
Decoupling of mRNA and Protein Expression in Aging Brains Reveals the Age-Dependent Adaptation of Specific Gene Subsets

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Histology
2.3. RNA and Protein Extraction
2.4. RNA-Seq Library Preparation
2.5. Data Analysis
2.6. Quantitative PCR
2.7. Sample Preparation for Mass Spectrometry
2.8. Data-Dependent Acquisition (DDA) Mass Spectrometry
2.9. Parallel Reaction Monitoring (PRM) Mass Spectrometry
2.10. Immunoblotting
2.11. Immunofluorescence
3. Results
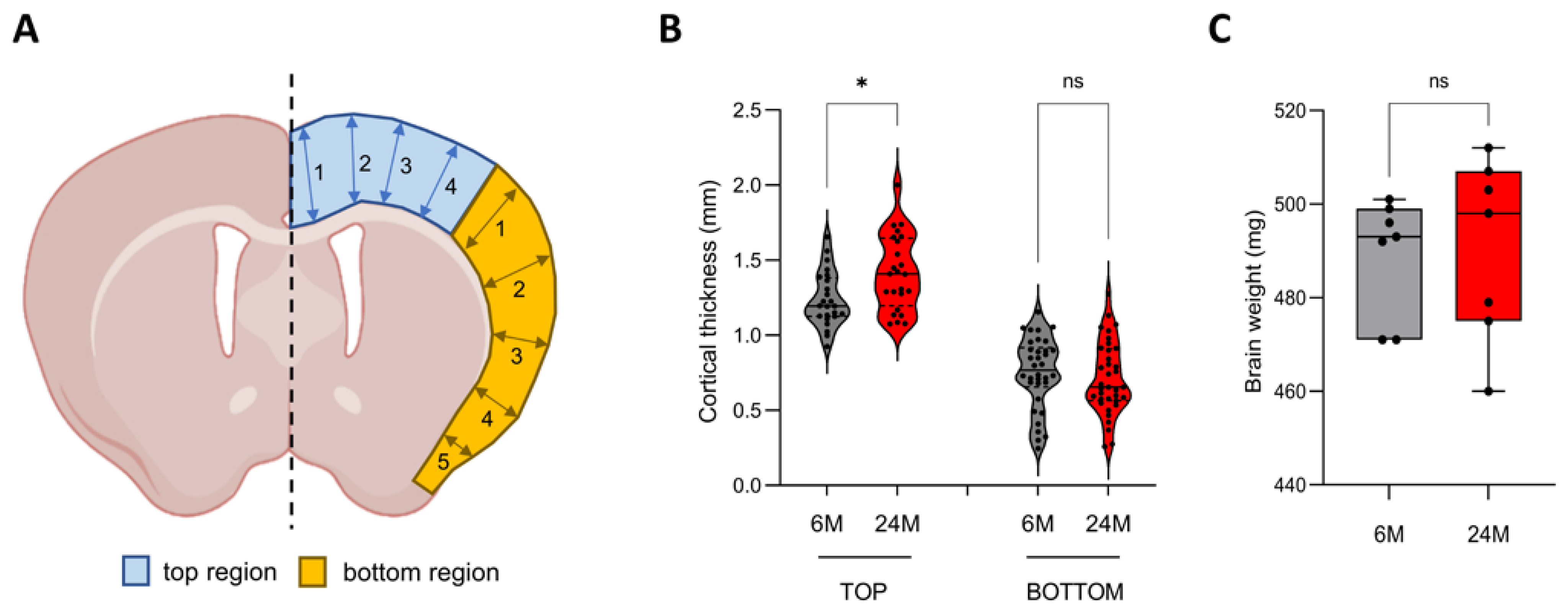
