
KV Channel-Interacting Proteins in the Neurological and Cardiovascular Systems: An Updated Review


Abstract
:1. Introduction

{kind=link}
{kind=link}
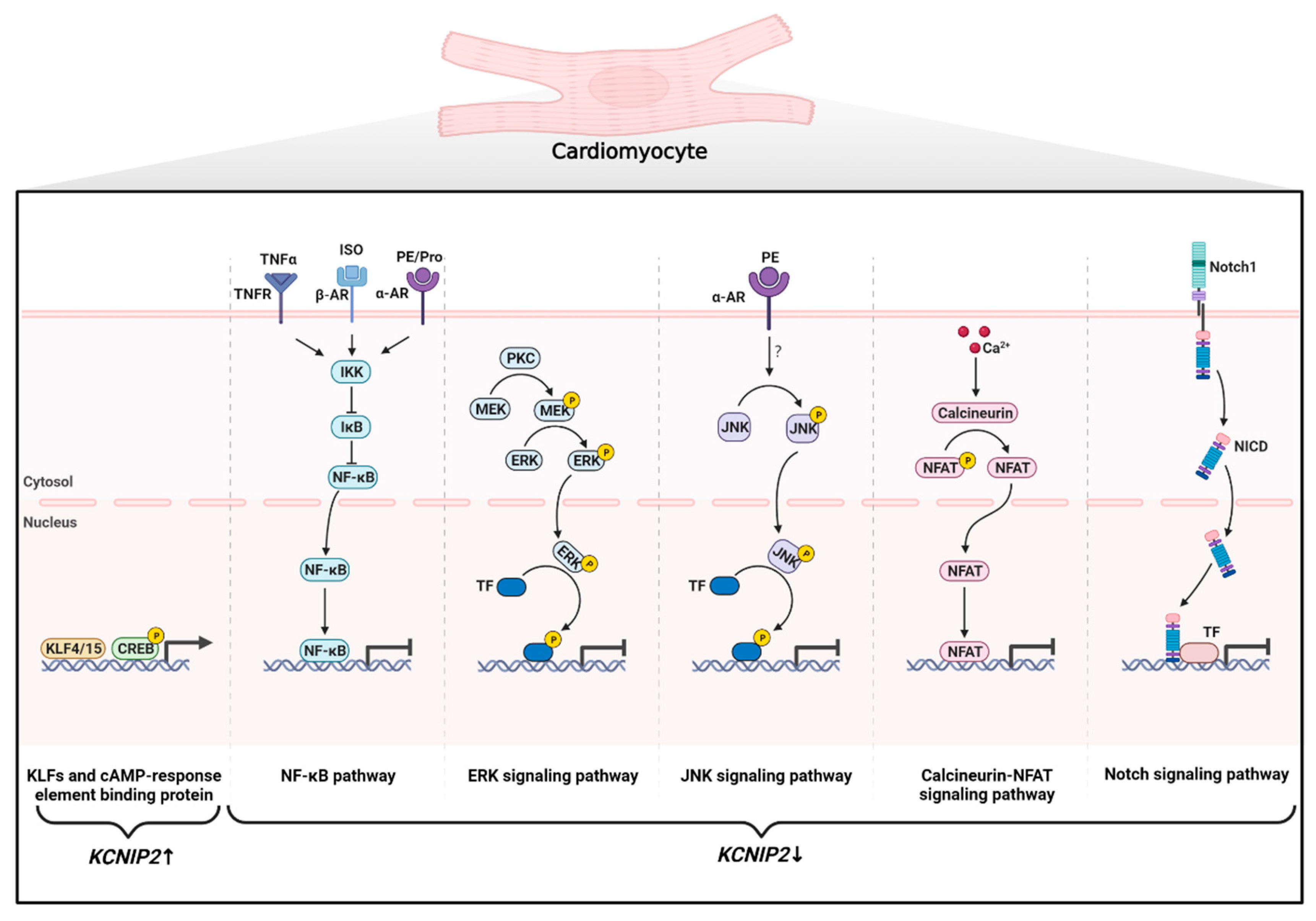{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KChIP1 | KChIP2 | KChIP3 | KChIP4 | |
|---|---|---|---|---|
| Aliases | NA | NA | DREAM [8]; Calsenilin [7] | CALP [10] |
| Tissue distribution | Brain [1]; Heart [13]; Intestine [4,5]; Stomach [6]; Pancreas [14] | Brain [15]; Heart [1]; Lung [2]; Immune System [16] | Brain [1]; Heart [13,17]; Lung [2]; Immune System [16]; Nasal Mucosa [18] | Brain [1]; Kidney [3] |
| Post-translational modification | Myristoylation [19] | Palmitoylation [20] | Palmitoylation [20] Phosphorylation [21] Sumoylation [22] | Palmitoylation [20] |
| Interacted KV subtypes | KV4.1 [23,24] KV4.2 [9,24,25,26] KV4.3 [9,23,25,26] | KV4.1 [27] KV4.2 [9,27] KV4.3 [27] | KV4.1 [28] KV4.2 [9,29,30,31] KV4.3 [32,33,34] | KV4.2 [30] KV4.3 [25,35] |
2. The Molecular Properties of KChIPs
2.1. The Structure of KChIPs
2.2. Regulation of KChIPs Expression
2.2.1. Transcriptional Level
2.2.2. Protein Level
3. Biological Function of KChIPs
3.1. KChIPs Are Auxiliary Subunits of KV4 Channels
3.1.1. The Interaction of KChIPs with KV4 Channels
3.1.2. KChIPs Modulate the Gating Properties of KV4 Channels
3.1.3. KChIPs Modulate the Trafficking of KV4 Channels
3.1.4. KChIP Ligands Affect KChIP Regulation on KV4
| Compounds | KChIP1 | KChIP2 | KChIP3 | KChIP4 |
|---|---|---|---|---|
| Arachidonic acid | Accelerate KV4 inactivation; Reduce current amplitude [26] | NA | NA | NA |
| CL-888 | Reduce peak current amplitude; Accelerate inactivation kinetics [73] | NA | Reduce peak current amplitude; Accelerate inactivation kinetics [74] | NA |
| IQM-PC330 IQM-PC332 | NA | NA | Reduce peak current amplitude; Accelerate inactivation kinetics; Delay recovery from inactivation [75] | NA |
| IQM-266 | NA | Increase current [77] | Reduce peak current amplitude; Accelerate inactivation kinetics; Delay recovery from inactivation [76] | NA |
| NS5806 | NA | Play opposite roles in different species, even in different tissues from one species [79,80,81] | Activate neural KV4.3; Delay inactivation kinetics; Reduce the maximum peak current slightly [78] | NA |
| Repaglinide | NA | NA | Inhibit KV4 channels [74] | NA |
3.2. Role of KChIPs in Regulating Other Ion Channels
3.2.1. KV1.5
3.2.2. CaV1.2
3.2.3. NaV1.5
3.3. KChIPs Are Ca2+-Dependent Transcriptional Factors
4. KChIPs and Diseases
4.1. Neurological Diseases
4.1.1. Epilepsy
4.1.2. Pain
4.1.3. Memory Dysfunction
4.1.4. Alzheimer’s Disease
4.1.5. Other Neurodegenerative Disorders
4.2. Cardiovascular Diseases
4.2.1. Arrhythmias
4.2.2. Cardiac Remodeling
4.2.3. Heart Failure
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosati, B.; Pan, Z.; Lypen, S.; Wang, H.S.; Cohen, I.; Dixon, J.E.; McKinnon, D. Regulation of KChIP2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J. Physiol. 2001, 533, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Maniak, P.J.; Ingbar, D.H.; O’Grady, S.M. Adult alveolar epithelial cells express multiple subtypes of voltage-gated K+ channels that are located in apical membrane. Am. J. Physiol. Cell Physiol. 2003, 284, C1614–C1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonne, A.; Vreede, L.; Kuiper, R.P.; Bodmer, D.; Jansen, C.; Eleveld, M.; van Erp, F.; Arkesteijn, G.; Hoogerbrugge, N.; van Ravenswaaij, C.; et al. Mapping of constitutional translocation breakpoints in renal cell cancer patients: Identification of KCNIP4 as a candidate gene. Cancer Genet. Cytogenet. 2007, 179, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Amberg, G.C.; Koh, S.D.; Hatton, W.J.; Murray, K.J.; Monaghan, K.; Horowitz, B.; Sanders, K.M. Contribution of Kv4 channels toward the A-type potassium current in murine colonic myocytes. J. Physiol. 2002, 544, 403–415. [Google Scholar] [CrossRef]
- Beckett, E.A.; McCloskey, C.; O’Kane, N.; Sanders, K.M.; Koh, S.D. Effects of female steroid hormones on A-type K+ currents in murine colon. J. Physiol. 2006, 573, 453–468. [Google Scholar] [CrossRef]
- Zhang, Y.; MacLean, J.N.; An, W.F.; Lanning, C.C.; Harris-Warrick, R.M. KChIP1 and frequenin modify shal-evoked potassium currents in pyloric neurons in the lobster stomatogastric ganglion. J. Neurophysiol. 2003, 89, 1902–1909. [Google Scholar] [CrossRef] [Green Version]
- Buxbaum, J.D.; Choi, E.K.; Luo, Y.; Lilliehook, C.; Crowley, A.C.; Merriam, D.E.; Wasco, W. Calsenilin: A calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med. 1998, 4, 1177–1181. [Google Scholar] [CrossRef]
- Carrión, A.M.; Link, W.A.; Ledo, F.; Mellström, B.; Naranjo, J.R. DREAM is a Ca2+-regulated transcriptional repressor. Nature 1999, 398, 80–84. [Google Scholar] [CrossRef]
- An, W.F.; Bowlby, M.R.; Betty, M.; Cao, J.; Ling, H.P.; Mendoza, G.; Hinson, J.W.; Mattsson, K.I.; Strassle, B.W.; Trimmer, J.S.; et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature 2000, 403, 553–556. [Google Scholar] [CrossRef]
- Morohashi, Y.; Hatano, N.; Ohya, S.; Takikawa, R.; Watabiki, T.; Takasugi, N.; Imaizumi, Y.; Tomita, T.; Iwatsubo, T. Molecular cloning and characterization of CALP/KChIP4, a novel EF-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit Kv4. J. Biol. Chem. 2002, 277, 14965–14975. [Google Scholar] [CrossRef] [Green Version]
- Cho, J. Downstream Regulatory Element Antagonist Modulator (DREAM), a target for anti-thrombotic agents. Pharmacol. Res. 2017, 117, 283–287. [Google Scholar] [CrossRef]
- Cercós, P.; Peraza, D.A.; Benito-Bueno, A.; Socuéllamos, P.G.; Aziz-Nignan, A.; Arrechaga-Estévez, D.; Beato, E.; Peña-Acevedo, E.; Albert, A.; González-Vera, J.A.; et al. Pharmacological Approaches for the Modulation of the Potassium Channel K(V)4.x and KChIPs. Int. J. Mol. Sci. 2021, 22, 1419. [Google Scholar] [CrossRef]
- Tsai, C.T.; Hsieh, C.S.; Chang, S.N.; Chuang, E.Y.; Ueng, K.C.; Tsai, C.F.; Lin, T.H.; Wu, C.K.; Lee, J.K.; Lin, L.Y.; et al. Genome-wide screening identifies a KCNIP1 copy number variant as a genetic predictor for atrial fibrillation. Nat. Commun. 2016, 7, 10190. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Pinna, J.; Marroqui, L.; Hmadcha, A.; Lopez-Beas, J.; Soriano, S.; Villar-Pazos, S.; Alonso-Magdalena, P.; Dos Santos, R.S.; Quesada, I.; Martin, F.; et al. Oestrogen receptor β mediates the actions of bisphenol-A on ion channel expression in mouse pancreatic beta cells. Diabetologia 2019, 62, 1667–1680. [Google Scholar] [CrossRef] [Green Version]
