
Biomarker Development in Cardiology: Reviewing the Past to Inform the Future

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
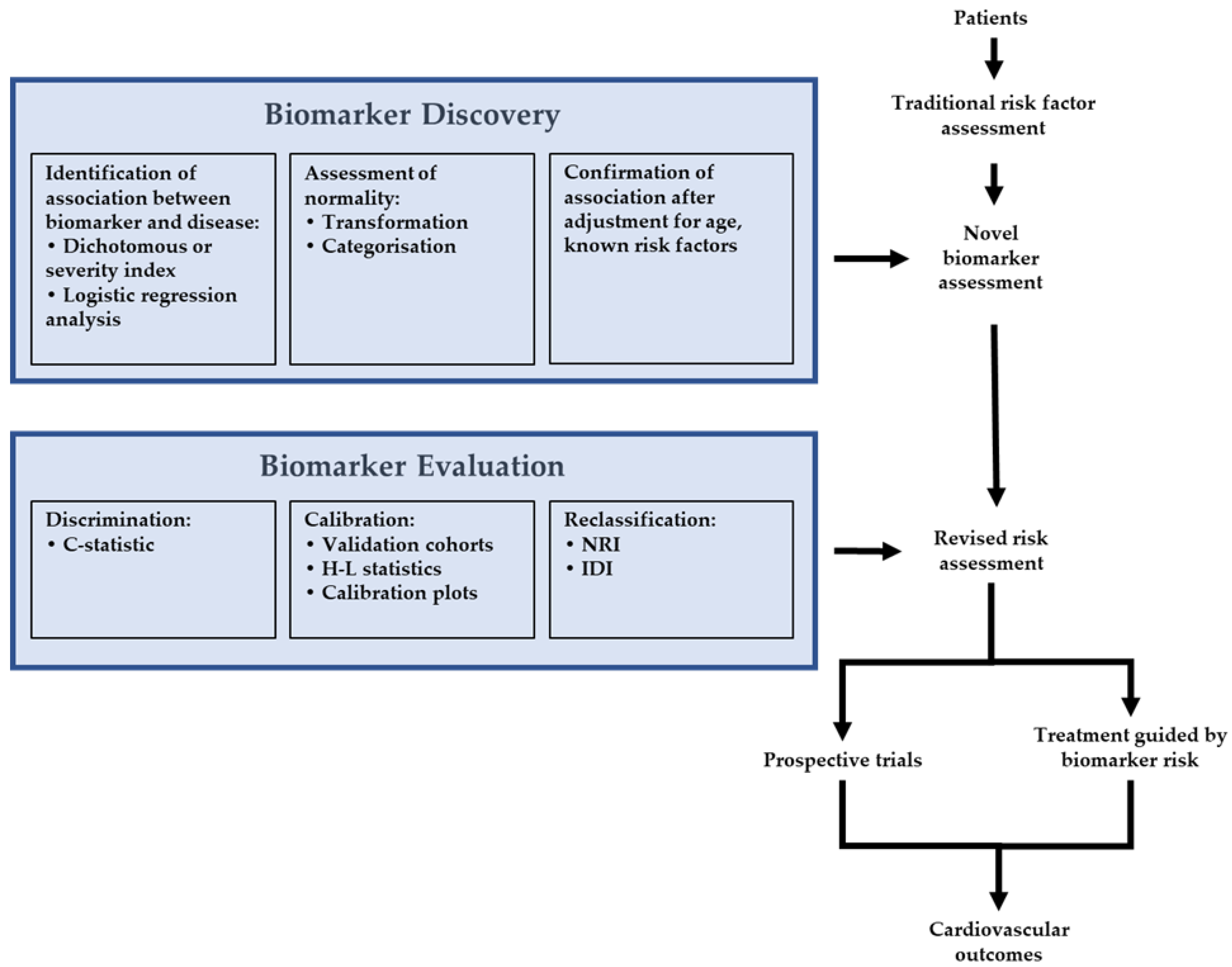
2. Statistical Approaches to Assessing New Biomarkers
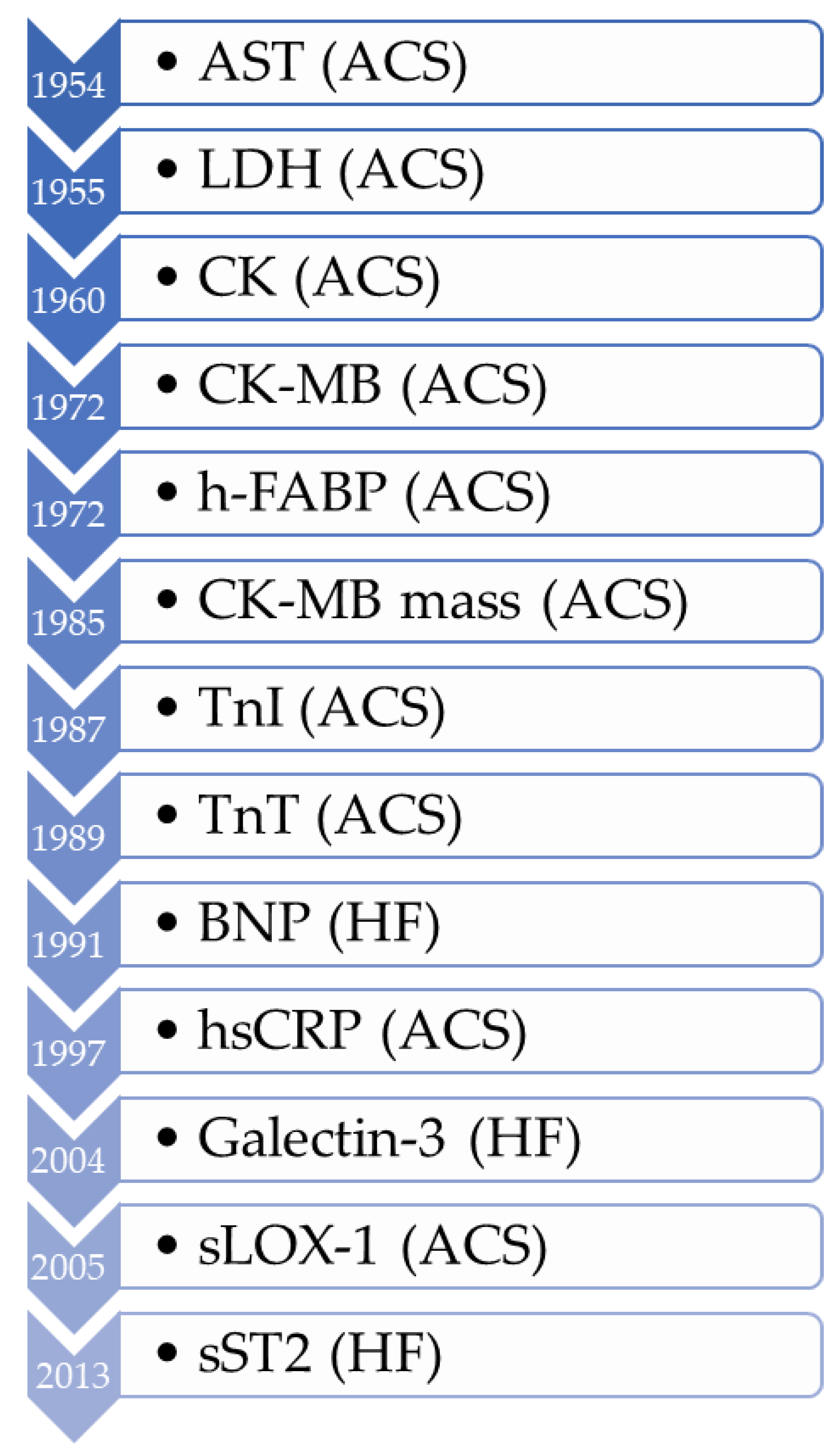
3. The History of Biomarker Development in Cardiovascular Disease
3.1. Acute Coronary Syndrome (ACS) Biomarkers
3.1.1. Creatine Kinase
3.1.2. Heart Fatty Acid-Binding Proteins (h-FABP)
3.1.3. Troponin
3.1.4. Soluble Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 (sLOX-1)
3.2. Heart Failure (HF) Biomarkers
3.2.1. Natriuretic Peptides
3.2.2. Galectin-3
3.2.3. Suppression of Tumorigenesis-2 (sST2)
3.3. Atherosclerosis Risk
High-Sensitivity C-Reactive Protein (hsCRP)

{kind=link}
{kind=link}
| Biomarker | Indication | Reference Range | Statistics |
|---|---|---|---|
| CK-MB | AMI | >99th percentile of upper reference limit of sex-specific controls for assay [65] >20–25 U/I [30,37] >30 U/I [29] CK-MB2:CK-MB1 ratio > 1.5 [34,35] | Analysis of frequency distribution [29], C-statistic [35] |
| Troponin | AMI/CV Risk | >99th percentile of upper reference limit for assay [65] | ≈3 SDs above the mean for normal range [63] |
| sLOX-1 | AMI | >91.0–131.7 pg/mL (suggested) [95,96,97] | C-statistic [95,96,97] |
| h-FABP | AMI | >4 ug/L [46,47] | C-statistic [43,44,47] |
| BNP | HF | Rule out: <100 ng/L [134] | C-statistic [114,116] |
| Rule in: >400 ng/L [134] | |||
| NT-proBNP | HF | Rule out: <300 ng/L [134] | C-statistic [198,199] |
| Rule in: age < 50; >450 ng/L [134] Rule in: age 50–75; >900 ng/L [134] Rule in: age > 75; >1800 ng/L [134] | |||
| Galectin-3 | HF | >17.8 ng/L [144,145] | C-statistic [144,145,146] |
| sST2 | HF | >35 ng/L [200,201] | C-statistic [202], NRI, and IDI [174] |
| hsCRP | CV Risk | High risk: >3 mg/L [192,193] | Tertiles [194,195], quartiles [196], quintiles [197], and the C-statistic [197] |
| Increased risk: >1 mg/L [203] |
| Biomarker | When to Take Sample (Time Sampling) | Biomarker Changes over Time (Time Dynamics) | Non-Cardiac Causes of Altered Levels |
|---|---|---|---|
| CK-MB | 4–6 h after symptom onset [34,35] | Peak occurs after 16–30 h, returns to baseline by 24–36 h [204] | Elevated in skeletal muscle injury [205], vigorous exercise [206], stroke [207], trauma patients [208], and kidney disease [209,210]; 1.2–2.6x higher 99th percentile in males [211] and post-operatively in spinal surgery [212] |
| h-FABP | 2–4 h after symptom onset [43,44] | Peak occurs 6 h after symptom onset, returns to baseline by 24 h [45] | Elevated in AKI [213], PE [214], stroke [215], sepsis [216], acute HF [217], NAFLD [218], smoking, and COPD [219] |
| Troponin | At presentation and then 2–6 h later if the first result is negative [65] | Peak occurs at 12–48 h [220], returns to baseline by 14 days [221] | Elevated in sepsis [222], critical illness [223], LVH [224], coronary vasospasm [225], stroke [226], AF [227], heart failure [228], myocarditis [229], dialysis patients [230], males, black people, DM, and HTN [231]; lower in smoking, alcohol use, and statin use [231] |
| sLOX-1 | At presentation [96] | Peak is maintained from presentation up to 24 h [96] | Conflicting association with smoking [95,232] and not significantly correlated with lipids, diabetes, or hypertension [94,95] |
| BNP | At presentation for acute dyspnea [116] as a screening tool [117,118,233] | Levels remain elevated in untreated HF; treatment may lower levels to normal range [234,235] | Elevated in smokers [236] and renal insufficiency [237,238]; lower in obesity [239], even in patients with HF [240]; degraded by neprilysin (ARNI therapy causes BNP elevation) [241] |
| NT-proBNP | At presentation for acute dyspnea [123,124,125,126] | Levels remain elevated in untreated HF; treatment may lower levels to normal range [234] | Elevated in smokers [242]; renal insufficiency (greater than BNP) [243]. Lower in obesity [239]. Not degraded by neprilysin (can be used to monitor ARNI therapy) [241]. |
| Galectin-3 | At presentation as a prognostic marker [144] | Levels remain stable over time [141] | Conflicting evidence for association with sex, age, DM, and HTN [244,245] |
| sST2 | Serially, as a prognostic marker [172,201,246] | Levels may remain elevated (indicating worse prognosis) or decrease by 48–72 h [247] | Elevated in smoking [248], males, DM [249], and ALD [250] |
| hsCRP | As a risk-enhancing factor at screening for patients at borderline or intermediate risk of atherosclerotic CVD [251] | Levels may fluctuate considerably over time [252], and statin therapy may reduce levels [189] | Elevated in smoking [248,253] and other inflammatory processes [254,255] |
| Biomarker | AHA/ACC | ESC | ||||
|---|---|---|---|---|---|---|
| Recommendation | COR | LOE | Recommendation | COR | LOE | |
| CK-MB | Not recommended for diagnosis of ACS [256] | III | A | Not recommended for diagnosis of ACS [257] | III | |
| h-FABP | Not in guidelines | Not recommended for diagnosis of ACS [257] | III | B | ||
| Troponin | Diagnosis of ACS [256] | I | A | Diagnosis of ACS [257] | I | B |
| Additive risk stratification in chronic HF (hscTn) [182] | IIb | B-NR | ||||
| sLOX-1 | Not in guidelines | Not in guidelines | ||||
| BNP and NT-proBNP | Screening for HF [182] | IIa | B-R | |||
| Diagnosis of HF [182] | I | A | Diagnosis of HF [135] | I | B | |
| Prognosis or disease severity in chronic HF [182] | I | A | ||||
| Prognosis in ADHF [182] | I | A | ||||
| Pre-discharge for prognosis [182] | IIa | B-NR | ||||
| Galectin-3 | Additive risk stratification in chronic HF [182] | IIb | B-NR | Not in guidelines | ||
| sST2 | Additive risk stratification in chronic HF [182] | IIb | B-NR | Not in guidelines | ||
| hsCRP | As a risk enhancing factor to aid discussion of statin therapy initiation [258] | Not recommended for risk stratification in CVD prevention [259,260] | III | B | ||
4. Prospective Biomarker Trials
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lloyd-Jones, D.M.; Liu, K.; Tian, L.; Greenland, P. Narrative Review: Assessment of C-Reactive Protein in Risk Prediction for Cardiovascular Disease. Ann. Intern. Med. 2006, 145, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, T.; Fiuzat, M.; Pencina, M.J.; Geller, N.L.; Zannad, F.; Cleland, J.G.; Snider, J.V.; Blankenberg, S.; Adams, K.F.; Redberg, R.F.; et al. Charting a roadmap for heart failure biomarker studies. JACC Heart Fail 2014, 2, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Pletcher, M.J.; Pignone, M. Evaluating the Clinical Utility of a Biomarker. Circulation 2011, 123, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Grund, B.; Sabin, C. Analysis of biomarker data: Logs, odds ratios, and receiver operating characteristic curves. Curr. Opin. HIV AIDS 2010, 5, 473–479. [Google Scholar] [CrossRef]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, N.R. Use and Misuse of the Receiver Operating Characteristic Curve in Risk Prediction. Circulation 2007, 115, 928–935. [Google Scholar] [CrossRef] [Green Version]
- McGeechan, K.; Macaskill, P.; Irwig, L.; Liew, G.; Wong, T.Y. Assessing New Biomarkers and Predictive Models for Use in Clinical Practice: A Clinician’s Guide. Arch. Intern. Med. 2008, 168, 2304–2310. [Google Scholar] [CrossRef]
- Hosmer, D.W.; Lemesbow, S. Goodness of fit tests for the multiple logistic regression model. Commun. Stat. Theory Methods 1980, 9, 1043–1069. [Google Scholar] [CrossRef]
- Hosmer, D.W.; Hosmer, T.; Le Cessie, S.; Lemeshow, S. A comparison of goodness-of-fit tests for the logistic regression model. Stat. Med. 1997, 16, 965–980. [Google Scholar] [CrossRef]
- Crowson, C.S.; Atkinson, E.J.; Therneau, T.M. Assessing calibration of prognostic risk scores. Stat. Methods Med. Res. 2016, 25, 1692–1706. [Google Scholar] [CrossRef] [Green Version]
- Pencina, M.J.; D’Agostino Sr, R.B.; D’Agostino, R.B., Jr.; Vasan, R.S. Evaluating the added predictive ability of a new marker: From area under the ROC curve to reclassification and beyond. Stat. Med. 2008, 27, 157–212. [Google Scholar] [CrossRef] [PubMed]
- Kerr, K.F.; McClelland, R.L.; Brown, E.R.; Lumley, T. Evaluating the Incremental Value of New Biomarkers with Integrated Discrimination Improvement. Am. J. Epidemiol. 2011, 174, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toraih, E.A.; Elshazli, R.M.; Hussein, M.H.; Elgaml, A.; Amin, M.; El-Mowafy, M.; El-Mesery, M.; Ellythy, A.; Duchesne, J.; Killackey, M.T.; et al. Association of cardiac biomarkers and comorbidities with increased mortality, severity, and cardiac injury in COVID-19 patients: A meta-regression and decision tree analysis. J. Med. Virol. 2020, 92, 2473–2488. [Google Scholar] [CrossRef] [PubMed]
- Danese, E.; Montagnana, M. An historical approach to the diagnostic biomarkers of acute coronary syndrome. Ann. Transl. Med. 2016, 4, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladue, J.S.; Wroblewski, F.; Karmen, A. Serum glutamic oxaloacetic transaminase activity in human acute transmural myocardial infarction. Science 1954, 120, 497–499. [Google Scholar] [CrossRef]
- Goldberg, D.M.; Winfield, D.A. Diagnostic accuracy of serum enzyme assays for myocardial infarction in a general hospital population. Br. Heart J. 1972, 34, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Coodley, E.L. Evaluation of enzyme diagnosis in myocardial infarction. Am. J. Med. Sci. 1968, 256, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Karmen, A.; Wroblewski, F.; Ladue, J.S. Transaminase activity in human blood. J. Clin. Investig. 1955, 34, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobel, B.E.; Shell, W.E. Serum Enzyme Determinations in the Diagnosis and Assessment of Myocardial Infarction. Circulation 1972, 45, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Johnston, C.C.; Bolton, E.C. Cardiac enzymes. Ann. Emerg. Med. 1982, 11, 27–35. [Google Scholar] [CrossRef]
