
Induction of Accelerated Aging in a Mouse Model


Abstract
:1. Introduction
2. Systemic-Induced Accelerated Aging Mouse Model
2.1. The D-Galactose-Induced Senescence Model
2.2. Senescence-Accelerated Mouse/Prone
2.3. Rps9 D95N Mouse
2.4. Progeria Syndrome Mouse
2.5. Mitochondrial DNA Polymerase Mutant Mouse
2.6. Total Body Irradiation (TBI) Model
2.7. Ozone-Induced Senescence Model
2.8. Chronic Jet-Lag Mouse
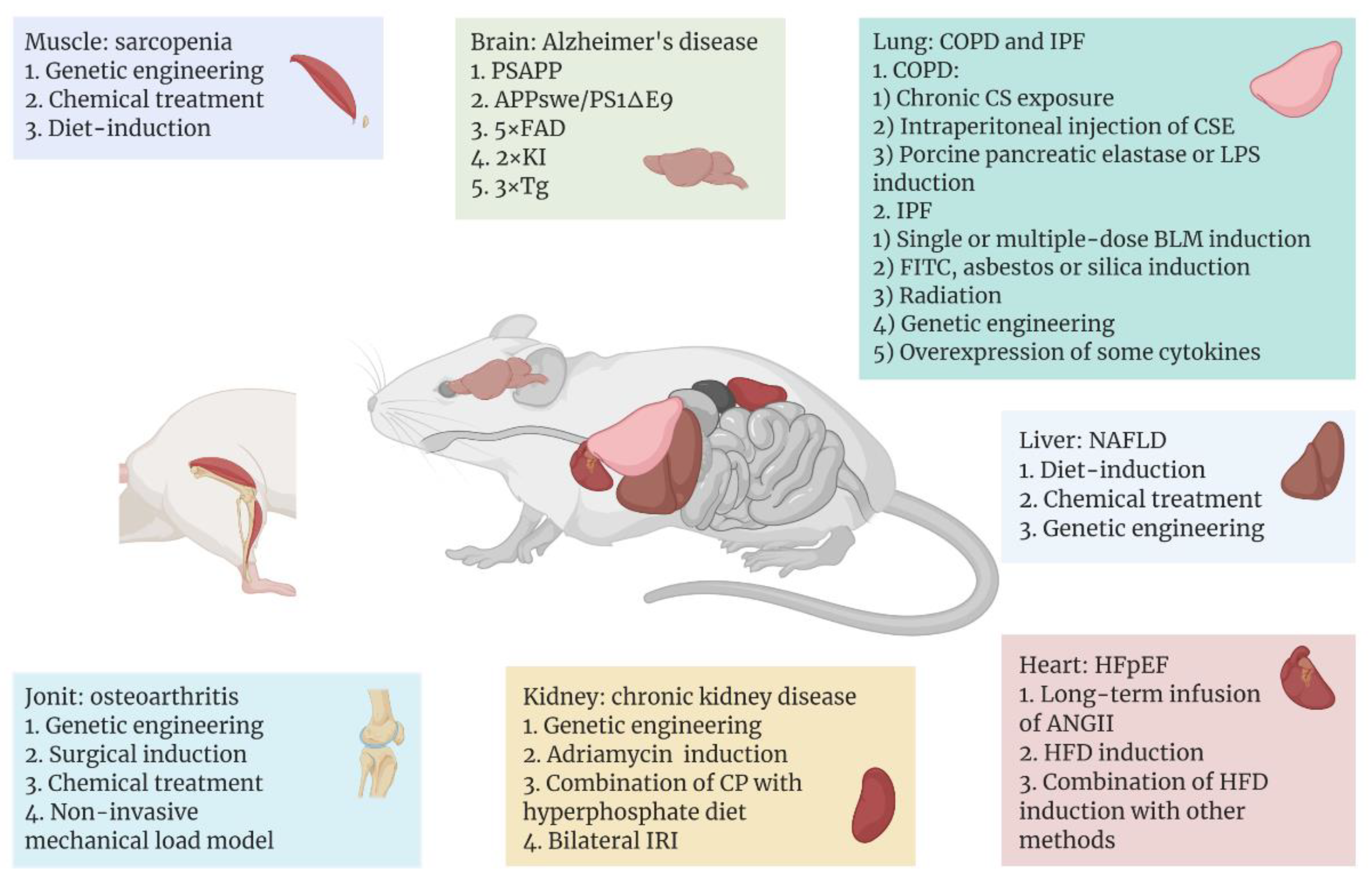
3. Tissue-, Organ-, or System-Specific Mouse Models of Aging-Related Diseases
3.1. Model of Aging Brain or Nerve System
3.2. Model of Aging Muscle
3.3. Model of Aging Heart
3.4. Model of Aging Liver
3.5. Model of Chronic Kidney Disease
3.6. Model of Osteoarthritis
3.7. Model of Aging Lung
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DESA|United Nations. Available online: https://www.un.org/en/desa (accessed on 20 March 2022).
- World Population Prospects 2019. Available online: https://population.un.org/wpp/ (accessed on 20 March 2022).
- Graw, J. Mouse models of cataract. J. Genet. 2009, 88, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.V.; Faulkner, J.A. Skeletal muscle weakness in old age: Underlying mechanisms. Med. Sci. Sports Exerc. 1994, 26, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Gurkar, A.U.; Niedernhofer, L.J. Comparison of mice with accelerated aging caused by distinct mechanisms. Exp. Gerontol. 2015, 68, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhooren, V.; Libert, C. The mouse as a model organism in aging research: Usefulness, pitfalls and possibilities. Ageing Res. Rev. 2013, 12, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Harkema, L.; Youssef, S.A.; de Bruin, A. Pathology of Mouse Models of Accelerated Aging. Vet. Pathol. 2016, 53, 366–389. [Google Scholar] [CrossRef] [PubMed]
- Acosta, P.B.; Gross, K.C. Hidden sources of galactose in the environment. Eur. J. Pediatrics 1995, 154, S87–S92. [Google Scholar] [CrossRef] [PubMed]
- Morava, E. Galactose supplementation in phosphoglucomutase-1 deficiency; review and outlook for a novel treatable CDG. Mol Genet. Metab. 2014, 112, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Ullah, F.; Ali, T.; Ullah, N.; Kim, M.O. Caffeine prevents d-galactose-induced cognitive deficits, oxidative stress, neuroinflammation and neurodegeneration in the adult rat brain. Neurochem. Int. 2015, 90, 114–124. [Google Scholar] [CrossRef]
- Wu, D.M.; Lu, J.; Zheng, Y.L.; Zhou, Z.; Shan, Q.; Ma, D.F. Purple sweet potato color repairs d-galactose-induced spatial learning and memory impairment by regulating the expression of synaptic proteins. Neurobiol. Learn Mem. 2008, 90, 19–27. [Google Scholar] [CrossRef]
- Azman, K.F.; Safdar, A.; Zakaria, R. D-galactose-induced liver aging model: Its underlying mechanisms and potential therapeutic interventions. Exp. Gerontol. 2021, 150, 111372. [Google Scholar] [CrossRef] [PubMed]
- Shwe, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. Role of D-galactose-induced brain aging and its potential used for therapeutic interventions. Exp. Gerontol. 2018, 101, 13–36. [Google Scholar] [CrossRef]
- Azman, K.F.; Zakaria, R. D-Galactose-induced accelerated aging model: An overview. Biogerontology 2019, 20, 763–782. [Google Scholar] [CrossRef]
- Li, W.; Wang, S.; Wang, H.; Wang, J.; Jin, F.; Fang, F.; Fang, C. Astragaloside IV Prevents Memory Impairment in D-galactose-induced Aging Rats Via the AGEs/RAGE/ NF-kappaB Axis. Arch. Med. Res. 2022, 53, 20–28. [Google Scholar] [CrossRef]
- Banji, O.J.; Banji, D.; Ch, K. Curcumin and hesperidin improve cognition by suppressing mitochondrial dysfunction and apoptosis induced by D-galactose in rat brain. Food Chem. Toxicol. 2014, 74, 51–59. [Google Scholar] [CrossRef]
- Khedr, N.F.; Werida, R.H.; Abo-Saif, M.A. Candesartan protects against d-galactose induced—Neurotoxicity and memory deficit via modulation of autophagy and oxidative stress. Toxicol. Appl. Pharm. 2022, 435, 115827. [Google Scholar] [CrossRef]
- Long, J.; Wang, X.; Gao, H.; Liu, Z.; Liu, C.; Miao, M.; Cui, X.; Packer, L.; Liu, J. D-galactose toxicity in mice is associated with mitochondrial dysfunction: Protecting effects of mitochondrial nutrient R-alpha-lipoic acid. Biogerontology 2007, 8, 373–381. [Google Scholar] [CrossRef]
- Kumar, A.; Dogra, S.; Prakash, A. Effect of carvedilol on behavioral, mitochondrial dysfunction, and oxidative damage against D-galactose induced senescence in mice. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 380, 431–441. [Google Scholar] [CrossRef]
- Haider, S.; Liaquat, L.; Shahzad, S.; Sadir, S.; Madiha, S.; Batool, Z.; Tabassum, S.; Saleem, S.; Naqvi, F.; Perveen, T. A high dose of short term exogenous D-galactose administration in young male rats produces symptoms simulating the natural aging process. Life Sci. 2015, 124, 110–119. [Google Scholar] [CrossRef]
- Sacco, R.L.; Roth, G.A.; Reddy, K.S.; Arnett, D.K.; Bonita, R.; Gaziano, T.A.; Heidenreich, P.A.; Huffman, M.D.; Mayosi, B.M.; Mendis, S.; et al. The Heart of 25 by 25: Achieving the Goal of Reducing Global and Regional Premature Deaths from Cardiovascular Diseases and Stroke: A Modeling Study from the American Heart Association and World Heart Federation. Circulation 2016, 133, e674–e690. [Google Scholar] [CrossRef]
- Cebe, T.; Yanar, K.; Atukeren, P.; Ozan, T.; Kuruç, A.I.; Kunbaz, A.; Sitar, M.E.; Mengi, M.; Aydın, M.S.; Eşrefoğlu, M.; et al. A comprehensive study of myocardial redox homeostasis in naturally and mimetically aged rats. Age 2014, 36, 9728. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.M.; Chang, H.H.; Lin, H.J.; Tsai, C.C.; Tsai, C.T.; Chang, H.N.; Lin, S.L.; PadmaViswanadha, V.; Chen, R.J.; Huang, C.Y. Inhibition of Cardiac Hypertrophy Effects in D-Galactose-Induced Senescent Hearts by Alpinate Oxyphyllae Fructus Treatment. Evid. Based Complement Altern. Med. 2017, 2017, 2624384. [Google Scholar] [CrossRef] [Green Version]
- Dehghani, A.; Hafizibarjin, Z.; Najjari, R.; Kaseb, F.; Safari, F. Resveratrol and 1,25-dihydroxyvitamin D co-administration protects the heart against D-galactose-induced aging in rats: Evaluation of serum and cardiac levels of klotho. Aging Clin. Exp. Res. 2019, 31, 1195–1205. [Google Scholar] [CrossRef]
- Wang, L.F.; Cao, Q.; Wen, K.; Xiao, Y.F.; Chen, T.T.; Guan, X.H.; Liu, Y.; Zuo, L.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Alleviates D-Galactose-Induced Myocardial Cell Senescence Through NAD(+)/Sirt1 Signaling Pathway. Front. Physiol. 2019, 10, 1125. [Google Scholar] [CrossRef] [Green Version]