- Norris, A.J.; Foeger, N.C.; Nerbonne, J.M. Interdependent roles for accessory KChIP2, KChIP3, and KChIP4 subunits in the generation of Kv4-encoded IA channels in cortical pyramidal neurons. J. Neurosci. 2010, 30, 13644–13655. [Google Scholar] [CrossRef] [Green Version]
- Savignac, M.; Mellström, B.; Bébin, A.G.; Oliveros, J.C.; Delpy, L.; Pinaud, E.; Naranjo, J.R. Increased B cell proliferation and reduced Ig production in DREAM transgenic mice. J. Immunol. 2010, 185, 7527–7536. [Google Scholar] [CrossRef] [Green Version]
- Ronkainen, J.J.; Hänninen, S.L.; Korhonen, T.; Koivumäki, J.T.; Skoumal, R.; Rautio, S.; Ronkainen, V.P.; Tavi, P. Ca2+-calmodulin-dependent protein kinase II represses cardiac transcription of the L-type calcium channel alpha(1C)-subunit gene (Cacna1c) by DREAM translocation. J. Physiol. 2011, 589, 2669–2686. [Google Scholar] [CrossRef]
- Lee, J.Y.; Byun, J.Y.; Lee, S.H. Proteomic analysis of normal human nasal mucosa: Establishment of a two-dimensional electrophoresis reference map. Clin. Biochem. 2009, 42, 692–700. [Google Scholar] [CrossRef]
- O’Callaghan, D.W.; Hasdemir, B.; Leighton, M.; Burgoyne, R.D. Residues within the myristoylation motif determine intracellular targeting of the neuronal Ca2+ sensor protein KChIP1 to post-ER transport vesicles and traffic of Kv4 K+ channels. J. Cell Sci. 2003, 116, 4833–4845. [Google Scholar] [CrossRef] [Green Version]
- Takimoto, K.; Yang, E.K.; Conforti, L. Palmitoylation of KChIP splicing variants is required for efficient cell surface expression of Kv4.3 channels. J. Biol. Chem. 2002, 277, 26904–26911. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Gomez, A.; Mellström, B.; Tornero, D.; Morato, E.; Savignac, M.; Holguín, H.; Aurrekoetxea, K.; González, P.; González-García, C.; Ceña, V.; et al. G protein-coupled receptor kinase 2-mediated phosphorylation of downstream regulatory element antagonist modulator regulates membrane trafficking of Kv4.2 potassium channel. J. Biol. Chem. 2007, 282, 1205–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palczewska, M.; Casafont, I.; Ghimire, K.; Rojas, A.M.; Valencia, A.; Lafarga, M.; Mellström, B.; Naranjo, J.R. Sumoylation regulates nuclear localization of repressor DREAM. Biochim. Biophys. Acta 2011, 1813, 1050–1058. [Google Scholar] [CrossRef]
- Beck, E.J.; Bowlby, M.; An, W.F.; Rhodes, K.J.; Covarrubias, M. Remodelling inactivation gating of Kv4 channels by KChIP1, a small-molecular-weight calcium-binding protein. J. Physiol. 2002, 538, 691–706. [Google Scholar] [CrossRef]
- Nakamura, T.Y.; Nandi, S.; Pountney, D.J.; Artman, M.; Rudy, B.; Coetzee, W.A. Different effects of the Ca(2+)-binding protein, KChIP1, on two Kv4 subfamily members, Kv4.1 and Kv4.2. FEBS Lett. 2001, 499, 205–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmqvist, M.H.; Cao, J.; Hernandez-Pineda, R.; Jacobson, M.D.; Carroll, K.I.; Sung, M.A.; Betty, M.; Ge, P.; Gilbride, K.J.; Brown, M.E.; et al. Elimination of fast inactivation in Kv4 A-type potassium channels by an auxiliary subunit domain. Proc. Natl. Acad. Sci. USA 2002, 99, 1035–1040. [Google Scholar] [CrossRef]
- Holmqvist, M.H.; Cao, J.; Knoppers, M.H.; Jurman, M.E.; Distefano, P.S.; Rhodes, K.J.; Xie, Y.; An, W.F. Kinetic modulation of Kv4-mediated A-current by arachidonic acid is dependent on potassium channel interacting proteins. J. Neurosci. 2001, 21, 4154–4161. [Google Scholar] [CrossRef]
- Bähring, R.; Dannenberg, J.; Peters, H.C.; Leicher, T.; Pongs, O.; Isbrandt, D. Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. J. Biol. Chem. 2001, 276, 23888–23894. [Google Scholar] [CrossRef] [Green Version]
- Matsuyoshi, H.; Takimoto, K.; Yunoki, T.; Erickson, V.L.; Tyagi, P.; Hirao, Y.; Wanaka, A.; Yoshimura, N. Distinct cellular distributions of Kv4 pore-forming and auxiliary subunits in rat dorsal root ganglion neurons. Life Sci. 2012, 91, 258–263. [Google Scholar] [CrossRef] [Green Version]
- Jerng, H.H.; Kunjilwar, K.; Pfaffinger, P.J. Multiprotein assembly of Kv4.2, KChIP3 and DPP10 produces ternary channel complexes with ISA-like properties. J. Physiol. 2005, 568, 767–788. [Google Scholar] [CrossRef] [PubMed]
- Jerng, H.H.; Pfaffinger, P.J. Multiple Kv channel-interacting proteins contain an N-terminal transmembrane domain that regulates Kv4 channel trafficking and gating. J. Biol. Chem. 2008, 283, 36046–36059. [Google Scholar] [CrossRef] [Green Version]
- Kunjilwar, K.; Strang, C.; DeRubeis, D.; Pfaffinger, P.J. KChIP3 rescues the functional expression of Shal channel tetramerization mutants. J. Biol. Chem. 2004, 279, 54542–54551. [Google Scholar] [CrossRef] [Green Version]
- Jerng, H.H.; Pfaffinger, P.J.; Covarrubias, M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol. Cell. Neurosci. 2004, 27, 343–369. [Google Scholar] [CrossRef]
- Alexander, J.C.; McDermott, C.M.; Tunur, T.; Rands, V.; Stelly, C.; Karhson, D.; Bowlby, M.R.; An, W.F.; Sweatt, J.D.; Schrader, L.A. The role of calsenilin/DREAM/KChIP3 in contextual fear conditioning. Learn. Mem. 2009, 16, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, W.G.; Pham, K.; Miksovska, J. Modulation of the voltage-gated potassium channel (Kv4.3) and the auxiliary protein (KChIP3) interactions by the current activator NS5806. J. Biol. Chem. 2014, 289, 32201–32213. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, J.; Zolles, G.; Kandias, N.G.; Neubauer, I.; Kalbacher, H.; Covarrubias, M.; Fakler, B.; Bentrop, D. NMR analysis of KChIP4a reveals structural basis for control of surface expression of Kv4 channel complexes. J. Biol. Chem. 2008, 283, 18937–18946. [Google Scholar] [CrossRef] [Green Version]
- Néant, I.; Haiech, J.; Kilhoffer, M.C.; Aulestia, F.J.; Moreau, M.; Leclerc, C. Ca(2+)-Dependent Transcriptional Repressors KCNIP and Regulation of Prognosis Genes in Glioblastoma. Front. Mol. Neurosci. 2018, 11, 472. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.Q.; Liang, P.; Zhou, J.; Lu, Y.; Lei, L.; Bian, X.; Wang, K. Auxiliary KChIP4a suppresses A-type K+ current through endoplasmic reticulum (ER) retention and promoting closed-state inactivation of Kv4 channels. J. Biol. Chem. 2013, 288, 14727–14741. [Google Scholar] [CrossRef] [Green Version]