- Dreyfus, J.C.; Schapira, G.; Resnais, J.; Scebat, L. Serum creatine kinase in the diagnosis of myocardial infarct. Rev. Fr. Etud. Clin. Biol. 1960, 5, 386–387. [Google Scholar] [PubMed]
- Sørensen, N.S. Creatine Phosphokinase in the Diagnosis of Myocardial Infarction. Acta Med. Scand. 1963, 174, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Duma, R.J.; Siegel, A.L. Serum Creatinine Phosphokinase in Acute Myocardial Infarction: Diagnostic Value. Arch. Intern. Med. 1965, 115, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Vincent, W.R.; Rapaport, E. Serum creatine phosphokinase in the diagnosis of acute myocardial infarction. Am. J. Cardiol. 1965, 15, 17–26. [Google Scholar] [CrossRef]
- Burger, A.; Richterich, R.; Aebi, H. Die Heterogenitat der Kreatin-Kinase. Biochemistry 1964, 2, 305. [Google Scholar]
- Jacobs, H.; Heldt, H.W.; Klingenberg, M. High activity of creatine kinase in mitochondria from muscle and brain and evidence for a separate mitochondrial isoenzyme of creatine kinase. Biochem. Biophys. Res. Commun. 1964, 16, 516–521. [Google Scholar] [CrossRef]
- Eppenberger, H.M.; Eppenberger, M.; Richterich, R.; Aebi, H. The ontogeny of creatine kinase isozymes. Dev. Biol. 1964, 10, 1–16. [Google Scholar] [CrossRef]
- Konttinen, A.; Somer, H. Determination of serum creatine kinase isoenzymes in myocardial infarction. Am. J. Cardiol. 1972, 29, 817–820. [Google Scholar] [CrossRef]
- Grande, P.; Christiansen, C.; Næstoft, J. Creatine kinase isoenzyme MB assay by electrophoresis. Scand. J. Clin. Lab. Investig. 1979, 39, 607–612. [Google Scholar] [CrossRef]
- Ljungdahl, L.; Gerhardt, W.; Hofvendahl, S. Serum creatine kinase B subunit activity in diagnosis of acute myocardial infarction. Br. Heart J. 1980, 43, 514–522. [Google Scholar] [CrossRef]
- Gerhardt, W.; Waldenström, J.; Hörder, M.; Hofvendahl, S.; Billström, R.; Ljungdahl, R.; Berning, H.; Bagger, P. Creatine kinase and creatine kinase B-subunit activity in serum in cases of suspected myocardial infarction. Clin. Chem. 1982, 28, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.; Gowda, K.S.; Ludbrook, P.A.; Sobel, B.E. Specificity of elevated serum MB creatine phosphokinase activity in the diagnosis of acute myocardial infarction. Am. J. Cardiol. 1975, 36, 433–437. [Google Scholar] [CrossRef]
- Blomberg, D.J.; Kimber, W.D.; Burke, M.D. Creatine kinase isoenzymes. Predictive value in the early diagnosis of acute myocardial infarction. Am. J. Med. 1975, 59, 464–469. [Google Scholar] [CrossRef]
- Puleo, P.R.; Guadagno, P.A.; Roberts, R.; Scheel, M.V.; Marian, A.J.; Churchill, D.; Perryman, M.B. Early diagnosis of acute myocardial infarction based on assay for subforms of creatine kinase-MB. Circulation 1990, 82, 759–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puleo, P.R.; Meyer, D.; Wathen, C.; Tawa, C.B.; Wheeler, S.; Hamburg, R.J.; Ali, N.; Obermueller, S.D.; Triana, F.J.; Zimmerman, J.L.; et al. Use of a Rapid Assay of Subforms of Creatine Kinase MB to Diagnose or Rule Out Acute Myocardial Infarction. N. Engl. J. Med. 1994, 331, 561–566. [Google Scholar] [CrossRef]
- Lee, T.H.; Goldman, L. Serum enzyme assays in the diagnosis of acute myocardial infarction. Recommendations based on a quantitative analysis. Ann. Intern. Med. 1986, 105, 221–233. [Google Scholar] [CrossRef] [PubMed]
- White, R.D.; Grande, P.; Califf, L.; Palmeri, S.T.; Califf, R.M.; Wagner, G.S. Diagnostic and prognostic significance of minimally elevated creatine kinase-mb in suspected acute myocardial infarction. Am. J. Cardiol. 1985, 55, 1478–1484. [Google Scholar] [CrossRef]
- Alpert, J.S.; Thygesen, K.; Antman, E.; Bassand, J.P. Myocardial infarction redefined--a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J. Am. Coll. Cardiol. 2000, 36, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Ockner, R.K.; Manning, J.A.; Poppenhausen, R.B.; Ho, W.K. A binding protein for fatty acids in cytosol of intestinal mucosa, liver, myocardium, and other tissues. Science 1972, 177, 56. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; van Bilsen, M.; Paulussen, R.J.A.; Veerkamp, J.H.; van Der Vusse, G.J.; Reneman, R.S. Release of fatty acid-binding protein from isolated rat heart subjected to ischemia and reperfusion or to the calcium paradox. Biochim. Et Biophys. Acta (BBA)/Lipids Lipid. Metab. 1988, 961, 148–152. [Google Scholar] [CrossRef]
- Tanaka, T.; Hirota, Y.; Sohmiya, K.-I.; Nishimura, S.; Kawamura, K. Serum and urinary human heart fatty acid-binding protein in acute myocardial infarction. Clin. Biochem. 1991, 24, 195–201. [Google Scholar] [CrossRef]
- Kleine, A.; Glatz, J.; Nieuwenhoven, F.; Vusse, G. Release of heart fatty acid-binding protein into plasma after acute myocardial infarction in man. Mol. Cell. Biochem. 1992, 116, 155–162. [Google Scholar] [CrossRef]
- Seino, Y.; Ogata, K.; Takano, T. Use of a whole blood rapid panel test for heart-type fatty acid–binding protein in patients with acute chest pain: Comparison with rapid troponin T and myoglobin tests. ACC Curr. J. Rev. 2004, 13, 12–13. [Google Scholar] [CrossRef]
- McCann, C.J.; Glover, B.M.; Menown, I.B.A.; Moore, M.J.; McEneny, J.; Owens, C.G.; Smith, B.; Sharpe, P.C.; Young, I.S.; Adgey, J.A. Novel biomarkers in early diagnosis of acute myocardial infarction compared with cardiac troponin T. Eur. Heart J. 2008, 29, 2843–2850. [Google Scholar] [CrossRef] [Green Version]
- Gururajan, P.; Gurumurthy, P.; Nayar, P.; Srinivasa Nageswara Rao, G.; Babu, S.; Cherian, K.M. Heart Fatty Acid Binding Protein (H-FABP) as a Diagnostic Biomarker in Patients with Acute Coronary Syndrome. Heart Lung Circ. 2010, 19, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Wodzig, K.W.; Pelsers, M.M.; van der Vusse, G.J.; Roos, W.; Glatz, J.F. One-step enzyme-linked immunosorbent assay (ELISA) for plasma fatty acid-binding protein. Ann. Clin. Biochem. 1997, 34 Pt 3, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.T.A.; van Severen, E.; Vandervoort, P.M.; Grieten, L.; Buntinx, F.; Glatz, J.F.; Dinant, G.J. Heart-type fatty acid binding protein (H-FABP) in patients in an emergency department setting, suspected of acute coronary syndrome: Optimal cut-off point, diagnostic value and future opportunities in primary care. Eur. J. Gen. Pract. 2015, 21, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Body, R.; Burrows, G.; Carley, S.; Lewis, P.S. The Manchester Acute Coronary Syndromes (MACS) decision rule: Validation with a new automated assay for heart-type fatty acid binding protein. Emerg. Med. J. 2015, 32, 769–774. [Google Scholar] [CrossRef] [Green Version]
- Bivona, G.; Agnello, L.; Bellia, C.; Lo Sasso, B.; Ciaccio, M. Diagnostic and prognostic value of H-FABP in acute coronary syndrome: Still evidence to bring. Clin. Biochem. 2018, 58, 1–4. [Google Scholar] [CrossRef]
- Willemsen, R.T.; Winkens, B.; Kietselaer, B.L.; Smolinska, A.; Buntinx, F.; Glatz, J.F.; Dinant, G.-J. Evaluating possible acute coronary syndrome in primary care: The value of signs, symptoms, and plasma heart-type fatty acid-binding protein (H-FABP). A diagnostic study. BJGP Open 2019, 3, bjgpopen19X101652. [Google Scholar] [CrossRef]
- Ebashi, S.; Kodama, A. A new protein factor promoting aggregation of tropomyosin. J. Biochem. 1965, 58, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Greaser, M.L.; Gergely, J. Reconstitution of troponin activity from three protein components. J. Biol. Chem. 1971, 246, 4226–4233. [Google Scholar] [CrossRef]
- Cummins, B.; Auckland, M.L.; Cummins, P. Cardiac-specific troponin-I radioimmunoassay in the diagnosis of acute myocardial infarction. Am. Heart J. 1987, 113, 1333–1344. [Google Scholar] [CrossRef]
- Katus, H.A.; Remppis, A.; Looser, S.; Hallermeier, K.; Scheffold, T.; Kubler, W. Enzyme linked immuno assay of cardiac troponin T for the detection of acute myocardial infarction in patients. J. Mol. Cell Cardiol. 1989, 21, 1349–1353. [Google Scholar] [CrossRef]
- Ravkilde, J.; Hørder, M.; Gerhardt, W.; Ljungdahl, L.; Pettersson, T.; Tryding, N.; Møller, B.H.; Hamfelt, A.; Graven, T.; Åsberg, A.; et al. Diagnostic performance and prognostic value of serum troponin T in suspected acute myocardial infarction. Scand. J. Clin. Lab. Investig. 1993, 53, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Ohman, E.M.; Armstrong, P.W.; Christenson, R.H.; Granger, C.B.; Katus, H.A.; Hamm, C.W.; O’Hanesian, M.A.; Wagner, G.S.; Kleiman, N.S.; Harrell, F.E.; et al. Cardiac Troponin T Levels for Risk Stratification in Acute Myocardial Ischemia. N. Engl. J. Med. 1996, 335, 1333–1342. [Google Scholar] [CrossRef]
- Hamm, C.W.; Ravkilde, J.; Gerhardt, W.; Jørgensen, P.; Peheim, E.; Ljungdahl, L.; Goldmann, B.; Katus, H.A. The Prognostic Value of Serum Troponin T in Unstable Angina. N. Engl. J. Med. 1992, 327, 146–150. [Google Scholar] [CrossRef]
- Collinson, P.; Gerhardt, W.; Katus, H.; Müller-Bardorff, M.; Braun, S.; Schricke, U.; Vogt, W.; Nagel, D.; Zander, M.; Leinberger, R.; et al. Multicentre evaluation of an immunological rapid test for the detection of troponin T in whole blood samples. Ren. Fail. 1996, 34, 591–598. [Google Scholar]
- Hamm, C.W.; Goldmann, B.U.; Heeschen, C.; Kreymann, G.; Berger, J.; Meinertz, T. Emergency room triage of patients with acute chest pain by means of rapid testing for cardiac troponin T or troponin I. N. Engl. J. Med. 1997, 337, 1648–1653. [Google Scholar] [CrossRef]