- Lei, L.; Ou, L.; Yu, X. The antioxidant effect of Asparagus cochinchinensis (Lour.) Merr. shoot in D-galactose induced mice aging model and in vitro. J. Chin. Med. Assoc. 2016, 79, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.L.; Guo, L.; Ren, Y.C.; Wang, B.; Li, R.H.; Qi, Y.S.; Yu, H.; Chang, N.D.; Li, M.H.; Peng, H.S. Anti-apoptosis effect of polysaccharide isolated from the seeds of Cuscuta chinensis Lam on cardiomyocytes in aging rats. Mol. Biol. Rep. 2014, 41, 6117–6124. [Google Scholar] [CrossRef]
- Hegab, Z.; Gibbons, S.; Neyses, L.; Mamas, M.A. Role of advanced glycation end products in cardiovascular disease. World J. Cardiol. 2012, 4, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Bo-Htay, C.; Palee, S.; Apaijai, N.; Chattipakorn, S.C.; Chattipakorn, N. Effects of d-galactose-induced ageing on the heart and its potential interventions. J. Cell. Mol. Med. 2018, 22, 1392–1410. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.M.; Tamilselvi, S.; Lin, H.J.; Tsai, C.C.; Lin, Y.M.; Day, C.H.; Viswanadha, V.P.; Chang, H.N.; Kuo, W.W.; Huang, C.Y. Alpinia oxyphylla Miq extract ameliorates cardiac fibrosis associated with D-galactose induced aging in rats. Environ. Toxicol. 2019, 34, 172–178. [Google Scholar] [CrossRef]
- Yeh, S.L.; Wu, T.C.; Chan, S.T.; Hong, M.J.; Chen, H.L. Fructo-oligosaccharide attenuates the production of pro-inflammatory cytokines and the activation of JNK/Jun pathway in the lungs of D-galactose-treated Balb/cJ mice. Eur. J. Nutr. 2014, 53, 449–456. [Google Scholar] [CrossRef]
- Feng, Y.; Yu, Y.H.; Wang, S.T.; Ren, J.; Camer, D.; Hua, Y.Z.; Zhang, Q.; Huang, J.; Xue, D.L.; Zhang, X.F.; et al. Chlorogenic acid protects D-galactose-induced liver and kidney injury via antioxidation and anti-inflammation effects in mice. Pharm. Biol. 2016, 54, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.L.; Wang, C.H.; Kuo, Y.W.; Tsai, C.H. Antioxidative and hepatoprotective effects of fructo-oligosaccharide in d-galactose-treated Balb/cJ mice. Br. J. Nutr. 2011, 105, 805–809. [Google Scholar] [CrossRef] [Green Version]
- Shahroudi, M.J.; Mehri, S.; Hosseinzadeh, H. Anti-Aging Effect of Nigella Sativa Fixed Oil on D-Galactose-Induced Aging in Mice. J. Pharmacopunct. 2017, 20, 29–35. [Google Scholar] [CrossRef]
- Fan, Y.; Xia, J.; Jia, D.; Zhang, M.; Zhang, Y.; Huang, G.; Wang, Y. Mechanism of ginsenoside Rg1 renal protection in a mouse model of d-galactose-induced subacute damage. Pharm. Biol. 2016, 54, 1815–1821. [Google Scholar] [CrossRef]
- Miller, M.R. Structural and physiological age-associated changes in aging lungs. Semin. Respir. Crit. Care Med. 2010, 31, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yu, Z.; Jing, S.; Jiang, W.; Liu, C.; Yu, C.; Sun, J.; Wang, C.; Chen, J.; Li, H. Protective effect of Anwulignan against D-galactose-induced hepatic injury through activating p38 MAPK-Nrf2-HO-1 pathway in mice. Clin. Interv. Aging 2018, 13, 1859–1869. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.Q.; Xie, Y.L.; Gui, S.H.; Zhang, X.; Mo, Z.Z.; Sun, C.Y.; Li, C.L.; Luo, D.D.; Zhang, Z.B.; Su, Z.R.; et al. Polydatin attenuates d-galactose-induced liver and brain damage through its anti-oxidative, anti-inflammatory and anti-apoptotic effects in mice. Food Funct. 2016, 7, 4545–4555. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, J.Z.; Lin, Z.X.; Yuan, Q.J.; Li, Y.C.; Liang, J.L.; Zhan, J.Y.; Xie, Y.L.; Su, Z.R.; Liu, Y.H. Ameliorative effect of supercritical fluid extract of Chrysanthemum indicum Linnén against D-galactose induced brain and liver injury in senescent mice via suppression of oxidative stress, inflammation and apoptosis. J. Ethnopharmacol. 2019, 234, 44–56. [Google Scholar] [CrossRef]
- Huang, C.C.; Chiang, W.D.; Huang, W.C.; Huang, C.Y.; Hsu, M.C.; Lin, W.T. Hepatoprotective Effects of Swimming Exercise against D-Galactose-Induced Senescence Rat Model. Evid. Based Complement Altern. Med 2013, 2013, 275431. [Google Scholar] [CrossRef]
- Wang, H.; Hu, L.; Li, L.; Wu, X.; Fan, Z.; Zhang, C.; Wang, J.; Jia, J.; Wang, S. Inorganic nitrate alleviates the senescence-related decline in liver function. Sci. China Life Sci. 2018, 61, 24–34. [Google Scholar] [CrossRef]
- Mo, Z.Z.; Liu, Y.H.; Li, C.L.; Xu, L.Q.; Wen, L.L.; Xian, Y.F.; Lin, Z.X.; Zhan, J.Y.; Chen, J.N.; Xu, F.F.; et al. Protective Effect of SFE-CO2 of Ligusticum chuanxiong Hort Against d-Galactose-Induced Injury in the Mouse Liver and Kidney. Rejuvenation Res. 2017, 20, 231–243. [Google Scholar] [CrossRef]
- Liu, C.M.; Ma, J.Q.; Lou, Y. Chronic administration of troxerutin protects mouse kidney against D-galactose-induced oxidative DNA damage. Food Chem. Toxicol. 2010, 48, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Ma, Q.; Guo, Y.; Sun, L. Protective effects of rambutan (Nephelium lappaceum) peel phenolics on H2O2-induced oxidative damages in HepG2 cells and d-galactose-induced aging mice. Food Chem. Toxicol. 2017, 108, 554–562. [Google Scholar] [CrossRef]
- Liao, C.H.; Chen, B.H.; Chiang, H.S.; Chen, C.W.; Chen, M.F.; Ke, C.C.; Wang, Y.Y.; Lin, W.N.; Wang, C.C.; Lin, Y.H. Optimizing a Male Reproductive Aging Mouse Model by D-Galactose Injection. Int. J. Mol. Sci. 2016, 17, 98. [Google Scholar] [CrossRef] [Green Version]
- Djahanbakhch, O.; Ezzati, M.; Zosmer, A. Reproductive ageing in women. J. Pathol. 2007, 211, 219–231. [Google Scholar] [CrossRef]
- Ahangarpour, A.; Najimi, S.A.; Farbood, Y. Effects of Vitex agnus-castus fruit on sex hormones and antioxidant indices in a d-galactose-induced aging female mouse model. J. Chin. Med. Assoc. 2016, 79, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhou, H.; Deng, L.; Wang, L.; Chen, J.; Zhou, X. Serine Deficiency Exacerbates Inflammation and Oxidative Stress via Microbiota-Gut-Brain Axis in D-Galactose-Induced Aging Mice. Mediat. Inflamm 2020, 2020, 5821428. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Han, D.; Liu, J.; Liu, H.; Jiang, Z. Partially Hydrolyzed Guar Gum Attenuates d-Galactose-Induced Oxidative Stress and Restores Gut Microbiota in Rats. Int. J. Mol. Sci. 2019, 20, 4861. [Google Scholar] [CrossRef] [Green Version]
- Fransen, F.; van Beek, A.A.; Borghuis, T.; Aidy, S.E.; Hugenholtz, F.; van der Gaast-de Jongh, C.; Savelkoul, H.F.J.; De Jonge, M.I.; Boekschoten, M.V.; Smidt, H.; et al. Aged Gut Microbiota Contributes to Systemical Inflammaging after Transfer to Germ-Free Mice. Front. Immunol. 2017, 8, 1385. [Google Scholar] [CrossRef] [Green Version]