- Jerng, H.H.; Pfaffinger, P.J. Modulatory mechanisms and multiple functions of somatodendritic A-type K(+) channel auxiliary subunits. Front. Cell. Neurosci. 2014, 8, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bähring, R. Kv channel-interacting proteins as neuronal and non-neuronal calcium sensors. Channels 2018, 12, 187–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murthy, A.; Workman, S.W.; Jiang, M.; Hu, J.; Sifa, I.; Bernas, T.; Tang, W.; Deschenes, I.; Tseng, G.N. Dynamic palmitoylation regulates trafficking of K channel interacting protein 2 (KChIP2) across multiple subcellular compartments in cardiac myocytes. J. Mol. Cell. Cardiol. 2019, 135, 1–9. [Google Scholar] [CrossRef]
- Choi, E.K.; Miller, J.S.; Zaidi, N.F.; Salih, E.; Buxbaum, J.D.; Wasco, W. Phosphorylation of calsenilin at Ser63 regulates its cleavage by caspase-3. Mol. Cell. Neurosci. 2003, 23, 495–506. [Google Scholar] [CrossRef]
- Liang, P.; Wang, H.; Chen, H.; Cui, Y.; Gu, L.; Chai, J.; Wang, K. Structural Insights into KChIP4a Modulation of Kv4.3 Inactivation. J. Biol. Chem. 2009, 284, 4960–4967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, G.; Zhang, C.; Ding, W.; Cheng, W.; Luo, Y.; Wei, C.; Liu, J. MG53, A Novel Regulator of KChIP2 and I(to,f), Plays a Critical Role in Electrophysiological Remodeling in Cardiac Hypertrophy. Circulation 2019, 139, 2142–2156. [Google Scholar] [CrossRef]
- Wagner, S.; Hacker, E.; Grandi, E.; Weber, S.L.; Dybkova, N.; Sossalla, S.; Sowa, T.; Fabritz, L.; Kirchhof, P.; Bers, D.M.; et al. Ca/calmodulin kinase II differentially modulates potassium currents. Circ. Arrhythmia Electrophysiol. 2009, 2, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossow, C.F.; Dilly, K.W.; Santana, L.F. Differential calcineurin/NFATc3 activity contributes to the Ito transmural gradient in the mouse heart. Circ. Res. 2006, 98, 1306–1313. [Google Scholar] [CrossRef]
- Jia, Y.; Takimoto, K. Mitogen-activated protein kinases control cardiac KChIP2 gene expression. Circ. Res. 2006, 98, 386–393. [Google Scholar] [CrossRef]
- Huang, H.; Tang, Y.; Wu, G.; Mei, Y.; Liu, W.; Liu, X.; Wan, N.; Liu, Y.; Huang, C. ALK7 protects against pathological cardiac hypertrophy in mice. Cardiovasc. Res. 2015, 108, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Ying, S.; Cao, H.; Hu, H.; Wang, X.; Tang, Y.; Huang, C. Alk7 Depleted Mice Exhibit Prolonged Cardiac Repolarization and Are Predisposed to Ventricular Arrhythmia. PLoS ONE 2016, 11, e0149205. [Google Scholar] [CrossRef]
- Borghetti, G.; Eisenberg, C.A.; Signore, S.; Sorrentino, A.; Kaur, K.; Andrade-Vicenty, A.; Edwards, J.G.; Nerkar, M.; Qanud, K.; Sun, D.; et al. Notch signaling modulates the electrical behavior of cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H68–H81. [Google Scholar] [CrossRef]
- Kaur, K.; Zarzoso, M.; Ponce-Balbuena, D.; Guerrero-Serna, G.; Hou, L.; Musa, H.; Jalife, J. TGF-β1, released by myofibroblasts, differentially regulates transcription and function of sodium and potassium channels in adult rat ventricular myocytes. PLoS ONE 2013, 8, e55391. [Google Scholar] [CrossRef] [PubMed]
- Panama, B.K.; Latour-Villamil, D.; Farman, G.P.; Zhao, D.; Bolz, S.S.; Kirshenbaum, L.A.; Backx, P.H. Nuclear factor kappaB downregulates the transient outward potassium current I(to,f) through control of KChIP2 expression. Circ. Res. 2011, 108, 537–543. [Google Scholar] [CrossRef]
- Panama, B.K.; Korogyi, A.S.; Aschar-Sobbi, R.; Oh, Y.; Gray, C.B.; Gang, H.; Brown, J.H.; Kirshenbaum, L.A.; Backx, P.H. Reductions in the Cardiac Transient Outward K+ Current Ito Caused by Chronic β-Adrenergic Receptor Stimulation Are Partly Rescued by Inhibition of Nuclear Factor κB. J. Biol. Chem. 2016, 291, 4156–4165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Mai, J.T.; Wang, F.; Lin, Y.Q.; Yuan, W.L.; Luo, N.S.; Fang, M.C.; Wang, J.F.; Chen, Y.X. Effects of C-reactive protein on K(+) channel interaction protein 2 in cardiomyocytes. Am. J. Transl. Res. 2015, 7, 922–931. [Google Scholar]
- Ozgen, N.; Lau, D.H.; Shlapakova, I.N.; Sherman, W.; Feinmark, S.J.; Danilo, P., Jr.; Rosen, M.R. Determinants of CREB degradation and KChIP2 gene transcription in cardiac memory. Heart Rhythm 2010, 7, 964–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, S.K.; Liu, W.; Zi, M.; Li, Y.; Wang, S.; Tsui, H.; Prehar, S.; Castro, S.; Zhang, H.; Ji, Y.; et al. Stress-Activated Kinase Mitogen-Activated Kinase Kinase-7 Governs Epigenetics of Cardiac Repolarization for Arrhythmia Prevention. Circulation 2017, 135, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraj, D.; Haldar, S.M.; Wan, X.; McCauley, M.D.; Ripperger, J.A.; Hu, K.; Lu, Y.; Eapen, B.L.; Sharma, N.; Ficker, E.; et al. Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature 2012, 483, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Menegola, M.; Trimmer, J.S. Unanticipated region- and cell-specific downregulation of individual KChIP auxiliary subunit isotypes in Kv4.2 knock-out mouse brain. J. Neurosci. 2006, 26, 12137–12142. [Google Scholar] [CrossRef]
- Nerbonne, J.M.; Gerber, B.R.; Norris, A.; Burkhalter, A. Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. J. Physiol. 2008, 586, 1565–1579. [Google Scholar] [CrossRef]
- Sun, W.; Maffie, J.K.; Lin, L.; Petralia, R.S.; Rudy, B.; Hoffman, D.A. DPP6 establishes the A-type K(+) current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron 2011, 71, 1102–1115. [Google Scholar] [CrossRef] [Green Version]
- Kuo, Y.L.; Cheng, J.K.; Hou, W.H.; Chang, Y.C.; Du, P.H.; Jian, J.J.; Rau, R.H.; Yang, J.H.; Lien, C.C.; Tsaur, M.L. K(+) Channel Modulatory Subunits KChIP and DPP Participate in Kv4-Mediated Mechanical Pain Control. J. Neurosci. 2017, 37, 4391–4404. [Google Scholar] [CrossRef]