- D’Costa, M.; Fleming, E.; Patterson, M.C. Cardiac Troponin I for the Diagnosis of Acute Myocardial Infarction in the Emergency Department. Am. J. Clin. Pathol. 1997, 108, 550–555. [Google Scholar] [CrossRef] [Green Version]
- Antman, E.M.; Tanasijevic, M.J.; Thompson, B.; Schactman, M.; McCabe, C.H.; Cannon, C.P.; Fischer, G.A.; Fung, A.Y.; Thompson, C.; Wybenga, D.; et al. Cardiac-Specific Troponin I Levels to Predict the Risk of Mortality in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 1996, 335, 1342–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.H.; Apple, F.S.; Gibler, W.B.; Jesse, R.L.; Warshaw, M.M.; Valdes, R., Jr. National Academy of Clinical Biochemistry Standards of Laboratory Practice: Recommendations for the Use of Cardiac Markers in Coronary Artery Diseases. Clin. Chem. 1999, 45, 1104–1121. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.S.; Ravkilde, J.; Roberts, R.; Naslund, U.; Apple, F.S.; Galvani, M.; Katus, H. It’s time for a change to a troponin standard. Circulation 2000, 102, 1216–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apple, F.S.; Jaffe, A.S.; Collinson, P.; Mockel, M.; Ordonez-Llanos, J.; Lindahl, B.; Hollander, J.; Plebani, M.; Than, M.; Chan, M.H.; et al. IFCC educational materials on selected analytical and clinical applications of high sensitivity cardiac troponin assays. Clin. Biochem 2015, 48, 201–203. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D.; Executive Group on behalf of the Joint European Society of Cardiology /American College of Cardiology /American Heart Association /World Heart Federation Task Force for the Universal Definition of Myocardial, I. Fourth Universal Definition of Myocardial Infarction (2018). Circulation 2018, 138, e618–e651. [Google Scholar] [CrossRef]
- Stengaard, C.; Sørensen, J.T.; Ladefoged, S.A.; Lassen, J.F.; Rasmussen, M.B.; Pedersen, C.K.; Ayer, A.; Bøtker, H.E.; Terkelsen, C.J.; Thygesen, K. The potential of optimizing prehospital triage of patients with suspected acute myocardial infarction using high-sensitivity cardiac troponin T and copeptin. Biomarkers 2017, 22, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Ishak, M.; Ali, D.; Fokkert, M.J.; Slingerland, R.J.; Tolsma, R.T.; Badings, E.; van der Sluis, A.; van Eenennaam, F.; Mosterd, A.; Ten Berg, J.M.; et al. Fast assessment and management of chest pain patients without ST-elevation in the pre-hospital gateway (FamouS Triage): Ruling out a myocardial infarction at home with the modified HEART score. Eur. Heart J. Acute Cardiovasc. Care 2018, 7, 102–110. [Google Scholar] [CrossRef]
- Neumann, J.T.; Twerenbold, R.; Ojeda, F.; Sörensen, N.A.; Chapman, A.R.; Shah, A.S.V.; Anand, A.; Boeddinghaus, J.; Nestelberger, T.; Badertscher, P.; et al. Application of High-Sensitivity Troponin in Suspected Myocardial Infarction. N. Engl. J. Med. 2019, 380, 2529–2540. [Google Scholar] [CrossRef]
- Chapman, A.R.; Lee, K.K.; McAllister, D.A.; Cullen, L.; Greenslade, J.H.; Parsonage, W.; Worster, A.; Kavsak, P.A.; Blankenberg, S.; Neumann, J.; et al. Association of High-Sensitivity Cardiac Troponin I Concentration with Cardiac Outcomes in Patients with Suspected Acute Coronary Syndrome. JAMA 2017, 318, 1913–1924. [Google Scholar] [CrossRef]
- Chapman, A.R.; Fujisawa, T.; Lee, K.K.; Andrews, J.P.; Anand, A.; Sandeman, D.; Ferry, A.V.; Stewart, S.; Marshall, L.; Strachan, F.E.; et al. Novel high-sensitivity cardiac troponin I assay in patients with suspected acute coronary syndrome. Heart 2019, 105, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Than, M.P.; Pickering, J.W.; Sandoval, Y.; Shah, A.S.V.; Tsanas, A.; Apple, F.S.; Blankenberg, S.; Cullen, L.; Mueller, C.; Neumann, J.T.; et al. Machine Learning to Predict the Likelihood of Acute Myocardial Infarction. Circulation 2019, 140, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.S.; Anand, A.; Sandoval, Y.; Lee, K.K.; Smith, S.W.; Adamson, P.D.; Chapman, A.R.; Langdon, T.; Sandeman, D.; Vaswani, A.; et al. High-sensitivity cardiac troponin I at presentation in patients with suspected acute coronary syndrome: A cohort study. Lancet 2015, 386, 2481–2488. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.S.V.; Anand, A.; Strachan, F.E.; Ferry, A.V.; Lee, K.K.; Chapman, A.R.; Sandeman, D.; Stables, C.L.; Adamson, P.D.; Andrews, J.P.M.; et al. High-sensitivity troponin in the evaluation of patients with suspected acute coronary syndrome: A stepped-wedge, cluster-randomised controlled trial. Lancet 2018, 392, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Anand, A.; Lee, K.K.; Chapman, A.R.; Ferry, A.V.; Adamson, P.D.; Strachan, F.E.; Berry, C.; Findlay, I.; Cruikshank, A.; Reid, A.; et al. High-Sensitivity Cardiac Troponin on Presentation to Rule Out Myocardial Infarction: A Stepped-Wedge Cluster Randomized Controlled Trial. Circulation 2021, 143, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Apple, F.S.; Ler, R.; Murakami, M.M. Determination of 19 cardiac troponin I and T assay 99th percentile values from a common presumably healthy population. Clin. Chem. 2012, 58, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Sedaghat-Hamedani, F.; Kayvanpour, E.; Frankenstein, L.; Mereles, D.; Amr, A.; Buss, S.; Keller, A.; Giannitsis, E.; Jensen, K.; Katus, H.A.; et al. Biomarker changes after strenuous exercise can mimic pulmonary embolism and cardiac injury--a metaanalysis of 45 studies. Clin. Chem. 2015, 61, 1246–1255. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Twerenbold, R.; Tanglay, Y.; Reichlin, T.; Honegger, U.; Wagener, M.; Jaeger, C.; Rubini Gimenez, M.; Hochgruber, T.; Puelacher, C.; et al. Clinical benefit of high-sensitivity cardiac troponin I in the detection of exercise-induced myocardial ischemia. Am. Heart J. 2016, 173, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Turer, A.T.; Addo, T.A.; Martin, J.L.; Sabatine, M.S.; Lewis, G.D.; Gerszten, R.E.; Keeley, E.C.; Cigarroa, J.E.; Lange, R.A.; Hillis, L.D.; et al. Myocardial ischemia induced by rapid atrial pacing causes troponin T release detectable by a highly sensitive assay: Insights from a coronary sinus sampling study. J. Am. Coll. Cardiol. 2011, 57, 2398–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKie, P.M.; Heublein, D.M.; Scott, C.G.; Gantzer, M.L.; Mehta, R.A.; Rodeheffer, R.J.; Redfield, M.M.; Burnett, J.C., Jr.; Jaffe, A.S. Defining high-sensitivity cardiac troponin concentrations in the community. Clin. Chem. 2013, 59, 1099–1107. [Google Scholar] [CrossRef]
- Collinson, P.O.; Heung, Y.M.; Gaze, D.; Boa, F.; Senior, R.; Christenson, R.; Apple, F.S. Influence of population selection on the 99th percentile reference value for cardiac troponin assays. Clin. Chem. 2012, 58, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Ford, I.; Shah, A.S.; Zhang, R.; McAllister, D.A.; Strachan, F.E.; Caslake, M.; Newby, D.E.; Packard, C.J.; Mills, N.L. High-Sensitivity Cardiac Troponin, Statin Therapy, and Risk of Coronary Heart Disease. J. Am. Coll. Cardiol. 2016, 68, 2719–2728. [Google Scholar] [CrossRef] [PubMed]
- Farmakis, D.; Mueller, C.; Apple, F.S. High-sensitivity cardiac troponin assays for cardiovascular risk stratification in the general population. Eur. Heart J. 2020, 41, 4050–4056. [Google Scholar] [CrossRef] [PubMed]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Navarra, T.; Del Turco, S.; Berti, S.; Basta, G. The lectin-like oxidized low-density lipoprotein receptor-1 and its soluble form: Cardiovascular implications. J. Atheroscler. Thromb. 2010, 17, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Dunn, S.; Vohra, R.S.; Murphy, J.E.; Homer-Vanniasinkam, S.; Walker, J.H.; Ponnambalam, S. The lectin-like oxidized low-density-lipoprotein receptor: A pro-inflammatory factor in vascular disease. Biochem. J. 2008, 409, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, D.J.; Seese, R.; Ponnambalam, S.; Ajjan, R. The role of lectin-like oxidised low-density lipoprotein receptor-1 in vascular pathology. Diab. Vasc. Dis. Res. 2014, 11, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Liu, S.; Wang, X.; Dai, Y.; Khaidakov, M.; Deng, X.; Fan, Y.; Xiang, D.; Mehta, J.L. LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: Implications in atherogenesis. Cardiovasc. Res. 2014, 103, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.C.; Chang, P.Y.; Lu, S.C. L5-LDL from ST-elevation myocardial infarction patients induces IL-1beta production via LOX-1 and NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H265–H274. [Google Scholar] [CrossRef] [Green Version]
- Kattoor, A.J.; Goel, A.; Mehta, J.L. LOX-1: Regulation, Signaling and Its Role in Atherosclerosis. Antioxidants 2019, 8, 218. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, H.; Kume, N.; Miyamoto, S.; Minami, M.; Moriwaki, H.; Murase, T.; Sawamura, T.; Masaki, T.; Hashimoto, N.; Kita, T. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation 1999, 99, 3110–3117. [Google Scholar] [CrossRef]
- Li, D.; Patel, A.R.; Klibanov, A.L.; Kramer, C.M.; Ruiz, M.; Kang, B.Y.; Mehta, J.L.; Beller, G.A.; Glover, D.K.; Meyer, C.H. Molecular imaging of atherosclerotic plaques targeted to oxidized LDL receptor LOX-1 by SPECT/CT and magnetic resonance. Circ. Cardiovasc. Imaging 2010, 3, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Q.; Zhang, M.W.; Wang, F.; Zhao, Y.X.; Li, J.J.; Wang, X.P.; Bu, P.L.; Yang, J.M.; Liu, X.L.; Zhang, M.X.; et al. CRP enhances soluble LOX-1 release from macrophages by activating TNF-alpha converting enzyme. J. Lipid Res. 2011, 52, 923–933. [Google Scholar] [CrossRef] [Green Version]