- Yin, R.; Liu, S.; Jiang, X.; Zhang, X.; Wei, F.; Hu, J. The Qingchangligan Formula Alleviates Acute Liver Failure by Regulating Galactose Metabolism and Gut Microbiota. Front. Cell Infect Microbiol. 2021, 11, 771483. [Google Scholar] [CrossRef]
- Kim, S.; Jazwinski, S.M. The Gut Microbiota and Healthy Aging: A Mini-Review. Gerontology 2018, 64, 513–520. [Google Scholar] [CrossRef]
- Takeda, T.; Hosokawa, M.; Higuchi, K. Senescence-accelerated mouse (SAM): A novel murine model of senescence. Exp. Gerontol. 1997, 32, 105–109. [Google Scholar] [CrossRef]
- Higuchi, K. Genetic characterization of senescence-accelerated mouse (SAM). Exp. Gerontol. 1997, 32, 129–138. [Google Scholar] [CrossRef]
- Garcia-Matas, S.; Gutierrez-Cuesta, J.; Coto-Montes, A.; Rubio-Acero, R.; Diez-Vives, C.; Camins, A.; Pallas, M.; Sanfeliu, C.; Cristofol, R. Dysfunction of astrocytes in senescence-accelerated mice SAMP8 reduces their neuroprotective capacity. Aging Cell 2008, 7, 630–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, J.; Horikawa, I.; Harris, C. Cellular Senescence: Mechanisms, Morphology, and Mouse Models. Vet. Pathol. 2020, 57, 747–757. [Google Scholar] [CrossRef]
- Lecka-Czernik, B.; Moerman, E.J.; Shmookler Reis, R.J.; Lipschitz, D.A. Cellular and molecular biomarkers indicate precocious in vitro senescence in fibroblasts from SAMP6 mice. Evidence supporting a murine model of premature senescence and osteopenia. J. Gerontol. Biol. Sci. Med. Sci. 1997, 52, B331–B336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Jung, Y.; Park, B.S.; Partovi, D.; Reddy, P.H. Time-course of mitochondrial gene expressions in mice brains: Implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J. Neurochem. 2005, 92, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Hosokawa, M.; Takeshita, S.; Irino, M.; Higuchi, K.; Matsushita, T.; Tomita, Y.; Yasuhira, K.; Hamamoto, H.; Shimizu, K.; et al. A new murine model of accelerated senescence. Mech. Ageing Dev. 1981, 17, 183–194. [Google Scholar] [CrossRef]
- Takeda, T.; Hosokawa, M.; Higuchi, K. Senescence-accelerated mouse (SAM): A novel murine model of accelerated senescence. J. Am. Geriatr. Soc. 1991, 39, 911–919. [Google Scholar] [CrossRef]
- Matsushita, M.; Tsuboyama, T.; Kasai, R.; Okumura, H.; Yamamuro, T.; Higuchi, K.; Higuchi, K.; Kohno, A.; Yonezu, T.; Utani, A.; et al. Age-related changes in bone mass in the senescence-accelerated mouse (SAM). SAM-R/3 and SAM-P/6 as new murine models for senile osteoporosis. Am. J. Pathol. 1986, 125, 276–283. [Google Scholar]
- Takada, K.; Inaba, M.; Ichioka, N.; Ueda, Y.; Taira, M.; Baba, S.; Mizokami, T.; Wang, X.; Hisha, H.; Iida, H.; et al. Treatment of senile osteoporosis in SAMP6 mice by intra-bone marrow injection of allogeneic bone marrow cells. Stem Cells 2006, 24, 399–405. [Google Scholar] [CrossRef] [Green Version]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulić, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Azab, M.; Wang, B.; Elkhider, A.; Walana, W.; Li, W.; Yuan, B.; Ye, Y.; Tang, Y.; Almoiliqy, M.; Adlat, S.; et al. Indian Hedgehog regulates senescence in bone marrow-derived mesenchymal stem cell through modulation of ROS/mTOR/4EBP1, p70S6K1/2 pathway. Aging 2020, 12, 5693–5715. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Armbrecht, H.J.; Farr, S.A.; Kumar, V.B. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 650–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karasawa, N.; Nagatsu, I.; Sakai, K.; Nagatsu, T.; Watanabe, K.; Onozuka, M. Immunocytochemical study of catecholaminergic neurons in the senescence-accelerated mouse (SAM-P8) brain. J. Neural Transm. 1997, 104, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Sureda, F.X.; Gutierrez-Cuesta, J.; Romeu, M.; Mulero, M.; Canudas, A.M.; Camins, A.; Mallol, J.; Pallas, M. Changes in oxidative stress parameters and neurodegeneration markers in the brain of the senescence-accelerated mice SAMP-8. Exp. Gerontol. 2006, 41, 360–367. [Google Scholar] [CrossRef]
- Manich, G.; Mercader, C.; del Valle, J.; Duran-Vilaregut, J.; Camins, A.; Pallàs, M.; Vilaplana, J.; Pelegrí, C. Characterization of amyloid-β granules in the hippocampus of SAMP8 mice. J. Alzheimers Dis. 2011, 25, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Alvarez-Garcia, O.; Vega-Naredo, I.; Sierra, V.; Caballero, B.; Tomas-Zapico, C.; Camins, A.; Garcia, J.J.; Pallas, M.; Coto-Montes, A. Elevated oxidative stress in the brain of senescence-accelerated mice at 5 months of age. Biogerontology 2006, 7, 43–52. [Google Scholar] [CrossRef]
- Ota, H.; Akishita, M.; Akiyoshi, T.; Kahyo, T.; Setou, M.; Ogawa, S.; Iijima, K.; Eto, M.; Ouchi, Y. Testosterone deficiency accelerates neuronal and vascular aging of SAMP8 mice: Protective role of eNOS and SIRT1. PLoS ONE 2012, 7, e29598. [Google Scholar] [CrossRef] [Green Version]
- Karuppagounder, V.; Arumugam, S.; Babu, S.S.; Palaniyandi, S.S.; Watanabe, K.; Cooke, J.P.; Thandavarayan, R.A. The senescence accelerated mouse prone 8 (SAMP8): A novel murine model for cardiac aging. Ageing Res. Rev. 2017, 35, 291–296. [Google Scholar] [CrossRef]
- Niimi, K.; Takahashi, E.; Itakura, C. Adiposity-related biochemical phenotype in senescence-accelerated mouse prone 6 (SAMP6). Comp. Med. 2009, 59, 431–436. [Google Scholar] [PubMed]
- Shimada, A.; Ohta, A.; Akiguchi, I.; Takeda, T. Inbred SAM-P/10 as a mouse model of spontaneous, inherited brain atrophy. J. Neuropathol. Exp. Neurol. 1992, 51, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Hosokawa, M.; Ohta, A.; Akiguchi, I.; Takeda, T. Localization of atrophy-prone areas in the aging mouse brain: Comparison between the brain atrophy model SAM-P/10 and the normal control SAM-R/1. Neuroscience 1994, 59, 859–869. [Google Scholar] [CrossRef]
- Shcherbakov, D.; Nigri, M.; Akbergenov, R.; Brilkova, M.; Mantovani, M.; Petit, P.I.; Grimm, A.; Karol, A.A.; Teo, Y.; Sanchón, A.C.; et al. Premature aging in mice with error-prone protein synthesis. Sci. Adv. 2022, 8, eabl9051. [Google Scholar] [CrossRef] [PubMed]