- Mellström, B.; Sahún, I.; Ruiz-Nuño, A.; Murtra, P.; Gomez-Villafuertes, R.; Savignac, M.; Oliveros, J.C.; Gonzalez, P.; Kastanauskaite, A.; Knafo, S.; et al. DREAM controls the on/off switch of specific activity-dependent transcription pathways. Mol. Cell. Biol. 2014, 34, 877–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kise, Y.; Kasuya, G.; Okamoto, H.H.; Yamanouchi, D.; Kobayashi, K.; Kusakizako, T.; Nishizawa, T.; Nakajo, K.; Nureki, O. Structural basis of gating modulation of Kv4 channel complexes. Nature 2021, 599, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Campbell, D.L. Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: Underlying molecular, cellular and biophysical mechanisms. J. Physiol. 2005, 569, 7–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.A.; Furst, J.; Gutierrez, D.; Butler, M.H.; Xu, S.; Goldstein, S.A.; Grigorieff, N. Three-dimensional structure of I(to); Kv4.2-KChIP2 ion channels by electron microscopy at 21 Angstrom resolution. Neuron 2004, 41, 513–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yan, Y.; Liu, Q.; Huang, Y.; Shen, Y.; Chen, L.; Chen, Y.; Yang, Q.; Hao, Q.; Wang, K.; et al. Structural basis for modulation of Kv4 K+ channels by auxiliary KChIP subunits. Nat. Neurosci. 2007, 10, 32–39. [Google Scholar] [CrossRef]
- Catte, A.; Ferbel, L.; Bhattacharjee, N.; Jan Akhunzada, M.; D’Agostino, T.; Brancato, G. In silico investigation of the interaction between the voltage-gated potassium channel Kv4.3 and its auxiliary protein KChIP1. Phys. Chem. Chem. Phys. 2019, 21, 25290–25301. [Google Scholar] [CrossRef]
- Fineberg, J.D.; Szanto, T.G.; Panyi, G.; Covarrubias, M. Closed-state inactivation involving an internal gate in Kv4.1 channels modulates pore blockade by intracellular quaternary ammonium ions. Sci. Rep. 2016, 6, 31131. [Google Scholar] [CrossRef]
- Patel, S.P.; Campbell, D.L.; Strauss, H.C. Elucidating KChIP effects on Kv4.3 inactivation and recovery kinetics with a minimal KChIP2 isoform. J. Physiol. 2002, 545, 5–11. [Google Scholar] [CrossRef]
- Ye, W.; Zhao, H.; Dai, Y.; Wang, Y.; Lo, Y.H.; Jan, L.Y.; Lee, C.H. Activation and closed-state inactivation mechanisms of the human voltage-gated K(V)4 channel complexes. Mol. Cell 2022, 82, 2427–2442.e2424. [Google Scholar] [CrossRef]
- Shibata, R.; Misonou, H.; Campomanes, C.R.; Anderson, A.E.; Schrader, L.A.; Doliveira, L.C.; Carroll, K.I.; Sweatt, J.D.; Rhodes, K.J.; Trimmer, J.S. A fundamental role for KChIPs in determining the molecular properties and trafficking of Kv4.2 potassium channels. J. Biol. Chem. 2003, 278, 36445–36454. [Google Scholar] [CrossRef] [Green Version]
- Foeger, N.C.; Marionneau, C.; Nerbonne, J.M. Co-assembly of Kv4 {alpha} subunits with K+ channel-interacting protein 2 stabilizes protein expression and promotes surface retention of channel complexes. J. Biol. Chem. 2010, 285, 33413–33422. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Duan, H.; Yi, J.; Wang, G.; Cheng, W.; Feng, L.; Liu, J. Kv4.2 phosphorylation by PKA drives Kv4.2-KChIP2 dissociation, leading to Kv4.2 out of lipid rafts and internalization. Am. J. Physiol. Cell Physiol. 2022, 323, C190–C201. [Google Scholar] [CrossRef]
- Bowlby, M.R.; Chanda, P.; Edris, W.; Hinson, J.; Jow, F.; Katz, A.H.; Kennedy, J.; Krishnamurthy, G.; Pitts, K.; Ryan, K.; et al. Identification and characterization of small molecule modulators of KChIP/Kv4 function. Bioorg. Med. Chem. 2005, 13, 6112–6119. [Google Scholar] [CrossRef]
- Naranjo, J.R.; Zhang, H.; Villar, D.; González, P.; Dopazo, X.M.; Morón-Oset, J.; Higueras, E.; Oliveros, J.C.; Arrabal, M.D.; Prieto, A.; et al. Activating transcription factor 6 derepression mediates neuroprotection in Huntington disease. J. Clin. Investig. 2016, 126, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Hurtado, A.; Peraza, D.A.; Cercos, P.; Lagartera, L.; Gonzalez, P.; Dopazo, X.M.; Herranz, R.; Gonzalez, T.; Martin-Martinez, M.; Mellström, B.; et al. Targeting the neuronal calcium sensor DREAM with small-molecules for Huntington’s disease treatment. Sci. Rep. 2019, 9, 7260. [Google Scholar] [CrossRef] [Green Version]
- Peraza, D.A.; Cercós, P.; Miaja, P.; Merinero, Y.G.; Lagartera, L.; Socuéllamos, P.G.; Izquierdo García, C.; Sánchez, S.A.; López-Hurtado, A.; Martín-Martínez, M.; et al. Identification of IQM-266, a Novel DREAM Ligand That Modulates K(V)4 Currents. Front. Mol. Neurosci. 2019, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- de Benito-Bueno, A.; Socuellamos, P.G.; Merinero, Y.G.; Cercos, P.; Izquierdo, C.; Daniel-Mozo, M.; Marín-Olivero, I.; Perez-Lara, A.; Gonzalez-Vera, J.A.; Orte, A.; et al. Modulation of K(V)4.3-KChIP2 Channels by IQM-266: Role of DPP6 and KCNE2. Int. J. Mol. Sci. 2022, 23, 9170. [Google Scholar] [CrossRef]
- Witzel, K.; Fischer, P.; Bähring, R. Hippocampal A-type current and Kv4.2 channel modulation by the sulfonylurea compound NS5806. Neuropharmacology 2012, 63, 1389–1403. [Google Scholar] [CrossRef]
- Calloe, K.; Soltysinska, E.; Jespersen, T.; Lundby, A.; Antzelevitch, C.; Olesen, S.P.; Cordeiro, J.M. Differential effects of the transient outward K(+) current activator NS5806 in the canine left ventricle. J. Mol. Cell. Cardiol. 2010, 48, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, H.; Wang, C.; Wang, Y.; Zou, R.; Shi, C.; Guan, B.; Gamper, N.; Xu, Y. Auxiliary subunits control biophysical properties and response to compound NS5806 of the Kv4 potassium channel complex. FASEB J. 2020, 34, 807–821. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Cannell, M.B.; Hancox, J.C. Differential responses of rabbit ventricular and atrial transient outward current (I(to)) to the I(to) modulator NS5806. Physiol. Rep. 2017, 5, e13172. [Google Scholar] [CrossRef] [PubMed]
- Maqoud, F.; Scala, R.; Tragni, V.; Pierri, C.L.; Perrone, M.G.; Scilimati, A.; Tricarico, D. Zoledronic Acid as a Novel Dual Blocker of KIR6.1/2-SUR2 Subunits of ATP-Sensitive K(+) Channels: Role in the Adverse Drug Reactions. Pharmaceutics 2021, 13, 1350. [Google Scholar] [CrossRef] [PubMed]
- Maqoud, F.; Scala, R.; Hoxha, M.; Zappacosta, B.; Tricarico, D. ATP-sensitive Potassium Channel Subunits in Neuroinflammation: Novel Drug Targets in Neurodegenerative Disorders. CNS Neurol. Disord. Drug Targets 2022, 21, 130–149. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Wahl, P.; Holmes, W.E.; Ashcroft, F.M. Effect of repaglinide on cloned beta cell, cardiac and smooth muscle types of ATP-sensitive potassium channels. Diabetologia 2001, 44, 747–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravens, U.; Wettwer, E. Ultra-rapid delayed rectifier channels: Molecular basis and therapeutic implications. Cardiovasc. Res. 2011, 89, 776–785. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Guo, W.; Mellor, R.L.; Nerbonne, J.M. KChIP2 modulates the cell surface expression of Kv 1.5-encoded K(+) channels. J. Mol. Cell. Cardiol. 2005, 39, 121–132. [Google Scholar] [CrossRef]