- Mitsuoka, H.; Kume, N.; Hayashida, K.; Inui-Hayashiada, A.; Aramaki, Y.; Toyohara, M.; Jinnai, T.; Nishi, E.; Kita, T. Interleukin 18 stimulates release of soluble lectin-like oxidized LDL receptor-1 (sLOX-1). Atherosclerosis 2009, 202, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, N.; Mitsuoka, H.; Hayashida, K.; Tanaka, M.; Kominami, G.; Kita, T. Soluble lectin-like oxidized LDL receptor-1 (sLOX-1) as a sensitive and specific biomarker for acute coronary syndrome--comparison with other biomarkers. J. Cardiol. 2010, 56, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashida, K.; Kume, N.; Murase, T.; Minami, M.; Nakagawa, D.; Inada, T.; Tanaka, M.; Ueda, A.; Kominami, G.; Kambara, H.; et al. Serum soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are elevated in acute coronary syndrome: A novel marker for early diagnosis. Circulation 2005, 112, 812–818. [Google Scholar] [CrossRef]
- Kobayashi, N.; Hata, N.; Kume, N.; Seino, Y.; Inami, T.; Yokoyama, S.; Shinada, T.; Tomita, K.; Kaneshige, T.; Mizuno, K. Soluble lectin-like oxidized low-density lipoprotein receptor-1 as an early biomarker for ST elevation myocardial infarction: Time-dependent comparison with other biomarkers: Time-dependent comparison with other biomarkers. Circ. J. 2011, 75, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Takano, M.; Hata, N.; Kume, N.; Yamamoto, M.; Yokoyama, S.; Shinada, T.; Tomita, K.; Shirakabe, A.; Otsuka, T.; et al. Soluble lectin-like oxidized LDL receptor-1 (sLOX-1) as a valuable diagnostic marker for rupture of thin-cap fibroatheroma: Verification by optical coherence tomography. Int. J. Cardiol. 2013, 168, 3217–3223. [Google Scholar] [CrossRef] [PubMed]
- Balin, M.; Celik, A.; Kobat, M.A. Circulating soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are associated with proximal/middle segment of the LAD lesions in patients with stable coronary artery disease. Clin. Res. Cardiol. 2012, 101, 247–253. [Google Scholar] [CrossRef]
- Zhao, Z.W.; Zhu, X.L.; Luo, Y.K.; Lin, C.G.; Chen, L.L. Circulating soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are associated with angiographic coronary lesion complexity in patients with coronary artery disease. Clin. Cardiol. 2011, 34, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, L.H.; Yang, X.G.; Liu, Y.; Liu, X.T.; Ren, Y.G. Postprocedural serum sLOX-1 levels are associated with coronary in-stent restenosis in patients with stable coronary artery disease. Coron. Artery Dis. 2011, 22, 259–263. [Google Scholar] [CrossRef]
- Balin, M.; Celik, A.; Kobat, M.A.; Baydas, A. Circulating soluble lectin-like oxidized low-density lipoprotein receptor-1 levels predict percutaneous coronary intervention-related periprocedural myocardial infarction in stable patients undergoing elective native single-vessel PCI. J. Thromb. Thrombolysis 2012, 34, 483–490. [Google Scholar] [CrossRef]
- Kume, N.; Mitsuoka, H.; Hayashida, K.; Tanaka, M.; Kita, T. Soluble lectin-like oxidized low-density lipoprotein receptor-1 predicts prognosis after acute coronary syndrome—A pilot study. Circ. J. 2010, 74, 1399–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bold, A.J.; Borenstein, H.B.; Veress, A.T.; Sonnenberg, H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sci. 1981, 28, 89–94. [Google Scholar] [CrossRef]
- de Bold, A.J. Atrial natriuretic factor: A hormone produced by the heart. Science 1985, 230, 767–770. [Google Scholar] [CrossRef]
- Burnett, J.C., Jr.; Kao, P.C.; Hu, D.C.; Heser, D.W.; Heublein, D.; Granger, J.P.; Opgenorth, T.J.; Reeder, G.S. Atrial natriuretic peptide elevation in congestive heart failure in the human. Science 1986, 231, 1145–1147. [Google Scholar] [CrossRef]
- Brandt, R.R.; Wright, R.S.; Redfield, M.M.; Burnett, J.C. Atrial natriuretic peptide in heart failure. J. Am. Coll. Cardiol. 1993, 22, A86–A92. [Google Scholar] [CrossRef] [Green Version]
- Sudoh, T.; Kangawa, K.; Minamino, N.; Matsuo, H. A new natriuretic peptide in porcine brain. Nature 1988, 332, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakao, K.; Itoh, H.; Yamada, T.; Mukoyama, M.; Arai, H.; Hosoda, K.; Shirakami, G.; Suga, S.-I.; Minamino, N.; et al. Brain natriuretic peptide is a novel cardiac hormone. Biochem. Biophys. Res. Commun. 1989, 158, 360–368. [Google Scholar] [CrossRef]
- Ogawa, Y.; Nakao, K.; Mukoyama, M.; Shirakami, G.; Itoh, H.; Hosoda, K.; Saito, Y.; Arai, H.; Suga, S.; Jougasaki, M.; et al. Rat brain natriuretic peptide--tissue distribution and molecular form. Endocrinology 1990, 126, 2225–2227. [Google Scholar] [CrossRef]
- Kambayashi, Y.; Nakao, K.; Mukoyama, M.; Saito, Y.; Ogawa, Y.; Shiono, S.; Inouye, K.; Yoshida, N.; Imura, H. Isolation and sequence determination of human brain natriuretic peptide in human atrium. FEBS Lett. 1990, 259, 341–345. [Google Scholar] [CrossRef] [Green Version]
- Mukoyama, M.; Nakao, K.; Hosoda, K.; Suga, S.; Saito, Y.; Ogawa, Y.; Shirakami, G.; Jougasaki, M.; Obata, K.; Yasue, H. Brain natriuretic peptide as a novel cardiac hormone in humans. Evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J. Clin. Investig. 1991, 87, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Yasue, H.; Yoshimura, M.; Sumida, H.; Kikuta, K.; Kugiyama, K.; Jougasaki, M.; Ogawa, H.; Okumura, K.; Mukoyama, M.; Nakao, K. Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation 1994, 90, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowie, M.R.; Struthers, A.D.; Wood, D.A.; Coats, A.J.; Thompson, S.G.; Poole-Wilson, P.A.; Sutton, G.C. Value of natriuretic peptides in assessment of patients with possible new heart failure in primary care. Lancet 1997, 350, 1349–1353. [Google Scholar] [CrossRef]
- Doust, J.A.; Glasziou, P.P.; Pietrzak, E.; Dobson, A.J. A Systematic Review of the Diagnostic Accuracy of Natriuretic Peptides for Heart Failure. Arch. Intern. Med. 2004, 164, 1978–1984. [Google Scholar] [CrossRef]
- Jensen, K.T.; Carstens, J.; Ivarsen, P.; Pedersen, E.B. A new, fast and reliable radioimmunoassay of brain natriuretic peptide in human plasma. Reference values in healthy subjects and in patients with different diseases. Scand. J. Clin. Lab. Investig. 1997, 57, 529–540. [Google Scholar] [CrossRef]
- Maisel, A.S.; Krishnaswamy, P.; Nowak, R.M.; McCord, J.; Hollander, J.E.; Duc, P.; Omland, T.; Storrow, A.B.; Abraham, W.T.; Wu, A.H.; et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N. Engl. J. Med. 2002, 347, 161–167. [Google Scholar] [CrossRef]
- Nakamura, M.; Endo, H.; Nasu, M.; Arakawa, N.; Segawa, T.; Hiramori, K. Value of plasma B type natriuretic peptide measurement for heart disease screening in a Japanese population. Heart 2002, 87, 131–135. [Google Scholar] [CrossRef] [Green Version]
- McDonagh, T.A.; Robb, S.D.; Murdoch, D.R.; Morton, J.J.; Ford, I.; Morrison, C.E.; Tunstall-Pedoe, H.; McMurray, J.J.; Dargie, H.J. Biochemical detection of left-ventricular systolic dysfunction. Lancet 1998, 351, 9–13. [Google Scholar] [CrossRef]
- Ng, L.L.; Loke, I.; Davies, J.E.; Khunti, K.; Stone, M.; Abrams, K.R.; Chin, D.T.; Squire, I.B. Identification of previously undiagnosed left ventricular systolic dysfunction: Community screening using natriuretic peptides and electrocardiography. Eur. J. Heart Fail. 2003, 5, 775–782. [Google Scholar] [CrossRef] [Green Version]
- Mair, J.; Friedl, W.; Thomas, S.; Puschendorf, B. Natriuretic peptides in assessment of left-ventricular dysfunction. Scand. J. Clin. Lab. Investig. 1999, 59, 132–142. [Google Scholar] [CrossRef]
- Hammerer-Lercher, A.; Neubauer, E.; Müller, S.; Pachinger, O.; Puschendorf, B.; Mair, J. Head-to-head comparison of N-terminal pro-brain natriuretic peptide, brain natriuretic peptide and N-terminal pro-atrial natriuretic peptide in diagnosing left ventricular dysfunction. Clin. Chim. Acta 2001, 310, 193–197. [Google Scholar] [CrossRef]
- Richards, A.M.; Nicholls, M.G.; Yandle, T.G.; Frampton, C.; Espiner, E.A.; Turner, J.G.; Buttimore, R.C.; Lainchbury, J.G.; Elliott, J.M.; Ikram, H.; et al. Plasma N-terminal pro brain natriuretic peptide and adrenomedullin: New neurohormonal predictors of left ventricular function and prognosis after myocardial infarction. Circulation 1998, 97, 1921–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Januzzi, J.L.; van Kimmenade, R.; Lainchbury, J.; Bayes-Genis, A.; Ordonez-Llanos, J.; Santalo-Bel, M.; Pinto, Y.M.; Richards, M. NT-proBNP testing for diagnosis and short-term prognosis in acute destabilized heart failure: An international pooled analysis of 1256 patients: The International Collaborative of NT-proBNP Study. Eur. Heart J. 2006, 27, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Lainchbury, J.G.; Campbell, E.; Frampton, C.M.; Yandle, T.G.; Nicholls, M.G.; Richards, A.M. Brain natriuretic peptide and n-terminal brain natriuretic peptide in the diagnosis of heart failure in patients with acute shortness of breath. J. Am. Coll. Cardiol. 2003, 42, 728–735. [Google Scholar] [CrossRef] [Green Version]
- Bayés-Genís, A.; Santaló-Bel, M.; Zapico-Muñiz, E.; López, L.; Cotes, C.; Bellido, J.; Leta, R.; Casan, P.; Ordóñez-Llanos, J. N-terminal probrain natriuretic peptide (NT-proBNP) in the emergency diagnosis and in-hospital monitoring of patients with dyspnoea and ventricular dysfunction. Eur. J. Heart Fail. 2004, 6, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.A.; Wood, M.J.; Krauser, D.G.; Baggish, A.L.; Tung, R.; Anwaruddin, S.; Picard, M.H.; Januzzi, J.L. NT-proBNP levels, echocardiographic findings, and outcomes in breathless patients: Results from the ProBNP Investigation of Dyspnoea in the Emergency Department (PRIDE) echocardiographic substudy. Eur. Heart J. 2006, 27, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Richards, A.M.; Doughty, R.; Nicholls, M.G.; MacMahon, S.; Sharpe, N.; Murphy, J.; Espiner, E.A.; Frampton, C.; Yandle, T.G. Plasma N-terminal pro-brain natriuretic peptide and adrenomedullin: Prognostic utility and prediction of benefit from carvedilol in chronic ischemic left ventricular dysfunction. Australia-New Zealand Heart Failure Group. J. Am. Coll. Cardiol. 2001, 37, 1781–1787. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, F.; Packer, M.; Coats, A.J.; Fowler, M.B.; Krum, H.; Mohacsi, P.; Rouleau, J.L.; Tendera, M.; Castaigne, A.; Anker, S.D.; et al. Prognostic impact of plasma N-terminal pro-brain natriuretic peptide in severe chronic congestive heart failure: A substudy of the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) trial. Circulation 2004, 110, 1780–1786. [Google Scholar] [CrossRef] [Green Version]