- Brennan, T.A.; Egan, K.P.; Lindborg, C.M.; Chen, Q.; Sweetwyne, M.T.; Hankenson, K.D.; Xie, S.X.; Johnson, F.B.; Pignolo, R.J. Mouse models of telomere dysfunction phenocopy skeletal changes found in human age-related osteoporosis. Dis. Model Mech. 2014, 7, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Botter, S.M.; Zar, M.; van Osch, G.J.; van Steeg, H.; Dolle, M.E.; Hoeijmakers, J.H.; Weinans, H.; van Leeuwen, J.P. Analysis of osteoarthritis in a mouse model of the progeroid human DNA repair syndrome trichothiodystrophy. Age 2011, 33, 247–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edstrom, E.; Altun, M.; Bergman, E.; Johnson, H.; Kullberg, S.; Ramirez-Leon, V.; Ulfhake, B. Factors contributing to neuromuscular impairment and sarcopenia during aging. Physiol. Behav. 2007, 92, 129–135. [Google Scholar] [CrossRef]
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Hartman, T.K.; Wengenack, T.M.; Poduslo, J.F.; van Deursen, J.M. Mutant mice with small amounts of BubR1 display accelerated age-related gliosis. Neurobiol. Aging 2007, 28, 921–927. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Cupit-Link, M.C.; Kirkland, J.L.; Ness, K.K.; Armstrong, G.T.; Tchkonia, T.; LeBrasseur, N.K.; Armenian, S.H.; Ruddy, K.J.; Hashmi, S.K. Biology of premature ageing in survivors of cancer. ESMO Open 2017, 2, e000250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robison, L.L.; Hudson, M.M. Survivors of childhood and adolescent cancer: Life-long risks and responsibilities. Nat. Rev. Cancer 2014, 14, 61–70. [Google Scholar] [CrossRef]
- Fielder, E.; Weigand, M.; Agneessens, J.; Griffin, B.; Parker, C.; Miwa, S.; von Zglinicki, T. Sublethal whole-body irradiation causes progressive premature frailty in mice. Mech. Ageing Dev. 2019, 180, 63–69. [Google Scholar] [CrossRef]
- Kasmann, L.; Dietrich, A.; Staab-Weijnitz, C.A.; Manapov, F.; Behr, J.; Rimner, A.; Jeremic, B.; Senan, S.; De Ruysscher, D.; Lauber, K.; et al. Radiation-induced lung toxicity—Cellular and molecular mechanisms of pathogenesis, management, and literature review. Radiat. Oncol. 2020, 15, 214. [Google Scholar] [CrossRef]
- Sun, L.; Inaba, Y.; Sogo, Y.; Ito, A.; Bekal, M.; Chida, K.; Moritake, T. Total body irradiation causes a chronic decrease in antioxidant levels. Sci. Rep. 2021, 11, 6716. [Google Scholar] [CrossRef]
- Xiao, S.; Shterev, I.D.; Zhang, W.; Young, L.; Shieh, J.H.; Moore, M.; van den Brink, M.; Sempowski, G.D.; Manley, N.R. Sublethal Total Body Irradiation Causes Long-Term Deficits in Thymus Function by Reducing Lymphoid Progenitors. J. Immunol. 2017, 199, 2701–2712. [Google Scholar] [CrossRef]
- Citrin, D.E.; Shankavaram, U.; Horton, J.A.; Shield, W., 3rd; Zhao, S.; Asano, H.; White, A.; Sowers, A.; Thetford, A.; Chung, E.J. Role of type II pneumocyte senescence in radiation-induced lung fibrosis. J. Natl. Cancer Inst. 2013, 105, 1474–1484. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Beattie, L.A.; Seed, T.M. Vitamin E: Tocopherols and tocotrienols as potential radiation countermeasures. J. Radiat. Res. 2013, 54, 973–988. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, I.D.; Olson, J.; Lindburg, C.A.; Payne, V.; Collins, B.; Smith, T.L.; Munley, M.T.; Wheeler, K.T.; Willey, J.S. Total-body irradiation produces late degenerative joint damage in rats. Int. J. Radiat. Biol. 2014, 90, 821–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornelissen, M.; de Ridder, L. Dose- and time-dependent increase of lysosomal enzymes in embryonic cartilage in vitro after ionizing radiation. Scanning Microsc. 1990, 4, 769–773; discussion 764–773. [Google Scholar] [PubMed]
- Cornelissen, M.; Thierens, H.; de Ridder, L. Effects of ionizing radiation on the size distribution of proteoglycan aggregates synthesized by chondrocytes in agarose. Scanning Microsc. 1993, 7, 1263–1267; discussion 1267–1268. [Google Scholar] [PubMed]
- Onder, G.O.; Balcioglu, E.; Baran, M.; Ceyhan, A.; Cengiz, O.; Suna, P.A.; Yıldız, O.G.; Yay, A. The different doses of radiation therapy-induced damage to the ovarian environment in rats. Int. J. Radiat. Biol. 2021, 97, 367–375. [Google Scholar] [CrossRef]
- Pesty, A.; Doussau, M.; Lahaye, J.B.; Lefèvre, B. Whole-body or isolated ovary (60)Co irradiation: Effects on in vivo and in vitro folliculogenesis and oocyte maturation. Reprod. Toxicol. 2010, 29, 93–98. [Google Scholar] [CrossRef]
- Chang, H.H.; Hao, H.; Sarnat, S.E. A Statistical Modeling Framework for Projecting Future Ambient Ozone and its Health Impact due to Climate Change. Atmos. Environ. 2014, 89, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Tyler, C.R.; Noor, S.; Young, T.L.; Rivero, V.; Sanchez, B.; Lucas, S.; Caldwell, K.K.; Milligan, E.D.; Campen, M.J. Aging Exacerbates Neuroinflammatory Outcomes Induced by Acute Ozone Exposure. Toxicol. Sci. 2018, 163, 123–139. [Google Scholar] [CrossRef]
- Stober, V.P.; Garantziotis, S. Assessment of Ozone-Induced Lung Injury in Mice. Methods Mol. Biol. 2018, 1809, 301–314. [Google Scholar] [CrossRef]
- Tomita, K.; Okawara, T.; Ohira, C.; Morimoto, A.; Aihara, R.; Kurihara, T.; Fukuyama, T. An Acceptable Concentration (0.1 ppm) of Ozone Exposure Exacerbates Lung Injury in a Mouse Model. Am. J. Respir. Cell Mol. Biol. 2021, 65, 674–676. [Google Scholar] [CrossRef]
- Hazucha, M.J.; Folinsbee, L.J.; Bromberg, P.A. Distribution and reproducibility of spirometric response to ozone by gender and age. J. Appl. Physiol. 2003, 95, 1917–1925. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, S.; Takeda, K.; Jin, N.; Okamoto, M.; Matsuda, H.; Shiraishi, Y.; Park, J.W.; McConville, G.; Joetham, A.; O’Brien, R.L.; et al. Vgamma1+ T cells and tumor necrosis factor-alpha in ozone-induced airway hyperresponsiveness. Am. J. Respir. Cell Mol. Biol. 2009, 40, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasu, V.T.; Oommen, S.; Lim, Y.; Valacchi, G.; Hobson, B.; Eirserich, J.P.; Leonard, S.W.; Traber, M.G.; Cross, C.E.; Gohil, K. Modulation of ozone-sensitive genes in alpha-tocopherol transfer protein null mice. Inhal. Toxicol. 2010, 22, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, A.J.; Sellix, M.T.; Daniel, J.; Yamazaki, S.; Menaker, M.; Block, G.D. Chronic jet-lag increases mortality in aged mice. Curr. Biol. 2006, 16, R914–R916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Tudela, E.; Bonmatí-Carrión Mde, L.; De la Fuente, M.; Mendiola, P. Chronodisruption and ageing. Rev. Esp. Geriatr. Gerontol. 2012, 47, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Castanon-Cervantes, O.; Wu, M.; Ehlen, J.C.; Paul, K.; Gamble, K.L.; Johnson, R.L.; Besing, R.C.; Menaker, M.; Gewirtz, A.T.; Davidson, A.J. Dysregulation of inflammatory responses by chronic circadian disruption. J. Immunol. 2010, 185, 5796–5805. [Google Scholar] [CrossRef] [Green Version]