- Thomsen, M.B.; Sosunov, E.A.; Anyukhovsky, E.P.; Ozgen, N.; Boyden, P.A.; Rosen, M.R. Deleting the accessory subunit KChIP2 results in loss of I(to,f) and increased I(K,slow) that maintains normal action potential configuration. Heart Rhythm 2009, 6, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, F.; Flockerzi, V.; Kahl, S.; Wegener, J.W. L-type CaV1.2 calcium channels: From in vitro findings to in vivo function. Physiol. Rev. 2014, 94, 303–326. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.B.; Wang, C.; Ozgen, N.; Wang, H.G.; Rosen, M.R.; Pitt, G.S. Accessory subunit KChIP2 modulates the cardiac L-type calcium current. Circ. Res. 2009, 104, 1382–1389. [Google Scholar] [CrossRef]
- Thomsen, M.B.; Foster, E.; Nguyen, K.H.; Sosunov, E.A. Transcriptional and electrophysiological consequences of KChIP2-mediated regulation of CaV1.2. Channels 2009, 3, 308–310. [Google Scholar] [CrossRef] [Green Version]
- Clatot, J.; Neyroud, N.; Cox, R.; Souil, C.; Huang, J.; Guicheney, P.; Antzelevitch, C. Inter-Regulation of K(v)4.3 and Voltage-Gated Sodium Channels Underlies Predisposition to Cardiac and Neuronal Channelopathies. Int. J. Mol. Sci. 2020, 21, 5057. [Google Scholar] [CrossRef]
- Deschênes, I.; Armoundas, A.A.; Jones, S.P.; Tomaselli, G.F. Post-transcriptional gene silencing of KChIP2 and Navbeta1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. J. Mol. Cell. Cardiol. 2008, 45, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Osawa, M.; Dace, A.; Tong, K.I.; Valiveti, A.; Ikura, M.; Ames, J.B. Mg2+ and Ca2+ differentially regulate DNA binding and dimerization of DREAM. J. Biol. Chem. 2005, 280, 18008–18014. [Google Scholar] [CrossRef] [Green Version]
- Lusin, J.D.; Vanarotti, M.; Li, C.; Valiveti, A.; Ames, J.B. NMR structure of DREAM: Implications for Ca(2+)-dependent DNA binding and protein dimerization. Biochemistry 2008, 47, 2252–2264. [Google Scholar] [CrossRef]
- Zaidi, N.F.; Kuplast, K.G.; Washicosky, K.J.; Kajiwara, Y.; Buxbaum, J.D.; Wasco, W. Calsenilin interacts with transcriptional co-repressor C-terminal binding protein(s). J. Neurochem. 2006, 98, 1290–1301. [Google Scholar] [CrossRef]
- Ledo, F.; Carrión, A.M.; Link, W.A.; Mellström, B.; Naranjo, J.R. DREAM-alphaCREM interaction via leucine-charged domains derepresses downstream regulatory element-dependent transcription. Mol. Cell. Biol. 2000, 20, 9120–9126. [Google Scholar] [CrossRef] [Green Version]
- Ledo, F.; Kremer, L.; Mellström, B.; Naranjo, J.R. Ca2+-dependent block of CREB-CBP transcription by repressor DREAM. EMBO J. 2002, 21, 4583–4592. [Google Scholar] [CrossRef] [Green Version]
- Rivas, M.; Mellström, B.; Naranjo, J.R.; Santisteban, P. Transcriptional repressor DREAM interacts with thyroid transcription factor-1 and regulates thyroglobulin gene expression. J. Biol. Chem. 2004, 279, 33114–33122. [Google Scholar] [CrossRef] [Green Version]
- Scsucova, S.; Palacios, D.; Savignac, M.; Mellström, B.; Naranjo, J.R.; Aranda, A. The repressor DREAM acts as a transcriptional activator on Vitamin D and retinoic acid response elements. Nucleic Acids Res. 2005, 33, 2269–2279. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.Y.; Pitcher, G.M.; Laviolette, S.R.; Whishaw, I.Q.; Tong, K.I.; Kockeritz, L.K.; Wada, T.; Joza, N.A.; Crackower, M.; Goncalves, J.; et al. DREAM is a critical transcriptional repressor for pain modulation. Cell 2002, 108, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Sanz, C.; Mellstrom, B.; Link, W.A.; Naranjo, J.R.; Fernandez-Luna, J.L. Interleukin 3-dependent activation of DREAM is involved in transcriptional silencing of the apoptotic Hrk gene in hematopoietic progenitor cells. EMBO J. 2001, 20, 2286–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera-Arconada, I.; Benedet, T.; Roza, C.; Torres, B.; Barrio, J.; Krzyzanowska, A.; Avendaño, C.; Mellström, B.; Lopez-Garcia, J.A.; Naranjo, J.R. DREAM regulates BDNF-dependent spinal sensitization. Mol. Pain 2010, 6, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savignac, M.; Pintado, B.; Gutierrez-Adan, A.; Palczewska, M.; Mellström, B.; Naranjo, J.R. Transcriptional repressor DREAM regulates T-lymphocyte proliferation and cytokine gene expression. EMBO J. 2005, 24, 3555–3564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiruppathi, C.; Soni, D.; Wang, D.M.; Xue, J.; Singh, V.; Thippegowda, P.B.; Cheppudira, B.P.; Mishra, R.K.; Debroy, A.; Qian, Z.; et al. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat. Immunol. 2014, 15, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, B.; Di Palma, T.; Mascia, A.; Motti, M.L.; Viglietto, G.; Nitsch, L.; Zannini, M. The transcriptional repressor DREAM is involved in thyroid gene expression. Exp. Cell Res. 2005, 305, 166–178. [Google Scholar] [CrossRef]
- Link, W.A.; Ledo, F.; Torres, B.; Palczewska, M.; Madsen, T.M.; Savignac, M.; Albar, J.P.; Mellström, B.; Naranjo, J.R. Day-night changes in downstream regulatory element antagonist modulator/potassium channel interacting protein activity contribute to circadian gene expression in pineal gland. J. Neurosci. 2004, 24, 5346–5355. [Google Scholar] [CrossRef]
- Leclerc, G.M.; Boockfor, F.R. Calcium influx and DREAM protein are required for GnRH gene expression pulse activity. Mol. Cell. Endocrinol. 2007, 267, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Cebolla, B.; Fernández-Pérez, A.; Perea, G.; Araque, A.; Vallejo, M. DREAM mediates cAMP-dependent, Ca2+-induced stimulation of GFAP gene expression and regulates cortical astrogliogenesis. J. Neurosci. 2008, 28, 6703–6713. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Villafuertes, R.; Torres, B.; Barrio, J.; Savignac, M.; Gabellini, N.; Rizzato, F.; Pintado, B.; Gutierrez-Adan, A.; Mellström, B.; Carafoli, E.; et al. Downstream regulatory element antagonist modulator regulates Ca2+ homeostasis and viability in cerebellar neurons. J. Neurosci. 2005, 25, 10822–10830. [Google Scholar] [CrossRef]