- Bettencourt, P.; Azevedo, A.; Pimenta, J.; Friões, F.; Ferreira, S.; Ferreira, A. N-terminal-pro-brain natriuretic peptide predicts outcome after hospital discharge in heart failure patients. Circulation 2004, 110, 2168–2174. [Google Scholar] [CrossRef] [Green Version]
- Schou, M.; Gustafsson, F.; Corell, P.; Kistorp, C.N.; Kjaer, A.; Hildebrandt, P.R. The relationship between N-terminal pro-brain natriuretic peptide and risk for hospitalization and mortality is curvilinear in patients with chronic heart failure. Am. Heart J. 2007, 154, 123–129. [Google Scholar] [CrossRef]
- Hunt, P.J.; Richards, A.M.; Nicholls, M.G.; Yandle, T.G.; Doughty, R.N.; Espiner, E.A. Immunoreactive amino-terminal pro-brain natriuretic peptide (NT-PROBNP): A new marker of cardiac impairment. Clin. Endocrinol. 1997, 47, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Mair, J. Biochemistry of B-type natriuretic peptide--where are we now? Clin. Chem. Lab. Med. 2008, 46, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Rørth, R.; Jhund, P.S.; Yilmaz, M.B.; Kristensen, S.L.; Welsh, P.; Desai, A.S.; Køber, L.; Prescott, M.F.; Rouleau, J.L.; Solomon, S.D.; et al. Comparison of BNP and NT-proBNP in Patients with Heart Failure and Reduced Ejection Fraction. Circ. Heart Fail. 2020, 13, e006541. [Google Scholar] [CrossRef] [Green Version]
- Atherton, J.J.; Sindone, A.; De Pasquale, C.G.; Driscoll, A.; MacDonald, P.S.; Hopper, I.; Kistler, P.M.; Briffa, T.; Wong, J.; Abhayaratna, W.; et al. National Heart Foundation of Australia and Cardiac Society of Australia and New Zealand: Guidelines for the Prevention, Detection, and Management of Heart Failure in Australia 2018. Heart Lung. Circ. 2018, 27, 1123–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Suthahar, N.; Meems, L.M.G.; Ho, J.E.; de Boer, R.A. Sex-related differences in contemporary biomarkers for heart failure: A review. Eur. J. Heart Fail. 2020, 22, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Felker, G.M.; Hasselblad, V.; Hernandez, A.F.; O’Connor, C.M. Biomarker-guided therapy in chronic heart failure: A meta-analysis of randomized controlled trials. Am. Heart J. 2009, 158, 422–430. [Google Scholar] [CrossRef]
- Pfisterer, M.; Buser, P.; Rickli, H.; Gutmann, M.; Erne, P.; Rickenbacher, P.; Vuillomenet, A.; Jeker, U.; Dubach, P.; Beer, H.; et al. BNP-Guided vs Symptom-Guided Heart Failure Therapy: The Trial of Intensified vs Standard Medical Therapy in Elderly Patients with Congestive Heart Failure (TIME-CHF) Randomized Trial. JAMA 2009, 301, 383–392. [Google Scholar] [CrossRef]
- Felker, G.M.; Anstrom, K.J.; Adams, K.F.; Ezekowitz, J.A.; Fiuzat, M.; Houston-Miller, N.; Januzzi, J.L., Jr.; Mark, D.B.; Piña, I.L.; Passmore, G.; et al. Effect of Natriuretic Peptide–Guided Therapy on Hospitalization or Cardiovascular Mortality in High-Risk Patients with Heart Failure and Reduced Ejection Fraction: A Randomized Clinical Trial. JAMA 2017, 318, 713–720. [Google Scholar] [CrossRef]
- Sharma, U.C.; Pokharel, S.; van Brakel, T.J.; van Berlo, J.H.; Cleutjens, J.P.; Schroen, B.; Andre, S.; Crijns, H.J.; Gabius, H.J.; Maessen, J.; et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004, 110, 3121–3128. [Google Scholar] [CrossRef]
- de Boer, R.A.; Lok, D.J.; Jaarsma, T.; van der Meer, P.; Voors, A.A.; Hillege, H.L.; van Veldhuisen, D.J. Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Ann. Med. 2011, 43, 60–68. [Google Scholar] [CrossRef]
- van Kimmenade, R.R.; Januzzi, J.L., Jr.; Ellinor, P.T.; Sharma, U.C.; Bakker, J.A.; Low, A.F.; Martinez, A.; Crijns, H.J.; MacRae, C.A.; Menheere, P.P.; et al. Utility of amino-terminal pro-brain natriuretic peptide, galectin-3, and apelin for the evaluation of patients with acute heart failure. J. Am. Coll. Cardiol. 2006, 48, 1217–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, D.J.; Van Der Meer, P.; de la Porte, P.W.; Lipsic, E.; Van Wijngaarden, J.; Hillege, H.L.; van Veldhuisen, D.J. Prognostic value of galectin-3, a novel marker of fibrosis, in patients with chronic heart failure: Data from the DEAL-HF study. Clin. Res. Cardiol. 2010, 99, 323–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, T.F.; Shin, H.J.; Mathenge, N.; Wang, F.; Kim, B.; Joseph, J.; Gaziano, J.M.; Djousse, L. Meta-Analysis of the Usefulness of Plasma Galectin-3 to Predict the Risk of Mortality in Patients with Heart Failure and in the General Population. Am. J. Cardiol 2017, 119, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Meijers, W.C.; Januzzi, J.L.; deFilippi, C.; Adourian, A.S.; Shah, S.J.; van Veldhuisen, D.J.; de Boer, R.A. Elevated plasma galectin-3 is associated with near-term rehospitalization in heart failure: A pooled analysis of 3 clinical trials. Am. Heart J. 2014, 167, 853–860.e854. [Google Scholar] [CrossRef] [Green Version]
- French, B.; Wang, L.; Ky, B.; Brandimarto, J.; Basuray, A.; Fang, J.C.; Sweitzer, N.K.; Cappola, T.P. Prognostic Value of Galectin-3 for Adverse Outcomes in Chronic Heart Failure. J. Card Fail. 2016, 22, 256–262. [Google Scholar] [CrossRef] [Green Version]
- van Vark, L.C.; Lesman-Leegte, I.; Baart, S.J.; Postmus, D.; Pinto, Y.M.; de Boer, R.A.; Asselbergs, F.W.; Wajon, E.; Orsel, J.G.; Boersma, E.; et al. Prognostic Value of Serial Galectin-3 Measurements in Patients with Acute Heart Failure. J. Am. Heart Assoc. 2017, 6, e003700. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, Q.; Li, Y.; Jing, X.; Liang, T.; Yang, J. Prognostic value of galectin-3 on admission in Chinese patients with heart failure: A prospective observational study. Acta Cardiol. 2017, 72, 188–195. [Google Scholar] [CrossRef]
- Yu, X.; Sun, Y.; Zhao, Y.; Zhang, W.; Yang, Z.; Gao, Y.; Cai, H.; Li, Y.; Wang, Q.; Bian, B.; et al. Prognostic value of plasma galectin-3 levels in patients with coronary heart disease and chronic heart failure. Int. Heart J. 2015, 56, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-j.; Hao, W.-R.; Chang, K.-C.; Liu, J.-C. LBOS 02-03 the infiltrating macrophage-secreted galectin-3 plays an essential role in cardiac fibrosis and diastolic function in murine pressure-overload model. J. Hypertens. 2016, 34, e549. [Google Scholar] [CrossRef]
- Gonzalez, G.E.; Rhaleb, N.E.; D’Ambrosio, M.A.; Nakagawa, P.; Liao, T.D.; Peterson, E.L.; Leung, P.; Dai, X.; Janic, B.; Liu, Y.H.; et al. Cardiac-deleterious role of galectin-3 in chronic angiotensin II-induced hypertension. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1287–H1296. [Google Scholar] [CrossRef] [Green Version]
- Hsu, D.K.; Dowling, C.A.; Jeng, K.C.G.; Chen, J.T.; Yang, R.Y.; Liu, F.T. Galectin-3 expression is induced in cirrhotic liver and hepatocellular carcinoma. Int. J. Cancer 1999, 81, 519–526. [Google Scholar] [CrossRef]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Poirier, F.; Russo, F.P.; Iredale, J.P.; Haslett, C.; Simpson, K.J.; Sethi, T. Galectin-3 regulates myofibroblast activation and hepatic fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 5060–5065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, Y.; Sano, H.; Kawashima, T.; Okada, T.; Kuroda, T.; Kikkawa, K.; Kawashima, S.; Tanabe, M.; Goto, T.; Matsuzawa, Y.; et al. Role of Galectin-3 in Human Pulmonary Fibrosis. Allergol. Int. 2007, 56, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.E.; Liu, C.; Lyass, A.; Courchesne, P.; Pencina, M.J.; Vasan, R.S.; Larson, M.G.; Levy, D. Galectin-3, a Marker of Cardiac Fibrosis, Predicts Incident Heart Failure in the Community. J. Am. Coll. Cardiol. 2012, 60, 1249–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, S. A putative protein of a growth specific cDNA from BALB/c-3T3 cells is highly similar to the extracellular portion of mouse interleukin 1 receptor. FEBS Lett. 1989, 258, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Figal, D.A.; Januzzi, J.L. The Biology of ST2: The International ST2 Consensus Panel. Am. J. Cardiol. 2015, 115, 3B–7B. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an Interleukin-1-like Cytokine that Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Kakkar, R.; Hei, H.; Dobner, S.; Lee, R.T. Interleukin 33 as a Mechanically Responsive Cytokine Secreted by Living Cells. J. Biol. Chem. 2012, 287, 6941–6948. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, E.O.; Shimpo, M.; De Keulenaer, G.W.; MacGillivray, C.; Tominaga, S.-I.; Solomon, S.D.; Rouleau, J.-L.; Lee, R.T. Expression and Regulation of ST2, an Interleukin-1 Receptor Family Member, in Cardiomyocytes and Myocardial Infarction. Circulation 2002, 106, 2961–2966. [Google Scholar] [CrossRef] [Green Version]