- Filipski, E.; Delaunay, F.; King, V.M.; Wu, M.W.; Claustrat, B.; Gréchez-Cassiau, A.; Guettier, C.; Hastings, M.H.; Francis, L. Effects of chronic jet lag on tumor progression in mice. Cancer Res. 2004, 64, 7879–7885. [Google Scholar] [CrossRef] [Green Version]
- Filipski, E.; Li, X.M.; Lévi, F. Disruption of circadian coordination and malignant growth. Cancer Causes Control 2006, 17, 509–514. [Google Scholar] [CrossRef]
- Filipski, E.; Lévi, F. Circadian disruption in experimental cancer processes. Integr. Cancer Ther. 2009, 8, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Papagiannakopoulos, T.; Bauer, M.R.; Davidson, S.M.; Heimann, M.; Subbaraj, L.; Bhutkar, A.; Bartlebaugh, J.; Vander Heiden, M.G.; Jacks, T. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 2016, 24, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Kettner, N.M.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Q.; Wu, J.; Ni, Y.; Xie, X.; Yu, C.; Xiao, Q.; Zhou, J.; Wang, X.; Fu, Z. Exposure to jet lag aggravates depression-like behaviors and age-related phenotypes in rats subject to chronic corticosterone. Acta Biochim. Biophys. Sin. 2019, 51, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Van Dycke, K.C.; Rodenburg, W.; van Oostrom, C.T.; van Kerkhof, L.W.; Pennings, J.L.; Roenneberg, T.; van Steeg, H.; van der Horst, G.T. Chronically Alternating Light Cycles Increase Breast Cancer Risk in Mice. Curr. Biol. 2015, 25, 1932–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, I.; van der Eerden, B.; Boers, R.; Boers, J.; Streng, A.A.; Ridwan, Y.; Schreuders-Koedam, M.; Vermeulen, M.; van der Pluijm, I.; Essers, J.; et al. Gestational jet lag predisposes to later-life skeletal and cardiac disease. Chronobiol. Int. 2019, 36, 657–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, M.; Korf, H.W.; Wicht, H. Synchronizing effects of melatonin on diurnal and circadian rhythms. Gen. Comp. Endocrinol. 2018, 258, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Oike, H.; Sakurai, M.; Ippoushi, K.; Kobori, M. Time-fixed feeding prevents obesity induced by chronic advances of light/dark cycles in mouse models of jet-lag/shift work. Biochem. Biophys. Res. Commun. 2015, 465, 556–561. [Google Scholar] [CrossRef]
- Matenchuk, B.A.; Mandhane, P.J.; Kozyrskyj, A.L. Sleep, circadian rhythm, and gut microbiota. Sleep Med. Rev. 2020, 53, 101340. [Google Scholar] [CrossRef]
- Doyle, S.E.; Feng, H.; Garber, G.; Menaker, M.; Lynch, W.J. Effects of circadian disruption on methamphetamine consumption in methamphetamine-exposed rats. Psychopharmacology 2015, 232, 2169–2179. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wu, T.; Jin, Y.; Fu, Z. Effects of age and jet lag on D-galactose induced aging process. Biogerontology 2009, 10, 153–161. [Google Scholar] [CrossRef]
- Loerch, P.M.; Lu, T.; Dakin, K.A.; Vann, J.M.; Isaacs, A.; Geula, C.; Wang, J.; Pan, Y.; Gabuzda, D.H.; Li, C.; et al. Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE 2008, 3, e3329. [Google Scholar] [CrossRef] [Green Version]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Güell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Teich, A.F.; Patel, M.; Arancio, O. A reliable way to detect endogenous murine β-amyloid. PLoS ONE 2013, 8, e55647. [Google Scholar] [CrossRef]
- Myers, A.; McGonigle, P. Overview of Transgenic Mouse Models for Alzheimer’s Disease. Curr. Protoc. Neurosci. 2019, 89, e81. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Hall, A.M.; Roberson, E.D. Mouse models of Alzheimer’s disease. Brain Res. Bull 2012, 88, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Luhrs, K.J.; Ryff, K.A.; Malik, W.T.; Driscoll, M.J.; Culver, B. Suppression of nuclear factor kappa B ameliorates astrogliosis but not amyloid burden in APPswe/PS1dE9 mice. Neuroscience 2009, 161, 53–58. [Google Scholar] [CrossRef]
- Sun, X.-Y.; Li, L.-J.; Dong, Q.-X.; Zhu, J.; Huang, Y.-R.; Hou, S.-J.; Yu, X.-L.; Liu, R.-T. Rutin prevents tau pathology and neuroinflammation in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2021, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.-Q.; He, M.; Yu, D.-J.; Wu, Y.-X.; Wang, X.-H.; Lv, S.; Xiao, W.-F.; Li, Y.-S. Mouse models of sarcopenia: Classification and evaluation. J. Cachexia Sarcopenia Muscle 2021, 12, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, D.; Moriggi, M.; De Palma, S.; Bizzotto, D.; Molon, S.; Torretta, E.; Fania, C.; Bonaldo, P.; Gelfi, C.; Braghetta, P. Collagen VI Null Mice as a Model for Early Onset Muscle Decline in Aging. Front. Mol. Neurosci. 2017, 10, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, F.; Abadir, P.; Marx, R.; Westbrook, R.; Cooke, C.; Yang, H.; Walston, J. Impaired mitochondrial degradation by autophagy in the skeletal muscle of the aged female interleukin 10 null mouse. Exp. Gerontol. 2016, 73, 23–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, B.; Smith, N.; Saunders, D.; Ranjit, R.; Kneis, P.; Towner, R.A.; Van Remmen, H. Using MRI to measure in vivo free radical production and perfusion dynamics in a mouse model of elevated oxidative stress and neurogenic atrophy. Redox. Biol. 2019, 26, 101308. [Google Scholar] [CrossRef]
- Sayed, R.K.A.; Fernández-Ortiz, M.; Diaz-Casado, M.E.; Aranda-Martínez, P.; Fernández-Martínez, J.; Guerra-Librero, A.; Escames, G.; López, L.C.; Alsaadawy, R.M.; Acuña-Castroviejo, D. Lack of NLRP3 Inflammasome Activation Reduces Age-Dependent Sarcopenia and Mitochondrial Dysfunction, Favoring the Prophylactic Effect of Melatonin. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2019, 74, 1699–1708. [Google Scholar] [CrossRef]
- Li, J.; Yi, X.; Yao, Z.; Chakkalakal, J.V.; Xing, L.; Boyce, B.F. TNF Receptor-Associated Factor 6 Mediates TNFα-Induced Skeletal Muscle Atrophy in Mice During Aging. J. Bone Min. Res. 2020, 35, 1535–1548. [Google Scholar] [CrossRef]
- Sun, H.; Gong, Y.; Qiu, J.; Chen, Y.; Ding, F.; Zhao, Q. TRAF6 inhibition rescues dexamethasone-induced muscle atrophy. Int. J. Mol. Sci. 2014, 15, 11126–11141. [Google Scholar] [CrossRef]
- Wada, S.; Kato, Y.; Okutsu, M.; Miyaki, S.; Suzuki, K.; Yan, Z.; Schiaffino, S.; Asahara, H.; Ushida, T.; Akimoto, T. Translational suppression of atrophic regulators by microRNA-23a integrates resistance to skeletal muscle atrophy. J. Biol. Chem. 2011, 286, 38456–38465. [Google Scholar] [CrossRef] [Green Version]