- Dierssen, M.; Fedrizzi, L.; Gomez-Villafuertes, R.; de Lagran, M.M.; Gutierrez-Adan, A.; Sahún, I.; Pintado, B.; Oliveros, J.C.; Dopazo, X.M.; Gonzalez, P.; et al. Reduced Mid1 Expression and Delayed Neuromotor Development in daDREAM Transgenic Mice. Front. Mol. Neurosci. 2012, 5, 58. [Google Scholar] [CrossRef] [Green Version]
- Calì, T.; Fedrizzi, L.; Ottolini, D.; Gomez-Villafuertes, R.; Mellström, B.; Naranjo, J.R.; Carafoli, E.; Brini, M. Ca2+-activated nucleotidase 1, a novel target gene for the transcriptional repressor DREAM (downstream regulatory element antagonist modulator), is involved in protein folding and degradation. J. Biol. Chem. 2012, 287, 18478–18491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontán-Lozano, A.; Capilla-Gonzalez, V.; Aguilera, Y.; Mellado, N.; Carrión, A.M.; Soria, B.; Hmadcha, A. Impact of transient down-regulation of DREAM in human embryonic stem cell pluripotency: The role of DREAM in the maintenance of hESCs. Stem Cell Res. 2016, 16, 568–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baczyk, D.; Kibschull, M.; Mellstrom, B.; Levytska, K.; Rivas, M.; Drewlo, S.; Lye, S.J.; Naranjo, J.R.; Kingdom, J.C. DREAM mediated regulation of GCM1 in the human placental trophoblast. PLoS ONE 2013, 8, e51837. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, D.A.; Cho, J.; Landa, L.R., Jr.; Tamarina, N.A.; Roe, M.W.; Buxbaum, J.D.; Philipson, L.H. Downstream regulatory element antagonistic modulator regulates islet prodynorphin expression. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E587–E595. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Kumari, T.; Barazia, A.; Jha, V.; Jeong, S.Y.; Olson, A.; Kim, M.; Lee, B.K.; Manickam, V.; Song, Z.; et al. Neutrophil DREAM promotes neutrophil recruitment in vascular inflammation. J. Exp. Med. 2022, 219, e20211083. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Maizuliana, H.; Usui, N.; Terada, K.; Kondo, A.; Inoue, Y. Clinical, semiological, electroencephalographic, and neuropsychological features of "pure" neocortical temporal lobe epilepsy. Epileptic Disord. 2020, 22, 55–65. [Google Scholar] [CrossRef]
- Maillard, L.; Vignal, J.P.; Gavaret, M.; Guye, M.; Biraben, A.; McGonigal, A.; Chauvel, P.; Bartolomei, F. Semiologic and electrophysiologic correlations in temporal lobe seizure subtypes. Epilepsia 2004, 45, 1590–1599. [Google Scholar] [CrossRef]
- Villa, C.; Combi, R. Potassium Channels and Human Epileptic Phenotypes: An Updated Overview. Front. Cell. Neurosci. 2016, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.M.; Jo, D.G.; Lee, M.C.; Kim, S.Y.; Jung, Y.K. Reduced expression of calsenilin/DREAM/KChIP3 in the brains of kainic acid-induced seizure and epilepsy patients. Neurosci. Lett. 2003, 340, 33–36. [Google Scholar] [CrossRef]
- Monaghan, M.M.; Menegola, M.; Vacher, H.; Rhodes, K.J.; Trimmer, J.S. Altered expression and localization of hippocampal A-type potassium channel subunits in the pilocarpine-induced model of temporal lobe epilepsy. Neuroscience 2008, 156, 550–562. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.G.; He, X.P.; Li, Q.; Madison, R.D.; Moore, S.D.; McNamara, J.O.; Pitt, G.S. The auxiliary subunit KChIP2 is an essential regulator of homeostatic excitability. J. Biol. Chem. 2013, 288, 13258–13268. [Google Scholar] [CrossRef] [Green Version]
- Kuner, R. Central mechanisms of pathological pain. Nat. Med. 2010, 16, 1258–1266. [Google Scholar] [CrossRef]
- Costigan, M.; Woolf, C.J. No DREAM, No pain. Closing the spinal gate. Cell 2002, 108, 297–300. [Google Scholar] [CrossRef] [Green Version]
- Tseng, P.Y.; Hoon, M.A. Molecular Genetics of Kappa Opioids in Pain and Itch Sensations. Handb. Exp. Pharmacol. 2022, 271, 255–274. [Google Scholar] [CrossRef]
- Ren, K.; Dubner, R. Pain facilitation and activity-dependent plasticity in pain modulatory circuitry: Role of BDNF-TrkB signaling and NMDA receptors. Mol. Neurobiol. 2007, 35, 224–235. [Google Scholar] [CrossRef]
- Benedet, T.; Gonzalez, P.; Oliveros, J.C.; Dopazo, J.M.; Ghimire, K.; Palczewska, M.; Mellstrom, B.; Naranjo, J.R. Transcriptional repressor DREAM regulates trigeminal noxious perception. J. Neurochem. 2017, 141, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Long, I.; Suppian, R.; Ismail, Z. Increases in mRNA and DREAM protein expression in the rat spinal cord after formalin induced pain. Neurochem. Res. 2011, 36, 533–539. [Google Scholar] [CrossRef]
- Ismail, C.A.N.; Suppian, R.; Aziz, C.B.A.; Long, I. Minocycline attenuates the development of diabetic neuropathy by modulating DREAM and BDNF protein expression in rat spinal cord. J. Diabetes Metab. Disord. 2019, 18, 181–190. [Google Scholar] [CrossRef]
- Guo, Y.P.; Zhi, Y.R.; Liu, T.T.; Wang, Y.; Zhang, Y. Global Gene Knockout of Kcnip3 Enhances Pain Sensitivity and Exacerbates Negative Emotions in Rats. Front. Mol. Neurosci. 2019, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Tian, N.X.; Xu, Y.; Yang, J.Y.; Li, L.; Sun, X.H.; Wang, Y.; Zhang, Y. KChIP3 N-Terminal 31-50 Fragment Mediates Its Association with TRPV1 and Alleviates Inflammatory Hyperalgesia in Rats. J. Neurosci. 2018, 38, 1756–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.F.; Wang, W.C.; Huang, C.Y.; Du, P.H.; Yang, J.H.; Tsaur, M.L. Coexpression of auxiliary subunits KChIP and DPPL in potassium channel Kv4-positive nociceptors and pain-modulating spinal interneurons. J. Comp. Neurol. 2016, 524, 846–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, P.; Liang, P.; Liu, T.; Liu, X.; Liu, X.Y.; Zhang, B.; Han, T.; Zhu, Y.B.; Yin, D.M.; et al. The DREAM protein negatively regulates the NMDA receptor through interaction with the NR1 subunit. J. Neurosci. 2010, 30, 7575–7586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koivisto, A.P.; Belvisi, M.G.; Gaudet, R.; Szallasi, A. Advances in TRP channel drug discovery: From target validation to clinical studies. Nat. Rev. Drug Discov. 2022, 21, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.; Mehaffey, W.H.; Iftinca, M.; Rehak, R.; Engbers, J.D.; Hameed, S.; Zamponi, G.W.; Turner, R.W. Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat. Neurosci. 2010, 13, 333–337. [Google Scholar] [CrossRef]
- Socuéllamos, P.G.; Olivos-Oré, L.A.; Barahona, M.V.; Cercós, P.; Pérez Pascual, M.; Arribas-Blázquez, M.; Naranjo, J.R.; Valenzuela, C.; Gutiérrez-Rodríguez, M.; Artalejo, A.R. IQM-PC332, a Novel DREAM Ligand with Antinociceptive Effect on Peripheral Nerve Injury-Induced Pain. Int. J. Mol. Sci. 2022, 23, 2142. [Google Scholar] [CrossRef]