- Sanada, S.; Hakuno, D.; Higgins, L.J.; Schreiter, E.R.; McKenzie, A.N.J.; Lee, R.T. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J. Clin. Investig. 2007, 117, 1538–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, K.; Sanada, S.; Kudinova, A.Y.; Steinhauser, M.L.; Handa, V.; Gannon, J.; Lee, R.T. Interleukin-33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ. Heart Fail. 2009, 2, 684–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, R.A.P.; Miller, A.M.; Murphy, G.E.J.; Clements, S.; Steedman, T.; Connell, J.M.C.; McInnes, I.B.; Dargie, H.J.; McMurray, J.J.V. Serum Soluble ST2: A Potential Novel Mediator in Left Ventricular and Infarct Remodeling After Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2010, 55, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.V.; Chen-Tournoux, A.A.; Picard, M.H.; Van Kimmenade, R.R.J.; Januzzi, J.L. Serum levels of the interleukin-1 receptor family member ST2, cardiac structure and function, and long-term mortality in patients with acute dyspnea. Circ. Heart Fail. 2009, 2, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Januzzi, J.L., Jr.; Rehman, S.; Mueller, T.; van Kimmenade, R.R.J.; Lloyd-Jones, D.M. Importance of Biomarkers for Long-Term Mortality Prediction in Acutely Dyspneic Patients. Clin. Chem. 2010, 56, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; Daniels, L.B.; Maisel, A.S.; Januzzi, J.L., Jr. State of the Art: Newer biomarkers in heart failure. Eur. J. Heart Fail. 2015, 17, 559–569. [Google Scholar] [CrossRef]
- Shah, R.V.; Januzzi, J.L. Soluble ST2 and Galectin-3 in Heart Failure. Clin. Lab. Med. 2014, 34, 87–97. [Google Scholar] [CrossRef]
- Weinberg, E.O.; Shimpo, M.; Hurwitz, S.; Tominaga, S.I.; Rouleau, J.L.; Lee, R.T. Identification of serum soluble ST2 receptor as a novel heart failure biomarker. Circulation 2003, 107, 721–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayes-Genis, A.; Zhang, Y.; Ky, B. ST2 and Patient Prognosis in Chronic Heart Failure. Am. J. Cardiol. 2015, 115, 64B–69B. [Google Scholar] [CrossRef]
- Aimo, A.; Vergaro, G.; Passino, C.; Ripoli, A.; Ky, B.; Miller, W.L.; Bayes-Genis, A.; Anand, I.; Januzzi, J.L.; Emdin, M. Prognostic Value of Soluble Suppression of Tumorigenicity-2 in Chronic Heart Failure: A Meta-Analysis. JACC Heart Fail. 2017, 5, 280–286. [Google Scholar] [CrossRef]
- Januzzi, J.L.; Peacock, W.F.; Maisel, A.S.; Chae, C.U.; Jesse, R.L.; Baggish, A.L.; O’Donoghue, M.; Sakhuja, R.; Chen, A.A.; van Kimmenade, R.R.J.; et al. Measurement of the Interleukin Family Member ST2 in Patients with Acute Dyspnea: Results from the PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) Study. J. Am. Coll. Cardiol. 2007, 50, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzano-Fernández, S.; Mueller, T.; Pascual-Figal, D.; Truong, Q.A.; Januzzi, J.L. Usefulness of Soluble Concentrations of Interleukin Family Member ST2 as Predictor of Mortality in Patients with Acutely Decompensated Heart Failure Relative to Left Ventricular Ejection Fraction. Am. J. Cardiol. 2011, 107, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, M.F.; Appelbaum, S.; Havulinna, A.S.; Jagodzinski, A.; Zeller, T.; Kee, F.; Blankenberg, S.; Salomaa, V. ST2 may not be a useful predictor for incident cardiovascular events, heart failure and mortality. Heart 2014, 100, 1715. [Google Scholar] [CrossRef] [PubMed]
- Lassus, J.; Gayat, E.; Mueller, C.; Peacock, W.F.; Spinar, J.; Harjola, V.-P.; van Kimmenade, R.; Pathak, A.; Mueller, T.; diSomma, S.; et al. Incremental value of biomarkers to clinical variables for mortality prediction in acutely decompensated heart failure: The Multinational Observational Cohort on Acute Heart Failure (MOCA) study. Int. J. Cardiol. 2013, 168, 2186–2194. [Google Scholar] [CrossRef]
- Gaggin, H.K.; Motiwala, S.; Bhardwaj, A.; Parks, K.A.; Januzzi, J.L. Soluble Concentrations of the Interleukin Receptor Family Member ST2 and β-Blocker Therapy in Chronic Heart Failure. Circ. Heart Fail. 2013, 6, 1206–1213. [Google Scholar] [CrossRef] [Green Version]
- Anand, I.S.; Rector, T.S.; Kuskowski, M.; Snider, J.; Cohn, J.N. Prognostic Value of Soluble ST2 in the Valsartan Heart Failure Trial. Circ. Heart Fail. 2014, 7, 418–426. [Google Scholar] [CrossRef] [Green Version]
- Maisel, A.; Xue, Y.; van Veldhuisen, D.J.; Voors, A.A.; Jaarsma, T.; Pang, P.S.; Butler, J.; Pitt, B.; Clopton, P.; de Boer, R.A. Effect of Spironolactone on 30-Day Death and Heart Failure Rehospitalization (from the COACH Study). Am. J. Cardiol. 2014, 114, 737–742. [Google Scholar] [CrossRef]
- Rehman, S.U.; Mueller, T.; Januzzi, J.L. Characteristics of the Novel Interleukin Family Biomarker ST2 in Patients with Acute Heart Failure. J. Am. Coll. Cardiol. 2008, 52, 1458–1465. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; de Antonio, M.; Vila, J.; Peñafiel, J.; Galán, A.; Barallat, J.; Zamora, E.; Urrutia, A.; Lupón, J. Head-to-Head Comparison of 2 Myocardial Fibrosis Biomarkers for Long-Term Heart Failure Risk Stratification. J. Am. Coll. Cardiol. 2014, 63, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Gaggin Hanna, K.; Szymonifka, J.; Bhardwaj, A.; Belcher, A.; De Berardinis, B.; Motiwala, S.; Wang Thomas, J.; Januzzi James, L. Head-to-Head Comparison of Serial Soluble ST2, Growth Differentiation Factor-15, and Highly-Sensitive Troponin T Measurements in Patients with Chronic Heart Failure. JACC Heart Fail. 2014, 2, 65–72. [Google Scholar] [CrossRef]
- Ahmad, T.; Fiuzat, M.; Neely, B.; Neely Megan, L.; Pen.ncina Michael, J.; Kraus William, E.; Zannad, F.; Whellan David, J.; Donahue Mark, P.; Piña Ileana, L.; et al. Biomarkers of Myocardial Stress and Fibrosis as Predictors of Mode of Death in Patients with Chronic Heart Failure. JACC Heart Fail. 2014, 2, 260–268. [Google Scholar] [CrossRef]
- Yancy Clyde, W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey Donald, E.; Colvin Monica, M.; Drazner Mark, H.; Filippatos Gerasimos, S.; Fonarow Gregg, C.; Givertz Michael, M.; et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure. J. Am. Coll. Cardiol. 2017, 70, 776–803. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- de Beer, F.C.; Hind, C.R.; Fox, K.M.; Allan, R.M.; Maseri, A.; Pepys, M.B. Measurement of serum C-reactive protein concentration in myocardial ischaemia and infarction. Br. Heart J. 1982, 47, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, Aspirin, and the Risk of Cardiovascular Disease in Apparently Healthy Men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Ridker, P.M.; Hennekens, C.H.; Buring, J.E.; Rifai, N. C-Reactive Protein and Other Markers of Inflammation in the Prediction of Cardiovascular Disease in Women. N. Engl. J. Med. 2000, 342, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Koenig, W.; Sund, M.; Fröhlich, M.; Fischer, H.-G.; Löwel, H.; Döring, A.; Hutchinson, W.L.; Pepys, M.B. C-Reactive Protein, a Sensitive Marker of Inflammation, Predicts Future Risk of Coronary Heart Disease in Initially Healthy Middle-Aged Men. Circulation 1999, 99, 237–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rost, N.S.; Wolf, P.A.; Kase, C.S.; Kelly-Hayes, M.; Silbershatz, H.; Massaro, J.M.; D’Agostino, R.B.; Franzblau, C.; Wilson, P.W.F. Plasma Concentration of C-Reactive Protein and Risk of Ischemic Stroke and Transient Ischemic Attack. Stroke 2001, 32, 2575–2579. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.H.; Genest, J.; Antonio, M.; Gotto, J.; Kastelein, J.J.P.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; et al. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Buhaescu, I.; Izzedine, H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.A.; Mensah, G.A.; Alexander, R.W.; Anderson, J.L.; Cannon, R.O., 3rd; Criqui, M.; Fadl, Y.Y.; Fortmann, S.P.; Hong, Y.; Myers, G.L.; et al. Markers of inflammation and cardiovascular disease: Application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003, 107, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.L. CDC/AHA Workshop on Markers of Inflammation and Cardiovascular Disease: Application to Clinical and Public Health Practice: Laboratory tests available to assess inflammation--performance and standardization: A background paper. Circulation 2004, 110, e572–e576. [Google Scholar] [CrossRef] [Green Version]
- Danesh, J.; Collins, R.; Appleby, P.; Peto, R. Association of Fibrinogen, C-reactive Protein, Albumin, or Leukocyte Count with Coronary Heart DiseaseMeta-analyses of Prospective Studies. JAMA 1998, 279, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Danesh, J.; Whincup, P.; Walker, M.; Lennon, L.; Thomson, A.; Appleby, P.; Gallimore, J.R.; Pepys, M.B. Low grade inflammation and coronary heart disease: Prospective study and updated meta-analyses. BMJ 2000, 321, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Koenig, W.; Löwel, H.; Baumert, J.; Meisinger, C. C-Reactive Protein Modulates Risk Prediction Based on the Framingham Score. Circulation 2004, 109, 1349–1353. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Rifai, N.; Rose, L.; Buring, J.E.; Cook, N.R. Comparison of C-Reactive Protein and Low-Density Lipoprotein Cholesterol Levels in the Prediction of First Cardiovascular Events. N. Engl. J. Med. 2002, 347, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Wright, S.P.; Doughty, R.N.; Pearl, A.; Gamble, G.D.; Whalley, G.A.; Walsh, H.J.; Gordon, G.; Bagg, W.; Oxenham, H.; Yandle, T.; et al. Plasma amino-terminal pro-brain natriuretic peptide and accuracy of heart-failure diagnosis in primary care: A randomized, controlled trial. J. Am. Coll. Cardiol. 2003, 42, 1793–1800. [Google Scholar] [CrossRef] [Green Version]
- Januzzi, J.L.; Camargo, C.A.; Anwaruddin, S.; Baggish, A.L.; Chen, A.A.; Krauser, D.G.; Tung, R.; Cameron, R.; Nagurney, J.T.; Chae, C.U.; et al. The N-terminal Pro-BNP Investigation of Dyspnea in the Emergency department (PRIDE) study. Am. J. Cardiol. 2005, 95, 948–954. [Google Scholar] [CrossRef]