- Campos, F.; Abrigo, J.; Aguirre, F.; Garcés, B.; Arrese, M.; Karpen, S.; Cabrera, D.; Andía, M.E.; Simon, F.; Cabello-Verrugio, C. Sarcopenia in a mice model of chronic liver disease: Role of the ubiquitin-proteasome system and oxidative stress. Pflug. Arch. Eur. J. Physiol. 2018, 470, 1503–1519. [Google Scholar] [CrossRef] [PubMed]
- Noll, N.A.; Lal, H.; Merryman, W.D. Mouse Models of Heart Failure with Preserved or Reduced Ejection Fraction. Am. J. Pathol. 2020, 190, 1596–1608. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Lai, Y.C.; Kelly, N.J.; Bueno, M.; Baust, J.J.; Bachman, T.N.; Goncharov, D.; Vanderpool, R.R.; Radder, J.E.; Hu, J.; et al. Development of a Mouse Model of Metabolic Syndrome, Pulmonary Hypertension, and Heart Failure with Preserved Ejection Fraction. Am. J. Respir. Cell Mol. Biol. 2017, 56, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manrique, C.; DeMarco, V.G.; Aroor, A.R.; Mugerfeld, I.; Garro, M.; Habibi, J.; Hayden, M.R.; Sowers, J.R. Obesity and insulin resistance induce early development of diastolic dysfunction in young female mice fed a Western diet. Endocrinology 2013, 154, 3632–3642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aroor, A.R.; Habibi, J.; Kandikattu, H.K.; Garro-Kacher, M.; Barron, B.; Chen, D.; Hayden, M.R.; Whaley-Connell, A.; Bender, S.B.; Klein, T.; et al. Dipeptidyl peptidase-4 (DPP-4) inhibition with linagliptin reduces western diet-induced myocardial TRAF3IP2 expression, inflammation and fibrosis in female mice. Cardiovasc. Diabetol. 2017, 16, 61. [Google Scholar] [CrossRef] [Green Version]
- Cannon, M.V.; Silljé, H.H.; Sijbesma, J.W.; Khan, M.A.; Steffensen, K.R.; van Gilst, W.H.; de Boer, R.A. LXRα improves myocardial glucose tolerance and reduces cardiac hypertrophy in a mouse model of obesity-induced type 2 diabetes. Diabetologia 2016, 59, 634–643. [Google Scholar] [CrossRef] [Green Version]
- Withaar, C.; Meems, L.M.G.; Markousis-Mavrogenis, G.; Boogerd, C.J.; Silljé, H.H.W.; Schouten, E.M.; Dokter, M.M.; Voors, A.A.; Westenbrink, B.D.; Lam, C.S.P.; et al. The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 2108–2124. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef]
- Mikolasevic, I.; Filipec-Kanizaj, T.; Mijic, M.; Jakopcic, I.; Milic, S.; Hrstic, I.; Sobocan, N.; Stimac, D.; Burra, P. Nonalcoholic fatty liver disease and liver transplantation—Where do we stand? World J. Gastroenterol. 2018, 24, 1491–1506. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Barrera, F.; George, J. The role of diet and nutritional intervention for the management of patients with NAFLD. Clin. Liver Dis. 2014, 18, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Denda, A.; Kitayama, W.; Kishida, H.; Murata, N.; Tamura, K.; Kusuoka, O.; Tsutsumi, M.; Nishikawa, F.; Kita, E.; Nakae, D.; et al. Expression of inducible nitric oxide (NO) synthase but not prevention by its gene ablation of hepatocarcinogenesis with fibrosis caused by a choline-deficient, L-amino acid-defined diet in rats and mice. Nitric Oxide Biol. Chem. 2007, 16, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Soejima, Y.; Fukusato, T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2300–2308. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007, 46, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Kaji, K.; Kitade, M.; Kubo, T.; Furukawa, M.; Saikawa, S.; Shimozato, N.; Sato, S.; Seki, K.; Kawaratani, H.; et al. Exogenous Administration of Low-Dose Lipopolysaccharide Potentiates Liver Fibrosis in a Choline-Deficient l-Amino-Acid-Defined Diet-Induced Murine Steatohepatitis Model. Int. J. Mol. Sci. 2019, 20, 2724. [Google Scholar] [CrossRef] [Green Version]
- Zhong, F.; Zhou, X.; Xu, J.; Gao, L. Rodent Models of Nonalcoholic Fatty Liver Disease. Digestion 2020, 101, 522–535. [Google Scholar] [CrossRef]
- Kubota, N.; Kado, S.; Kano, M.; Masuoka, N.; Nagata, Y.; Kobayashi, T.; Miyazaki, K.; Ishikawa, F. A high-fat diet and multiple administration of carbon tetrachloride induces liver injury and pathological features associated with non-alcoholic steatohepatitis in mice. Clin. Exp. Pharmacol. Physiol. 2013, 40, 422–430. [Google Scholar] [CrossRef]
- Hainer, V.; Aldhoon Hainerová, I.; Kunešová, M.; Taxová Braunerová, R.; Zamrazilová, H.; Bendlová, B. Melanocortin pathways: Suppressed and stimulated melanocortin-4 receptor (MC4R). Physiol. Res. 2020, 69, S245–S254. [Google Scholar] [CrossRef]
- Itoh, M.; Suganami, T.; Nakagawa, N.; Tanaka, M.; Yamamoto, Y.; Kamei, Y.; Terai, S.; Sakaida, I.; Ogawa, Y. Melanocortin 4 receptor-deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am. J. Pathol. 2011, 179, 2454–2463. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Qureshi, A.R.; Witasp, A.; Lindholm, B.; Stenvinkel, P. Early Vascular Ageing and Cellular Senescence in Chronic Kidney Disease. Comput. Struct. Biotechnol. J. 2019, 17, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Yang, M.; Ma, X. Implication of cellular senescence in the progression of chronic kidney disease and the treatment potencies. Biomed. Pharmacother. 2021, 135, 111191. [Google Scholar] [CrossRef] [PubMed]
- Deb, D.K.; Sun, T.; Wong, K.E.; Zhang, Z.; Ning, G.; Zhang, Y.; Kong, J.; Shi, H.; Chang, A.; Li, Y.C. Combined vitamin D analog and AT1 receptor antagonist synergistically block the development of kidney disease in a model of type 2 diabetes. Kidney Int. 2010, 77, 1000–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Østergaard, M.V.; Pinto, V.; Stevenson, K.; Worm, J.; Fink, L.N.; Coward, R.J. DBA2J db/db mice are susceptible to early albuminuria and glomerulosclerosis that correlate with systemic insulin resistance. Am. J. Physiol. Ren. Physiol. 2017, 312, F312–F321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogo, A.B. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat. Rev. Nephrol. 2015, 11, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Maimaitiyiming, H.; Zhou, Q.; Wang, S. Thrombospondin 1 Deficiency Ameliorates the Development of Adriamycin-Induced Proteinuric Kidney Disease. PLoS ONE 2016, 11, e0156144. [Google Scholar] [CrossRef] [Green Version]
