redondo, J.; Hares, K.; Rice, C.; Scolding, N.; Wilkins, A. Oxidative injury in multiple sclerosis cerebellar grey matter. Brain Res. 2016, 1642, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, I.Y.; Lee, P.; Hughes, A.J.; Denney, D.R.; Lynch, S.G. Longitudinal changes of cerebral glutathione (GSH) levels associated with the clinical course of disease progression in patients with secondary progressive multiple sclerosis. Mult. Scler. 2017, 23, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Lee, P.; Adany, P.; Hughes, A.J.; Belliston, S.; Denney, D.R.; Lynch, S.G. In vivo evidence of oxidative stress in brains of patients with progressive multiple sclerosis. Mult. Scler. 2018, 24, 1029–1038. [Google Scholar] [CrossRef]
- Scuderi, S.A.; Ardizzone, A.; Paterniti, I.; Esposito, E.; Campolo, M. Antioxidant and Anti-inflammatory Effect of Nrf2 Inducer Dimethyl Fumarate in Neurodegenerative Diseases. Antioxidants 2020, 9, 630. [Google Scholar] [CrossRef]
- Lee, N.J.; Ha, S.K.; Sati, P.; Absinta, M.; Nair, G.; Luciano, N.J.; Leibovitch, E.C.; Yen, C.C.; Rouault, T.A.; Silva, A.C.; et al. Potential role of iron in repair of inflammatory demyelinating lesions. J. Clin. Investig. 2019, 129, 4365–4376. [Google Scholar] [CrossRef]
- Cheli, V.T.; Correale, J.; Paez, P.M.; Pasquini, J.M. Iron Metabolism in Oligodendrocytes and Astrocytes, Implications for Myelination and Remyelination. ASN Neuro 2020, 12, 1759091420962681. [Google Scholar] [CrossRef]
- Hulet, S.W.; Powers, S.; Connor, J.R. Distribution of transferrin and ferritin binding in normal and multiple sclerotic human brains. J. Neurol. Sci. 1999, 165, 48–55. [Google Scholar] [CrossRef]
- Bagnato, F.; Hametner, S.; Yao, B.; van Gelderen, P.; Merkle, H.; Cantor, F.K.; Lassmann, H.; Duyn, J.H. Tracking iron in multiple sclerosis: A combined imaging and histopathological study at 7 Tesla. Brain 2011, 134, 3602–3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.L.; Nydes, M.; Shanley, K.L.; Morales Pantoja, I.E.; Howard, T.A.; Bizzozero, O.A. Reduced expression of the ferroptosis inhibitor glutathione peroxidase-4 in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurochem. 2019, 148, 426–439. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | HORAC (μmole/g) | ORAC (μmole/g) | Relative Protection against Iron Killing (%) |
|---|---|---|---|
| HS | 575 ± 75 (10 extracts) | 480 ± 45 (3 extracts) | 100 |
| Commercial hibiscus | >800 | Not determined | 0 |
| HS Fraction 1 | 0 | 0 | 100 |
| Alpha lipoic acid | 848 | 300 | ~50 |
| Ferulic acid | 4549 | 7451 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, M.K.; Wang, J.; Mirzaei, R.; Chan, R.; Melo, H.; Zhang, P.; Ling, C.-C.; Bruccoleri, A.; Tang, L.; Yong, V.W. A Distinct Hibiscus sabdariffa Extract Prevents Iron Neurotoxicity, a Driver of Multiple Sclerosis Pathology. Cells 2022, 11, 440. https://doi.org/10.3390/cells11030440
Mishra MK, Wang J, Mirzaei R, Chan R, Melo H, Zhang P, Ling C-C, Bruccoleri A, Tang L, Yong VW. A Distinct Hibiscus sabdariffa Extract Prevents Iron Neurotoxicity, a Driver of Multiple Sclerosis Pathology. Cells. 2022; 11(3):440. https://doi.org/10.3390/cells11030440
Chicago/Turabian StyleMishra, Manoj Kumar, Jianxiong Wang, Reza Mirzaei, Rigel Chan, Helvira Melo, Ping Zhang, Chang-Chun Ling, Aldo Bruccoleri, Lin Tang, and V. Wee Yong. 2022. "A Distinct Hibiscus sabdariffa Extract Prevents Iron Neurotoxicity, a Driver of Multiple Sclerosis Pathology" Cells 11, no. 3: 440. https://doi.org/10.3390/cells11030440
APA StyleMishra, M. K., Wang, J., Mirzaei, R., Chan, R., Melo, H., Zhang, P., Ling, C. -C., Bruccoleri, A., Tang, L., & Yong, V. W. (2022). A Distinct Hibiscus sabdariffa Extract Prevents Iron Neurotoxicity, a Driver of Multiple Sclerosis Pathology. Cells, 11(3), 440. https://doi.org/10.3390/cells11030440