- Dieplinger, B.; Januzzi, J.L.; Steinmair, M.; Gabriel, C.; Poelz, W.; Haltmayer, M.; Mueller, T. Analytical and clinical evaluation of a novel high-sensitivity assay for measurement of soluble ST2 in human plasma—The Presage™ ST2 assay. Clin. Chim. Acta 2009, 409, 33–40. [Google Scholar] [CrossRef]
- Januzzi, J.L.; Mebazaa, A.; Di Somma, S. ST2 and Prognosis in Acutely Decompensated Heart Failure: The International ST2 Consensus Panel. Am. J. Cardiol. 2015, 115, 26B–31B. [Google Scholar] [CrossRef] [PubMed]
- Maisel, A.S.; Di Somma, S. Do we need another heart failure biomarker: Focus on soluble suppression of tumorigenicity 2 (sST2). Eur. Heart J. 2016, 38, 2325–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laaksonen, D.E.; Niskanen, L.; Nyyssönen, K.; Punnonen, K.; Tuomainen, T.P.; Salonen, J.T. C-reactive protein in the prediction of cardiovascular and overall mortality in middle-aged men: A population-based cohort study. Eur. Heart J. 2005, 26, 1783–1789. [Google Scholar] [CrossRef] [Green Version]
- Cabaniss, C.D. Creatine Kinase. In Clinical Methods: The History, Physical, and Laboratory Examinations; Walker, H.K., Hall, W.D., Hurst, J.W., Eds.; Butterworth Publishers; A Division of Reed Publishing: Boston, MA, USA, 1990. [Google Scholar]
- Kiely, P.D.W.; Bruckner, F.E.; Nisbet, J.A.; Daghir, A. Serum skeletal troponin I in inflammatory muscle disease: Relation to creatine kinase, CKMB and cardiac troponin I. Ann. Rheum. Dis. 2000, 59, 750. [Google Scholar] [CrossRef] [Green Version]
- Siegel, A.J.; Silverman, L.M.; Evans, W.J. Elevated skeletal muscle creatine kinase MB isoenzyme levels in marathon runners. JAMA 1983, 250, 2835–2837. [Google Scholar] [CrossRef] [PubMed]
- Ay, H.; Arsava, E.M.; Sarıbaş, O. Creatine Kinase-MB Elevation After Stroke Is Not Cardiac in Origin. Stroke 2002, 33, 286–289. [Google Scholar] [CrossRef] [Green Version]
- Ruppert, M.; Van Hee, R. Creatinine-kinase-MB determination in non-cardiac trauma: Its difference with cardiac infarction and its restricted use in trauma situations. Eur. J. Emerg. Med. 2001, 8, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.S.; Ritter, C.; Meltzer, V.; Harter, H.; Roberts, R. Unmasking artifactual increases in creatine kinase isoenzymes in patients with renal failure. J. Lab. Clin. Med. 1984, 104, 193–202. [Google Scholar]
- Medeiros, L.J.; Schotte, D.; Gerson, B. Reliability and significance of increased creatine kinase MB isoenzyme in the serum of uremic patients. Am. J. Clin Pathol. 1987, 87, 103–108. [Google Scholar] [CrossRef]
- Apple, F.S.; Quist, H.E.; Doyle, P.J.; Otto, A.P.; Murakami, M.M. Plasma 99th percentile reference limits for cardiac troponin and creatine kinase MB mass for use with European Society of Cardiology/American College of Cardiology consensus recommendations. Clin. Chem. 2003, 49, 1331–1336. [Google Scholar] [CrossRef] [Green Version]
- Lenke, L.G.; Bridwell, K.H.; Jaffe, A.S. Increase in creatine kinase MB isoenzyme levels after spinal surgery. J Spinal Disord. 1994, 7, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Kobayashi, N.; Hata, N.; Shinada, T.; Tomita, K.; Tsurumi, M.; Okazaki, H.; Matsushita, M.; Yamamoto, Y.; Yokoyama, S.; et al. The serum heart-type fatty acid-binding protein (HFABP) levels can be used to detect the presence of acute kidney injury on admission in patients admitted to the non-surgical intensive care unit. BMC Cardiovasc. Disord. 2016, 16, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, M.; Forkmann, M.; Richter, U.; Tausche, A.-K.; Sveric, K.; Christoph, M.; Ibrahim, K.; Günther, M.; Kolschmann, S.; Boscheri, A.; et al. Heart-type fatty acid-binding protein and myocardial creatine kinase enable rapid risk stratification in normotensive patients with pulmonary embolism. J. Crit. Care 2016, 35, 174–179. [Google Scholar] [CrossRef]
- Wunderlich, M.T.; Hanhoff, T.; Goertler, M.; Spener, F.; Glatz, J.F.C.; Wallesch, C.W.; Pelsers, M.M.A.L. Release of brain–type and heart–type fatty acid–binding proteins in serum after acute ischaemic stroke. J. Neurol. 2005, 252, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-C.; Dai, H.-W.; Yu, Y.-H.; Yang, J.-D.; Hu, C.-B. Usefulness of heart-type fatty acid–binding protein in patients with severe sepsis. J. Crit. Care 2012, 27, 415.e413–415.e418. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, U.; Espeter, F.; Weiß, C.; Ahmad-Nejad, P.; Lang, S.; Brueckmann, M.; Akin, I.; Neumaier, M.; Borggrefe, M.; Behnes, M. Ischemic biomarker heart-type fatty acid binding protein (hFABP) in acute heart failure—Diagnostic and prognostic insights compared to NT-proBNP and troponin I. BMC Cardiovasc. Disord. 2015, 15, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Başar, O.; Akbal, E.; Köklü, S.; Tuna, Y.; Koçak, E.; Başar, N.; Tok, D.; Erbiş, H.; Senes, M. Increased H-FABP concentrations in nonalcoholic fatty liver disease. Possible marker for subclinical myocardial damage and subclinical atherosclerosis. Herz 2013, 38, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Aslani, M.R.; Ghobadi, H.; Sarikhani, K.; Hosseininia, S.; Sadeghieh-Ahari, S. Comparison of Serum Heart-Type Fatty Acid Binding Protein Levels in Stable Chronic Obstructive Pulmonary Disease and Healthy Subjects. Tanaffos 2020, 19, 208–215. [Google Scholar]
- Newby, L.K.; Goldmann, B.U.; Ohman, E.M. Troponin: An important prognostic marker and risk-stratification tool in non–ST-segment elevation acute coronary syndromes. J. Am. Coll. Cardiol. 2003, 41, S31–S36. [Google Scholar] [CrossRef] [Green Version]
- Lewandrowski, K.; Chen, A.; Januzzi, J. Cardiac Markers for Myocardial Infarction: A Brief Review. Pathol. Patterns Rev. 2002, 118, S93–S99. [Google Scholar] [CrossRef]
- Ammann, P.; Fehr, T.; Minder, E.I.; Günter, C.; Bertel, O. Elevation of troponin I in sepsis and septic shock. Intensive Care Med. 2001, 27, 965–969. [Google Scholar] [CrossRef]
- Lim, W.; Qushmaq, I.; Devereaux, P.J.; Heels-Ansdell, D.; Lauzier, F.; Ismaila, A.S.; Crowther, M.A.; Cook, D.J. Elevated cardiac troponin measurements in critically ill patients. Arch. Intern. Med. 2006, 166, 2446–2454. [Google Scholar] [CrossRef] [Green Version]
- Hamwi, S.M.; Sharma, A.K.; Weissman, N.J.; Goldstein, S.A.; Apple, S.; Caños, D.A.; Pinnow, E.E.; Lindsay, J. Troponin-I elevation in patients with increased left ventricular mass. Am. J. Cardiol. 2003, 92, 88–90. [Google Scholar] [CrossRef]
- Wang, C.H.; Kuo, L.T.; Hung, M.J.; Cherng, W.J. Coronary vasospasm as a possible cause of elevated cardiac troponin I in patients with acute coronary syndrome and insignificant coronary artery disease. Am. Heart J. 2002, 144, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Troøyen, M.; Indredavik, B.; Rossvoll, O.; Slørdahl, S.A. Myocardial injury in acute stroke assessed by troponin I. Tidsskr Nor Laegeforen 2001, 121, 421–425. [Google Scholar] [PubMed]
- Hijazi, Z.; Wallentin, L.; Siegbahn, A.; Andersson, U.; Alexander, J.H.; Atar, D.; Gersh, B.J.; Hanna, M.; Harjola, V.P.; Horowitz, J.D.; et al. High-sensitivity troponin T and risk stratification in patients with atrial fibrillation during treatment with apixaban or warfarin. J. Am. Coll. Cardiol. 2014, 63, 52–61. [Google Scholar] [CrossRef]
- Horwich, T.B.; Patel, J.; MacLellan, W.R.; Fonarow, G.C. Cardiac troponin I is associated with impaired hemodynamics, progressive left ventricular dysfunction, and increased mortality rates in advanced heart failure. Circulation 2003, 108, 833–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.C.; Ladenson, J.H.; Mason, J.W.; Jaffe, A.S. Elevations of cardiac troponin I associated with myocarditis. Experimental and clinical correlates. Circulation 1997, 95, 163–168. [Google Scholar] [CrossRef]
- Hassan, H.C.; Howlin, K.; Jefferys, A.; Spicer, S.T.; Aravindan, A.N.; Suryanarayanan, G.; Hall, B.M.; Cleland, B.D.; Wong, J.K.; Suranyi, M.G.; et al. High-sensitivity troponin as a predictor of cardiac events and mortality in the stable dialysis population. Clin. Chem. 2014, 60, 389–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, J.; Matsushita, K.; Lazo, M.; Ballantyne, C.M.; Nambi, V.; Hoogeveen, R.; Sharrett, A.R.; Blumenthal, R.S.; Coresh, J.; Selvin, E. Determinants of minimal elevation in high-sensitivity cardiac troponin T in the general population. Clin. Biochem. 2016, 49, 657–662. [Google Scholar] [CrossRef] [Green Version]
- Takanabe-Mori, R.; Ono, K.; Wada, H.; Takaya, T.; Ura, S.; Yamakage, H.; Satoh-Asahara, N.; Shimatsu, A.; Takahashi, Y.; Fujita, M.; et al. Lectin-like oxidized low-density lipoprotein receptor-1 plays an important role in vascular inflammation in current smokers. J. Atheroscler Thromb. 2013, 20, 585–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledwidge, M.; Gallagher, J.; Conlon, C.; Tallon, E.; O’Connell, E.; Dawkins, I.; Watson, C.; O’Hanlon, R.; Bermingham, M.; Patle, A.; et al. Natriuretic Peptide–Based Screening and Collaborative Care for Heart Failure: The STOP-HF Randomized Trial. JAMA 2013, 310, 66–74. [Google Scholar] [CrossRef]
- Troughton, R.W.; Mark Richards, A.; Yandle, T.G.; Frampton, C.M.; Gary Nicholls, M. The effects of medications on circulating levels of cardiac natriuretic peptides. Ann. Med. 2007, 39, 242–260. [Google Scholar] [CrossRef]
- Latini, R.; Masson, S.; Anand, I.; Judd, D.; Maggioni, A.P.; Chiang, Y.-T.; Bevilacqua, M.; Salio, M.; Cardano, P.; Dunselman, P.H.J.M.; et al. Effects of Valsartan on Circulating Brain Natriuretic Peptide and Norepinephrine in Symptomatic Chronic Heart Failure. Circulation 2002, 106, 2454–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamimura, D.; Cain, L.R.; Mentz, R.J.; White, W.B.; Blaha, M.J.; DeFilippis, A.P.; Fox, E.R.; Rodriguez, C.J.; Keith, R.J.; Benjamin, E.J.; et al. Cigarette Smoking and Incident Heart Failure. Circulation 2018, 137, 2572–2582. [Google Scholar] [CrossRef] [PubMed]