- Chawla, L.S.; Kimmel, P.L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 2012, 82, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; McMillan, K.L.; Wu, J.; Gillings, N.; Flores, B.; Moe, O.W.; Hu, M.C. Cisplatin nephrotoxicity as a model of chronic kidney disease. Lab. Investig. 2018, 98, 1105–1121. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, J.; Wang, L.; Jiang, S.; Fu, L.; Buggs, J.; Liu, R. New mouse model of chronic kidney disease transitioned from ischemic acute kidney injury. Am. J. Physiol. Ren. Physiol. 2019, 317, F286–F295. [Google Scholar] [CrossRef]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, V.L.; Hunter, D.J. The epidemiology of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2014, 28, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Coryell, P.R.; Diekman, B.O.; Loeser, R.F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 2021, 17, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Hunter, D.J. Post-traumatic osteoarthritis: From mouse models to clinical trials. Nat. Rev. Rheumatol. 2013, 9, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Staines, K.A.; Poulet, B.; Wentworth, D.N.; Pitsillides, A.A. The STR/ort mouse model of spontaneous osteoarthritis—An update. Osteoarthr. Cartil. 2017, 25, 802–808. [Google Scholar] [CrossRef] [Green Version]
- Bapat, S.; Hubbard, D.; Munjal, A.; Hunter, M.; Fulzele, S. Pros and cons of mouse models for studying osteoarthritis. Clin. Transl. Med. 2018, 7, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulloch, K.; Huesa, C.; Dunning, L.; Litherland, G.J.; Van ‘T Hof, R.J.; Lockhart, J.C.; Goodyear, C.S. Accelerated post traumatic osteoarthritis in a dual injury murine model. Osteoarthr. Cartil. 2019, 27, 1800–1810. [Google Scholar] [CrossRef] [Green Version]
- Alves-Simões, M. Rodent models of knee osteoarthritis for pain research. Osteoarthr. Cartil. 2022. [Google Scholar] [CrossRef]
- Poulet, B. Non-invasive Loading Model of Murine Osteoarthritis. Curr. Rheumatol. Rep. 2016, 18, 40. [Google Scholar] [CrossRef] [Green Version]
- Stiffel, V.; Rundle, C.H.; Sheng, M.H.C.; Das, S.; Lau, K.-H.W. A Mouse Noninvasive Intraarticular Tibial Plateau Compression Loading-Induced Injury Model of Posttraumatic Osteoarthritis. Calcif. Tissue Int. 2020, 106, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Easter, M.; Bollenbecker, S.; Barnes, J.W.; Krick, S. Targeting Aging Pathways in Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2020, 21, 6924. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-M.; Liu, G. Cell senescence and fibrotic lung diseases. Exp. Gerontol. 2020, 132, 110836. [Google Scholar] [CrossRef]
- Vlahos, R.; Bozinovski, S. Recent advances in pre-clinical mouse models of COPD. Clin. Sci. 2013, 126, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Ghorani, V.; Boskabady, M.H.; Khazdair, M.R.; Kianmeher, M. Experimental animal models for COPD: A methodological review. Tob. Induc. Dis. 2017, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Bozinovski, S. Preclinical murine models of Chronic Obstructive Pulmonary Disease. Eur. J. Pharmacol. 2015, 759, 265–271. [Google Scholar] [CrossRef]
- He, S.; Li, L.; Sun, S.; Zeng, Z.; Lu, J.; Xie, L. A Novel Murine Chronic Obstructive Pulmonary Disease Model and the Pathogenic Role of MicroRNA-21. Front. Physiol. 2018, 9, 503. [Google Scholar] [CrossRef]
- Serban, K.A.; Petrache, I. Mouse Models of COPD. Methods Mol. Biol. 2018, 1809, 379–394. [Google Scholar] [CrossRef]
- Antunes, M.A.; Rocco, P.R. Elastase-induced pulmonary emphysema: Insights from experimental models. An. Acad. Bras. Cienc. 2011, 83, 1385–1396. [Google Scholar] [CrossRef] [Green Version]
- Brass, D.M.; Hollingsworth, J.W.; Cinque, M.; Li, Z.; Potts, E.; Toloza, E.; Foster, W.M.; Schwartz, D.A. Chronic LPS inhalation causes emphysema-like changes in mouse lung that are associated with apoptosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 584–590. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Fang, L.; Feng, D.; Tang, S.; Yue, S.; Huang, Y.; Han, J.; Lan, J.; Liu, W.; Gao, L.; et al. Memantine ameliorates pulmonary inflammation in a mice model of COPD induced by cigarette smoke combined with LPS. Biomed. Pharmacother. 2019, 109, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Mebratu, Y.A.; Tesfaigzi, Y. IL-17 Plays a Role in Respiratory Syncytial Virus-induced Lung Inflammation and Emphysema in Elastase and LPS-injured Mice. Am. J. Respir. Cell Mol. Biol. 2018, 58, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, J.; Sun, D.; Gong, X.; Jiang, H.; Shu, J.; Wang, Z.; Long, Z.; Chen, Y.; Zhang, Z.; et al. Tanshinone IIA sulfonate protects against cigarette smoke-induced COPD and down-regulation of CFTR in mice. Sci. Rep. 2018, 8, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, B.B.; Hogaboam, C.M. Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L152–L160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claussen, C.A.; Long, E.C. Nucleic Acid Recognition by Metal Complexes of Bleomycin. Chem. Rev. 1999, 99, 2797–2816. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Ma, T.; Cao, H.; Chen, Y.; Wang, C.; Chen, X.; Xiang, Z.; Han, X. TNF-α-induced NF-κB activation promotes myofibroblast differentiation of LR-MSCs and exacerbates bleomycin-induced pulmonary fibrosis. J. Cell. Physiol. 2018, 233, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Chitra, P.; Saiprasad, G.; Manikandan, R.; Sudhandiran, G. Berberine attenuates bleomycin induced pulmonary toxicity and fibrosis via suppressing NF-κB dependant TGF-β activation: A biphasic experimental study. Toxicol. Lett. 2013, 219, 178–193. [Google Scholar] [CrossRef]
- Carrington, R.; Jordan, S.; Pitchford, S.C.; Page, C.P. Use of animal models in IPF research. Pulm. Pharmacol. Ther. 2018, 51, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Redente, E.F.; Black, B.P.; Backos, D.S.; Bahadur, A.N.; Humphries, S.M.; Lynch, D.A.; Tuder, R.M.; Zemans, R.L.; Riches, D.W.H. Persistent, Progressive Pulmonary Fibrosis and Epithelial Remodeling in Mice. Am. J. Respir. Cell Mol. Biol. 2021, 64, 669–676. [Google Scholar] [CrossRef]
- Morgan, G.W.; Breit, S.N. Radiation and the lung: A reevaluation of the mechanisms mediating pulmonary injury. Int. J. Radiat. Oncol. Biol. Phys. 1995, 31, 361–369. [Google Scholar] [CrossRef]
- Degryse, A.L.; Lawson, W.E. Progress toward improving animal models for idiopathic pulmonary fibrosis. Am. J. Med. Sci. 2011, 341, 444–449. [Google Scholar] [CrossRef] [Green Version]