- Ma, S.; Zuo, Y. Synaptic modifications in learning and memory—A dendritic spine story. Semin. Cell Dev. Biol. 2022, 125, 84–90. [Google Scholar] [CrossRef]
- Bin Ibrahim, M.Z.; Benoy, A.; Sajikumar, S. Long-term plasticity in the hippocampus: Maintaining within and ‘tagging’ between synapses. FEBS J. 2022, 289, 2176–2201. [Google Scholar] [CrossRef]
- Lisman, J.; Buzsáki, G.; Eichenbaum, H.; Nadel, L.; Ranganath, C.; Redish, A.D. Viewpoints: How the hippocampus contributes to memory, navigation and cognition. Nat. Neurosci. 2017, 20, 1434–1447. [Google Scholar] [CrossRef]
- Fontán-Lozano, A.; Romero-Granados, R.; del-Pozo-Martín, Y.; Suárez-Pereira, I.; Delgado-García, J.M.; Penninger, J.M.; Carrión, A.M. Lack of DREAM protein enhances learning and memory and slows brain aging. Curr. Biol. 2009, 19, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.Y.; Li, X.H.; Zhuo, M. NMDA receptors and synaptic plasticity in the anterior cingulate cortex. Neuropharmacology 2021, 197, 108749. [Google Scholar] [CrossRef]
- Dore, K.; Malinow, R. Elevated PSD-95 Blocks Ion-flux Independent LTD: A Potential New Role for PSD-95 in Synaptic Plasticity. Neuroscience 2021, 456, 43–49. [Google Scholar] [CrossRef]
- Wu, L.J.; Mellström, B.; Wang, H.; Ren, M.; Domingo, S.; Kim, S.S.; Li, X.Y.; Chen, T.; Naranjo, J.R.; Zhuo, M. DREAM (downstream regulatory element antagonist modulator) contributes to synaptic depression and contextual fear memory. Mol. Brain 2010, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Lugo, J.N.; Brewster, A.L.; Spencer, C.M.; Anderson, A.E. Kv4.2 knockout mice have hippocampal-dependent learning and memory deficits. Learn. Mem. 2012, 19, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef]
- Robakis, N.K. Cell signaling abnormalities may drive neurodegeneration in familial Alzheimer disease. Neurochem. Res. 2014, 39, 570–575. [Google Scholar] [CrossRef]
- Pham, K.; Miksovska, J. Molecular insight of DREAM and presenilin 1 C-terminal fragment interactions. FEBS Lett. 2016, 590, 1114–1122. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef]
- Jo, D.G.; Lee, J.Y.; Hong, Y.M.; Song, S.; Mook-Jung, I.; Koh, J.Y.; Jung, Y.K. Induction of pro-apoptotic calsenilin/DREAM/KChIP3 in Alzheimer’s disease and cultured neurons after amyloid-beta exposure. J. Neurochem. 2004, 88, 604–611. [Google Scholar] [CrossRef]
- Zaidi, N.F.; Berezovska, O.; Choi, E.K.; Miller, J.S.; Chan, H.; Lilliehook, C.; Hyman, B.T.; Buxbaum, J.D.; Wasco, W. Biochemical and immunocytochemical characterization of calsenilin in mouse brain. Neuroscience 2002, 114, 247–263. [Google Scholar] [CrossRef]
- Jo, D.G.; Chang, J.W.; Hong, H.S.; Mook-Jung, I.; Jung, Y.K. Contribution of presenilin/gamma-secretase to calsenilin-mediated apoptosis. Biochem. Biophys. Res. Commun. 2003, 305, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.G.; Jang, J.; Kim, B.J.; Lundkvist, J.; Jung, Y.K. Overexpression of calsenilin enhances gamma-secretase activity. Neurosci. Lett. 2005, 378, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, R.; González, P.; Lopez-Hurtado, A.; Dopazo, X.M.; Mellström, B.; Naranjo, J.R. Inhibition of the Neuronal Calcium Sensor DREAM Modulates Presenilin-2 Endoproteolysis. Front. Mol. Neurosci. 2018, 11, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, E.; Resende, R.; Costa, R.; Oliveira, C.R.; Pereira, C.M. An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiol. Dis. 2006, 23, 669–678. [Google Scholar] [CrossRef] [Green Version]
- Lilliehook, C.; Chan, S.; Choi, E.K.; Zaidi, N.F.; Wasco, W.; Mattson, M.P.; Buxbaum, J.D. Calsenilin enhances apoptosis by altering endoplasmic reticulum calcium signaling. Mol. Cell. Neurosci. 2002, 19, 552–559. [Google Scholar] [CrossRef]
- Fedrizzi, L.; Lim, D.; Carafoli, E.; Brini, M. Interplay of the Ca2+-binding protein DREAM with presenilin in neuronal Ca2+ signaling. J. Biol. Chem. 2008, 283, 27494–27503. [Google Scholar] [CrossRef] [Green Version]
- Grillo, M.A.; Grillo, S.L.; Gerdes, B.C.; Kraus, J.G.; Koulen, P. Control of Neuronal Ryanodine Receptor-Mediated Calcium Signaling by Calsenilin. Mol. Neurobiol. 2019, 56, 525–534. [Google Scholar] [CrossRef]
- López-Hurtado, A.; Burgos, D.F.; González, P.; Dopazo, X.M.; González, V.; Rábano, A.; Mellström, B.; Naranjo, J.R. Inhibition of DREAM-ATF6 interaction delays onset of cognition deficit in a mouse model of Huntington’s disease. Mol. Brain 2018, 11, 13. [Google Scholar] [CrossRef]
- Larrodé, P.; Calvo, A.C.; Moreno-Martínez, L.; de la Torre, M.; Moreno-García, L.; Molina, N.; Castiella, T.; Iñiguez, C.; Pascual, L.F.; Mena, F.J.M.; et al. DREAM-Dependent Activation of Astrocytes in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 1–12. [Google Scholar] [CrossRef]
- Singh, D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J. Neuroinflam. 2022, 19, 206. [Google Scholar] [CrossRef]
- Tse, G. Mechanisms of cardiac arrhythmias. J. Arrhythmia 2016, 32, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Kant, R.; Hu, Z.; Malhotra, J.K.; Krogh-Madsen, T.; Christini, D.J.; Heerdt, P.M.; Abbott, G.W. NHE isoform switching and KChIP2 upregulation in aging porcine atria. PLoS ONE 2013, 8, e82951. [Google Scholar] [CrossRef]
- Monnerat-Cahli, G.; Alonso, H.; Gallego, M.; Alarcón, M.L.; Bassani, R.A.; Casis, O.; Medei, E. Toll-like receptor 4 activation promotes cardiac arrhythmias by decreasing the transient outward potassium current (Ito) through an IRF3-dependent and MyD88-independent pathway. J. Mol. Cell. Cardiol. 2014, 76, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Liu, T.; Li, Y.; Qin, M.; Tang, Y.; Shen, J.Y.; Liang, J.; Yang, B.; Huang, C. Chronic N-methyl-D-aspartate receptor activation induces cardiac electrical remodeling and increases susceptibility to ventricular arrhythmias. Pacing Clin. Electrophysiol. 2014, 37, 1367–1377. [Google Scholar] [CrossRef]
- Kuo, H.C.; Cheng, C.F.; Clark, R.B.; Lin, J.J.; Lin, J.L.; Hoshijima, M.; Nguyêñ-Trân, V.T.; Gu, Y.; Ikeda, Y.; Chu, P.H.; et al. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell 2001, 107, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Hlaing, T.; DiMino, T.; Kowey, P.R.; Yan, G.X. ECG repolarization waves: Their genesis and clinical implications. Ann. Noninvasive Electrocardiol. 2005, 10, 211–223. [Google Scholar] [CrossRef]