- Cataliotti, A.; Malatino, L.S.; Jougasaki, M.; Zoccali, C.; Castellino, P.; Giacone, G.; Bellanuova, I.; Tripepi, R.; Seminara, G.; Parlongo, S.; et al. Circulating natriuretic peptide concentrations in patients with end-stage renal disease: Role of brain natriuretic peptide as a biomarker for ventricular remodeling. Mayo Clin. Proc. 2001, 76, 1111–1119. [Google Scholar] [CrossRef]
- McCullough, P.A.; Duc, P.; Omland, T.; McCord, J.; Nowak, R.M.; Hollander, J.E.; Herrmann, H.C.; Steg, P.G.; Westheim, A.; Knudsen, C.W.; et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: An analysis from the Breathing Not Properly Multinational Study. Am. J. Kidney Dis. 2003, 41, 571–579. [Google Scholar] [CrossRef]
- Das, S.R.; Drazner, M.H.; Dries, D.L.; Vega, G.L.; Stanek, H.G.; Abdullah, S.M.; Canham, R.M.; Chung, A.K.; Leonard, D.; Wians, F.H., Jr.; et al. Impact of body mass and body composition on circulating levels of natriuretic peptides: Results from the Dallas Heart Study. Circulation 2005, 112, 2163–2168. [Google Scholar] [CrossRef] [Green Version]
- Mehra, M.R.; Uber, P.A.; Park, M.H.; Scott, R.L.; Ventura, H.O.; Harris, B.C.; Frohlich, E.D. Obesity and suppressed B-type natriuretic peptide levels in heart failure. J. Am. Coll Cardiol. 2004, 43, 1590–1595. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.; McMurray, J.J.V.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin Receptor Neprilysin Inhibition Compared with Enalapril on the Risk of Clinical Progression in Surviving Patients with Heart Failure. Circulation 2015, 131, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, T.; Kawada, T.; Seino, Y.; Ibuki, C.; Katsumata, M.; Kodani, E. Relation of smoking status to serum levels of N-terminal pro-brain natriuretic peptide in middle-aged men without overt cardiovascular disease. Am. J. Cardiol. 2010, 106, 1456–1460. [Google Scholar] [CrossRef] [PubMed]
- Anwaruddin, S.; Lloyd-Jones, D.M.; Baggish, A.; Chen, A.; Krauser, D.; Tung, R.; Chae, C.; Januzzi, J.L., Jr. Renal function, congestive heart failure, and amino-terminal pro-brain natriuretic peptide measurement: Results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) Study. J. Am. Coll. Cardiol. 2006, 47, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boer, R.A.; van Veldhuisen, D.J.; Gansevoort, R.T.; Muller Kobold, A.C.; van Gilst, W.H.; Hillege, H.L.; Bakker, S.J.L.; van der Harst, P. The fibrosis marker galectin-3 and outcome in the general population. J. Intern. Med. 2012, 272, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.V.; Chen-Tournoux, A.A.; Picard, M.H.; van Kimmenade, R.R.J.; Januzzi, J.L. Galectin-3, cardiac structure and function, and long-term mortality in patients with acutely decompensated heart failure. Eur. J. Heart Fail. 2010, 12, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Manzano-Fernández, S.; Januzzi, J.L.; Pastor-Pérez, F.J.; Bonaque-González, J.C.; Boronat-Garcia, M.; Pascual-Figal, D.A.; Montalban-Larrea, S.; Navarro-Peñalver, M.; Andreu-Cayuelas, J.M.; Valdés, M. Serial monitoring of soluble interleukin family member ST2 in patients with acutely decompensated heart failure. Cardiology 2012, 122, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wu, Y.; Grodin, J.L.; Hsu, A.P.; Hernandez, A.F.; Butler, J.; Metra, M.; Voors, A.A.; Felker, G.M.; Troughton, R.W.; et al. Prognostic Value of Baseline and Changes in Circulating Soluble ST2 Levels and the Effects of Nesiritide in Acute Decompensated Heart Failure. JACC Heart Fail. 2016, 4, 68–77. [Google Scholar] [CrossRef]
- Gottdiener, J.S.; Buzkova, P.; Kahn, P.A.; DeFilippi, C.; Shah, S.; Barasch, E.; Kizer, J.R.; Psaty, B.; Gardin, J.M. Relation of Cigarette Smoking and Heart Failure in Adults ≥65 Years of Age (From the Cardiovascular Health Study). Am. J. Cardiol. 2022; in press. [Google Scholar] [CrossRef]
- Filali, Y.; Kesäniemi, Y.A.; Ukkola, O. Soluble ST2, a biomarker of fibrosis, is associated with multiple risk factors, chronic diseases and total mortality in the OPERA study. Scand. J. Clin. Lab. Investig. 2021, 81, 324–331. [Google Scholar] [CrossRef]
- Sun, Z.; Chang, B.; Huang, A.; Hao, S.; Gao, M.; Sun, Y.; Shi, M.; Jin, L.; Zhang, W.; Zhao, J.; et al. Plasma levels of soluble ST2, but not IL-33, correlate with the severity of alcoholic liver disease. J. Cell Mol. Med. 2019, 23, 887–897. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e563–e595. [Google Scholar] [CrossRef]
- Bogaty, P.; Brophy, J.M.; Boyer, L.; Simard, S.; Joseph, L.; Bertrand, F.; Dagenais, G.R. Fluctuating inflammatory markers in patients with stable ischemic heart disease. Arch. Intern. Med. 2005, 165, 221–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kianoush, S.; Yakoob, M.Y.; Al-Rifai, M.; DeFilippis, A.P.; Bittencourt, M.S.; Duncan, B.B.; Bensenor, I.M.; Bhatnagar, A.; Lotufo, P.A.; Blaha, M.J. Associations of Cigarette Smoking with Subclinical Inflammation and Atherosclerosis: ELSA-Brasil (The Brazilian Longitudinal Study of Adult Health). J. Am. Heart Assoc. 2017, 6, e005088. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Kushner, I. Acute-Phase Proteins and Other Systemic Responses to Inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Kushner, I.; Samols, D.; Magrey, M. A unifying biologic explanation for “high-sensitivity” C-reactive protein and “low-grade” inflammation. Arthritis Care Res. 2010, 62, 442–446. [Google Scholar] [CrossRef]
- Amsterdam, E.A.; Wenger, N.K.; Brindis, R.G.; Casey, D.E.; Ganiats, T.G.; Holmes, D.R.; Jaffe, A.S.; Jneid, H.; Kelly, R.F.; Kontos, M.C.; et al. 2014 AHA/ACC guideline for the management of patients with non ST-elevation acute coronary syndromes. J. Am. Coll. Cardiol. 2014, 64, e139–e228. [Google Scholar] [CrossRef] [Green Version]
- Collet, J.-P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: The Task Force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 42, 1289–1367. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; Ferranti, S.D.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.-M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice: Developed by the Task Force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies With the special contribution of the European Association of Preventive Cardiology (EAPC). Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes: The Task Force for the diagnosis and management of chronic coronary syndromes of the European Society of Cardiology (ESC). Eur. Heart J. 2019, 41, 407–477. [Google Scholar] [CrossRef]
- Freidlin, B.; McShane, L.M.; Korn, E.L. Randomized clinical trials with biomarkers: Design issues. J. Natl. Cancer Inst. 2010, 102, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Kjekshus, J.; Apetrei, E.; Barrios, V.; Böhm, M.; Cleland, J.G.; Cornel, J.H.; Dunselman, P.; Fonseca, C.; Goudev, A.; Grande, P.; et al. Rosuvastatin in older patients with systolic heart failure. N. Engl. J. Med. 2007, 357, 2248–2261. [Google Scholar] [CrossRef] [PubMed]
- Gullestad, L.; Ueland, T.; Kjekshus, J.; Nymo, S.H.; Hulthe, J.; Muntendam, P.; Adourian, A.; Böhm, M.; van Veldhuisen, D.J.; Komajda, M.; et al. Galectin-3 predicts response to statin therapy in the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA). Eur. Heart J. 2012, 33, 2290–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.J.; Berger, P.B.; Teirstein, P.S.; Tanguay, J.F.; Angiolillo, D.J.; Spriggs, D.; Puri, S.; Robbins, M.; Garratt, K.N.; Bertrand, O.F.; et al. Standard- vs high-dose clopidogrel based on platelet function testing after percutaneous coronary intervention: The GRAVITAS randomized trial. JAMA 2011, 305, 1097–1105. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Januzzi, J.L.; Rehman, S.U.; Mohammed, A.A.; Bhardwaj, A.; Barajas, L.; Barajas, J.; Kim, H.-N.; Baggish, A.L.; Weiner, R.B.; Chen-Tournoux, A.; et al. Use of Amino-Terminal Pro–B-Type Natriuretic Peptide to Guide Outpatient Therapy of Patients with Chronic Left Ventricular Systolic Dysfunction. J. Am. Coll. Cardiol. 2011, 58, 1881–1889. [Google Scholar] [CrossRef] [Green Version]
- Newby, D.E.; Adamson, P.D.; Berry, C.; Boon, N.A.; Dweck, M.R.; Flather, M.; Forbes, J.; Hunter, A.; Lewis, S.; MacLean, S.; et al. Coronary CT Angiography and 5-Year Risk of Myocardial Infarction. N. Engl. J. Med. 2018, 379, 924–933. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kott, K.A.; Bishop, M.; Yang, C.H.J.; Plasto, T.M.; Cheng, D.C.; Kaplan, A.I.; Cullen, L.; Celermajer, D.S.; Meikle, P.J.; Vernon, S.T.; et al. Biomarker Development in Cardiology: Reviewing the Past to Inform the Future. Cells 2022, 11, 588. https://doi.org/10.3390/cells11030588
Kott KA, Bishop M, Yang CHJ, Plasto TM, Cheng DC, Kaplan AI, Cullen L, Celermajer DS, Meikle PJ, Vernon ST, et al. Biomarker Development in Cardiology: Reviewing the Past to Inform the Future. Cells. 2022; 11(3):588. https://doi.org/10.3390/cells11030588
Chicago/Turabian StyleKott, Katharine A., Michael Bishop, Christina H. J. Yang, Toby M. Plasto, Daniel C. Cheng, Adam I. Kaplan, Louise Cullen, David S. Celermajer, Peter J. Meikle, Stephen T. Vernon, and et al. 2022. "Biomarker Development in Cardiology: Reviewing the Past to Inform the Future" Cells 11, no. 3: 588. https://doi.org/10.3390/cells11030588
APA StyleKott, K. A., Bishop, M., Yang, C. H. J., Plasto, T. M., Cheng, D. C., Kaplan, A. I., Cullen, L., Celermajer, D. S., Meikle, P. J., Vernon, S. T., & Figtree, G. A. (2022). Biomarker Development in Cardiology: Reviewing the Past to Inform the Future. Cells, 11(3), 588. https://doi.org/10.3390/cells11030588