- Nogee, L.M.; Dunbar, A.E., 3rd; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 2001, 344, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.Q.; Lane, K.; Phillips, J., 3rd; Prince, M.; Markin, C.; Speer, M.; Schwartz, D.A.; Gaddipati, R.; Marney, A.; Johnson, J.; et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 2002, 165, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Nureki, S.-I.; Tomer, Y.; Venosa, A.; Katzen, J.; Russo, S.J.; Jamil, S.; Barrett, M.; Nguyen, V.; Kopp, M.; Mulugeta, S.; et al. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J. Clin. Investig. 2018, 128, 4008–4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naikawadi, R.P.; Disayabutr, S.; Mallavia, B.; Donne, M.L.; Green, G.; La, J.L.; Rock, J.R.; Looney, M.R.; Wolters, P.J. Telomere dysfunction in alveolar epithelial cells causes lung remodeling and fibrosis. JCI Insight 2016, 1, e86704. [Google Scholar] [CrossRef] [Green Version]
- Degryse, A.L.; Xu, X.C.; Newman, J.L.; Mitchell, D.B.; Tanjore, H.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Phillips, J.A., 3rd; et al. Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp. Lung Res. 2012, 38, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, L.A.; Hennessy, C.E.; Solomon, G.M.; Dobrinskikh, E.; Estrella, A.; Hara, N.; Hill, D.B.; Kissner, W.J.; Markovetz, M.R.; Grove Villalon, D.E.; et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat. Commun. 2018, 9, 5363. [Google Scholar] [CrossRef]
- He, H.; Huang, C.; Chen, Z.; Huang, H.; Wang, X.; Chen, J. An outlined review for the role of Nedd4-1 and Nedd4-2 in lung disorders. Biomed. Pharmacother. 2020, 125, 109983. [Google Scholar] [CrossRef]
- Duerr, J.; Leitz, D.H.W.; Szczygiel, M.; Dvornikov, D.; Fraumann, S.G.; Kreutz, C.; Zadora, P.K.; Seyhan Agircan, A.; Konietzke, P.; Engelmann, T.A.; et al. Conditional deletion of Nedd4-2 in lung epithelial cells causes progressive pulmonary fibrosis in adult mice. Nat. Commun. 2020, 11, 2012. [Google Scholar] [CrossRef] [PubMed]
- Beike, L.; Wrede, C.; Hegermann, J.; Lopez-Rodriguez, E.; Kloth, C.; Gauldie, J.; Kolb, M.; Maus, U.A.; Ochs, M.; Knudsen, L. Surfactant dysfunction and alveolar collapse are linked with fibrotic septal wall remodeling in the TGF-β1-induced mouse model of pulmonary fibrosis. Lab. Investig. J. Tech. Methods Pathol. 2019, 99, 830–852. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.C.; Kozlov, S.; Hernandez, L.; Sullivan, T.; Stewart, C.L. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature 2003, 423, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.; Magano, S.; Marrana, F.; Andrade, J.P. D-Galactose High-Dose Administration Failed to Induce Accelerated Aging Changes in Neurogenesis, Anxiety, and Spatial Memory on Young Male Wistar Rats. Rejuvenation Res. 2015, 18, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinney, S.E. Intrauterine Growth Retardation—A Developmental Model of Type 2 Diabetes. Drug Discov. Today. Dis. Models 2013, 10, e71–e77. [Google Scholar] [CrossRef] [Green Version]
- Bisht, K.; Sharma, K.; Tremblay, M.-È. Chronic stress as a risk factor for Alzheimer’s disease: Roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobiol Stress 2018, 9, 9–21. [Google Scholar] [CrossRef]
- Sueblinvong, V.; Neujahr, D.C.; Mills, S.T.; Roser-Page, S.; Ritzenthaler, J.D.; Guidot, D.; Rojas, M.; Roman, J. Predisposition for disrepair in the aged lung. Am. J. Med. Sci. 2012, 344, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Type | Subdivision | Phenotypes |
|---|---|---|
| D-galactose-induced senescence model | Brain | Cognitive impairment Mitochondrial dysfunction Neuronal degeneration Apoptosis Depressive and anxious |
| Heart | Cardiac fibrosis Collagen accumulation Fibroblasts disordered arrangement | |
| Kidney | Kidney index ↓ Uric acid & Cys-C ↑ Glomerular and tubular damage ↑ | |
| Liver | Liver fibrosis Glycogen levels ↓ Lipid deposition ↑ | |
| Reproductive system | Estrogen and progesterone ↓ Ovarian follicle regression Uterine wall endometrial gland atrophy Disrupt estrous cycles | |
| Intestinal flora | Disturbance | |
| Lung | Oxidative stress ↑ Fibrotic status Chronic inflammation | |
| SAMP mice | SAMP 1 | Aging amyloidosis Immune dysfunction Renal atrophy Hearing loss Senile pulmonary hyperinflation |
| SAMP 6 | Senile osteoporosis Myeloid progenitor cell senescence | |
| SAMP 8 | Astrogliosis Microgliosis Neurodegeneration Amyloid accumulation MAPT hyperphosphorylation | |
| SAMP 10 | Learning and memory impairment Cerebral cortex and limbic system atrophy | |
| Rps9 D95N mouse | Altered fur Cataracts Hunched posture Body composition function & body weight ↓ Fat mass & muscle strength ↓ Shortened lifespan Mouse urinary syndrome Extramedullary hematopoiesis | |
| Lama−/− | Short lifespan Growth retardation Muscular dystrophy Altered lipid metabolism | |
| Wrn∆hel/∆hel | Short lifespan Abnormal hyaluronic acid excretion Metabolic abnormalities Increased genomic instability and cancer incidence | |
| Csa−/−, Csb−/− | Short lifespan Reduced fat mass Photoreceptor cell loss Neural pathology | |
| Progeria syndrome mouse | XpdTTD/TTD | Short lifespan Trichothiodystrophy |
| Bub1bH/H, Xpg−/− | Brain atrophy Neuronal loss Neurofibrillary deposition of Aβ or senile plaques | |
| Bub1bH/H, Bub1b+/GTTA | Mean muscle fiber diameter ↓ Muscle fiber size variation ↑ Intermuscular fibrosis Regenerative capacity of skeletal muscles ↓ | |
| Mitochondrial DNA polymerase mutant mouse | Lifespan ↓ Weight loss Subcutaneous fat ↓ Hair loss Kyphosis Osteoporosis Anemia Fertility ↓ Spermatogonia depletion Heart enlargement | |
| Total body irradiation (TBI) model | Progressive premature frailty Cognitive decline Whole blood antioxidant capacity ↓ RBC glutathione ↓ Thymic involution Articular cartilage and bone degeneration Ovarian environment damage | |
| Ozone-induced senescence model | Cognitive decline Memory impairment AD symptoms Lung tumor growth ↑ | |
| Chronic jet-lag mouse | Accelerated initial tumor growth Shortened mouse survival Induce spontaneous hepatocellular carcinoma Obesity Depression Addiction Abnormal cardiac structure Impaired cardiac function |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, N.; Wu, Y.; Huang, Y. Induction of Accelerated Aging in a Mouse Model. Cells 2022, 11, 1418. https://doi.org/10.3390/cells11091418
Cai N, Wu Y, Huang Y. Induction of Accelerated Aging in a Mouse Model. Cells. 2022; 11(9):1418. https://doi.org/10.3390/cells11091418
Chicago/Turabian StyleCai, Nanshuo, Yifan Wu, and Yan Huang. 2022. "Induction of Accelerated Aging in a Mouse Model" Cells 11, no. 9: 1418. https://doi.org/10.3390/cells11091418
APA StyleCai, N., Wu, Y., & Huang, Y. (2022). Induction of Accelerated Aging in a Mouse Model. Cells, 11(9), 1418. https://doi.org/10.3390/cells11091418