- Foeger, N.C.; Wang, W.; Mellor, R.L.; Nerbonne, J.M. Stabilization of Kv4 protein by the accessory K(+) channel interacting protein 2 (KChIP2) subunit is required for the generation of native myocardial fast transient outward K(+) currents. J. Physiol. 2013, 591, 4149–4166. [Google Scholar] [CrossRef]
- Nassal, D.M.; Wan, X.; Liu, H.; Maleski, D.; Ramirez-Navarro, A.; Moravec, C.S.; Ficker, E.; Laurita, K.R.; Deschênes, I. KChIP2 is a core transcriptional regulator of cardiac excitability. eLife 2017, 6, e17304. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling--concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Jiang, M.; Zhang, M.; Tang, D.; Clemo, H.F.; Higgins, R.S.; Tseng, G.N. Electrical remodeling in a canine model of ischemic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H560–H571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakisaka, Y.; Niwano, S.; Niwano, H.; Saito, J.; Yoshida, T.; Hirasawa, S.; Kawada, H.; Izumi, T. Structural and electrical ventricular remodeling in rat acute myocarditis and subsequent heart failure. Cardiovasc. Res. 2004, 63, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.; Kim, K.B.; Ahn, H.; Cho, H.J.; Choi, Y.S. Remodeling of ion channel expression in patients with chronic atrial fibrillation and mitral valvular heart disease. Korean J. Intern. Med. 2010, 25, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Kawada, H.; Niwano, S.; Niwano, H.; Yumoto, Y.; Wakisaka, Y.; Yuge, M.; Kawahara, K.; Izumi, T. Tumor necrosis factor-alpha downregulates the voltage gated outward K+ current in cultured neonatal rat cardiomyocytes: A possible cause of electrical remodeling in diseased hearts. Circ. J. 2006, 70, 605–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.Z.; Jiang, H.; Li, L.; Xie, P.; Li, J.Y.; Lu, Z.B.; He, B. Semaphorin 3A attenuates electrical remodeling at infarct border zones in rats after myocardial infarction. Tohoku J. Exp. Med. 2011, 225, 51–57. [Google Scholar] [CrossRef]
- Sato, T.; Kobayashi, T.; Kuno, A.; Miki, T.; Tanno, M.; Kouzu, H.; Itoh, T.; Ishikawa, S.; Kojima, T.; Miura, T.; et al. Type 2 diabetes induces subendocardium predominant reduction in transient outward K+ current with downregulation of Kv4.2 and KChIP2. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1054–H1065. [Google Scholar] [CrossRef]
- Näbauer, M.; Kääb, S. Potassium channel down-regulation in heart failure. Cardiovasc. Res. 1998, 37, 324–334. [Google Scholar] [CrossRef]
- Jin, H.; Hadri, L.; Palomeque, J.; Morel, C.; Karakikes, I.; Kaprielian, R.; Hajjar, R.; Lebeche, D. KChIP2 attenuates cardiac hypertrophy through regulation of Ito and intracellular calcium signaling. J. Mol. Cell. Cardiol. 2010, 48, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Chiale, P.A.; Etcheverry, D.; Pastori, J.D.; Fernandez, P.A.; Garro, H.A.; González, M.D.; Elizari, M.V. The multiple electrocardiographic manifestations of ventricular repolarization memory. Curr. Cardiol. Rev. 2014, 10, 190–201. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; McKinnon, D.; Dixon, J.E.; Gao, J.; Wymore, R.; Cohen, I.S.; Danilo, P., Jr.; Shvilkin, A.; Anyukhovsky, E.P.; Sosunov, E.A.; et al. Transient outward current, Ito1, is altered in cardiac memory. Circulation 1999, 99, 1898–1905. [Google Scholar] [CrossRef] [Green Version]
- Radicke, S.; Cotella, D.; Graf, E.M.; Banse, U.; Jost, N.; Varró, A.; Tseng, G.N.; Ravens, U.; Wettwer, E. Functional modulation of the transient outward current Ito by KCNE beta-subunits and regional distribution in human non-failing and failing hearts. Cardiovasc. Res. 2006, 71, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, S.; Schnell, M.; Bühler, T.; Reinke, Y.; Lüdemann, J.; Nießner, F.; Brinkmeier, H.; Herda, L.R.; Staudt, A.; Kroemer, H.K.; et al. Antibodies against potassium channel interacting protein 2 induce necrosis in isolated rat cardiomyocytes. J. Cell. Biochem. 2014, 115, 678–689. [Google Scholar] [CrossRef]
- Odagiri, F.; Inoue, H.; Sugihara, M.; Suzuki, T.; Murayama, T.; Shioya, T.; Konishi, M.; Nakazato, Y.; Daida, H.; Sakurai, T.; et al. Effects of candesartan on electrical remodeling in the hearts of inherited dilated cardiomyopathy model mice. PLoS ONE 2014, 9, e101838. [Google Scholar] [CrossRef] [Green Version]
- Nassal, D.M.; Wan, X.; Liu, H.; Deschênes, I. Myocardial KChIP2 Expression in Guinea Pig Resolves an Expanded Electrophysiologic Role. PLoS ONE 2016, 11, e0146561. [Google Scholar] [CrossRef] [Green Version]
- Nassal, D.M.; Wan, X.; Liu, H.; Laurita, K.R.; Deschênes, I. KChIP2 regulates the cardiac Ca2+ transient and myocyte contractility by targeting ryanodine receptor activity. PLoS ONE 2017, 12, e0175221. [Google Scholar] [CrossRef] [Green Version]
- Speerschneider, T.; Grubb, S.; Metoska, A.; Olesen, S.P.; Calloe, K.; Thomsen, M.B. Development of heart failure is independent of K+ channel-interacting protein 2 expression. J. Physiol. 2013, 591, 5923–5937. [Google Scholar] [CrossRef]
- Grubb, S.; Speerschneider, T.; Occhipinti, D.; Fiset, C.; Olesen, S.P.; Thomsen, M.B.; Calloe, K. Loss of K+ currents in heart failure is accentuated in KChIP2 deficient mice. J. Cardiovasc. Electrophysiol. 2014, 25, 896–904. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, L.-Y.; Song, Y.-J.; Zhang, C.-L.; Liu, J. KV Channel-Interacting Proteins in the Neurological and Cardiovascular Systems: An Updated Review. Cells 2023, 12, 1894. https://doi.org/10.3390/cells12141894
Wu L-Y, Song Y-J, Zhang C-L, Liu J. KV Channel-Interacting Proteins in the Neurological and Cardiovascular Systems: An Updated Review. Cells. 2023; 12(14):1894. https://doi.org/10.3390/cells12141894
Chicago/Turabian StyleWu, Le-Yi, Yu-Juan Song, Cheng-Lin Zhang, and Jie Liu. 2023. "KV Channel-Interacting Proteins in the Neurological and Cardiovascular Systems: An Updated Review" Cells 12, no. 14: 1894. https://doi.org/10.3390/cells12141894
APA StyleWu, L. -Y., Song, Y. -J., Zhang, C. -L., & Liu, J. (2023). KV Channel-Interacting Proteins in the Neurological and Cardiovascular Systems: An Updated Review. Cells, 12(14), 1894. https://doi.org/10.3390/cells12141894