3.1. Cortical Thickness Increases during Aging
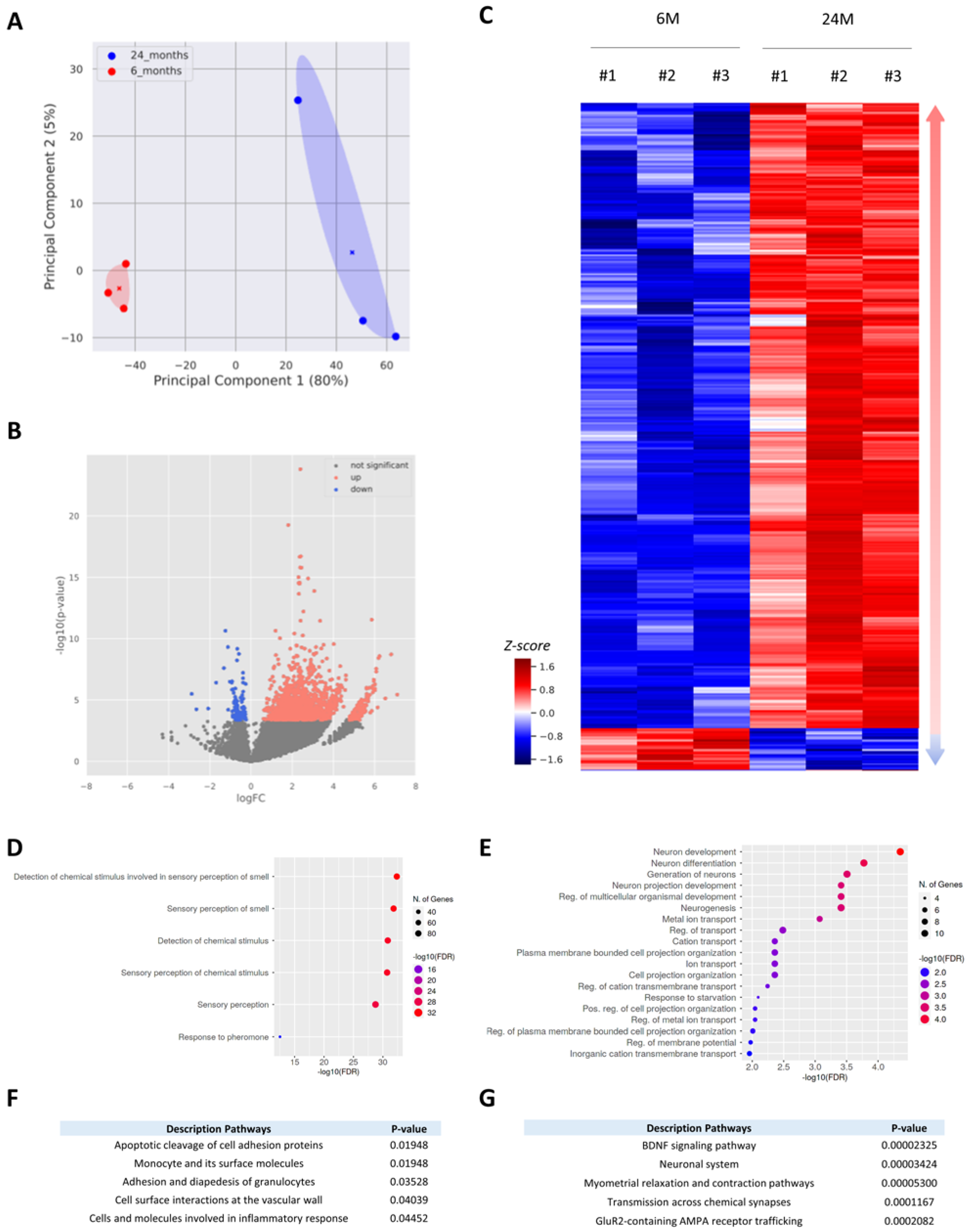
3.2. Aging Mainly Triggers Activation of Gene Expression in the Cortex
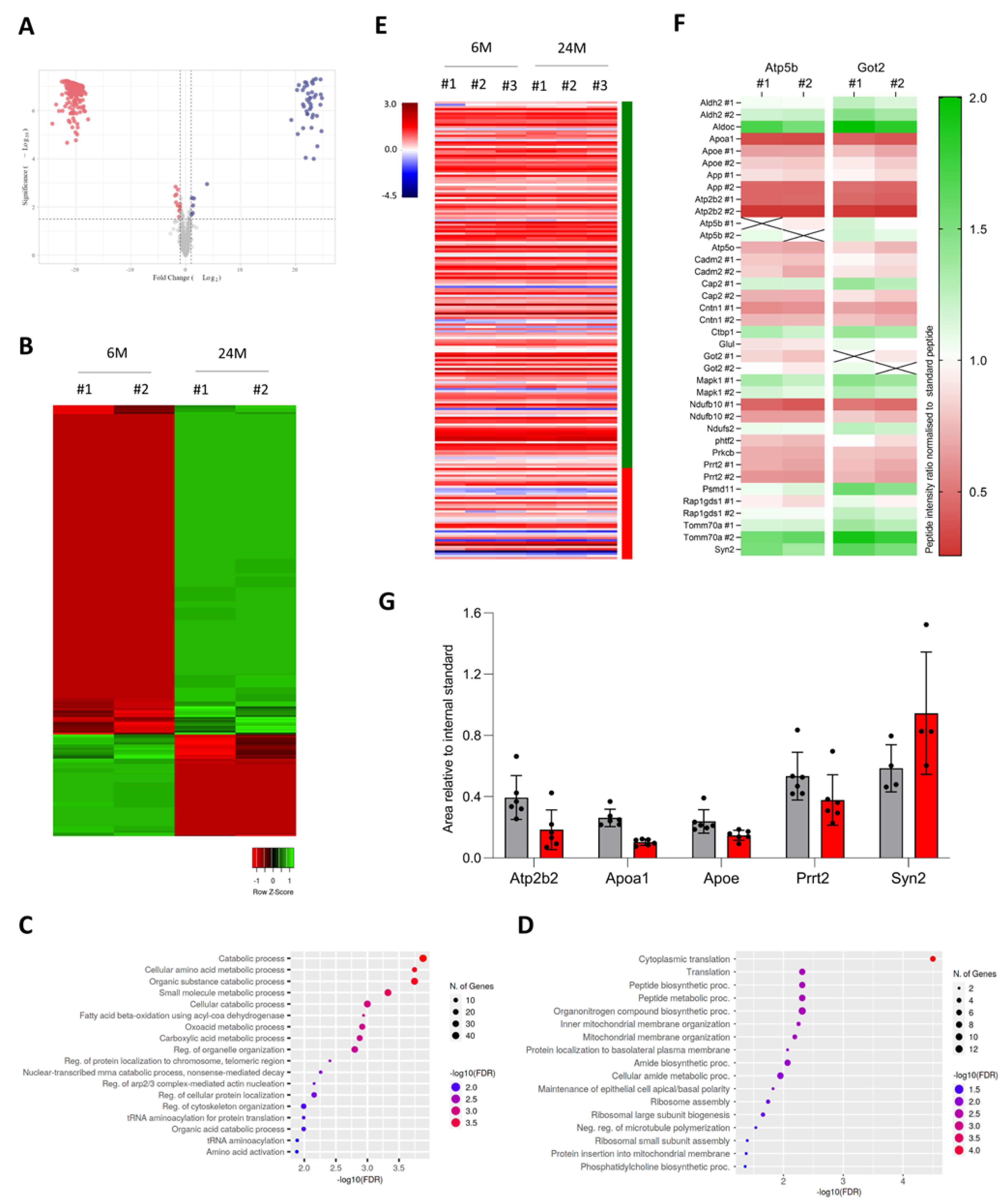
3.3. Increased mRNA Expression Correlates with Higher Levels of Proteins
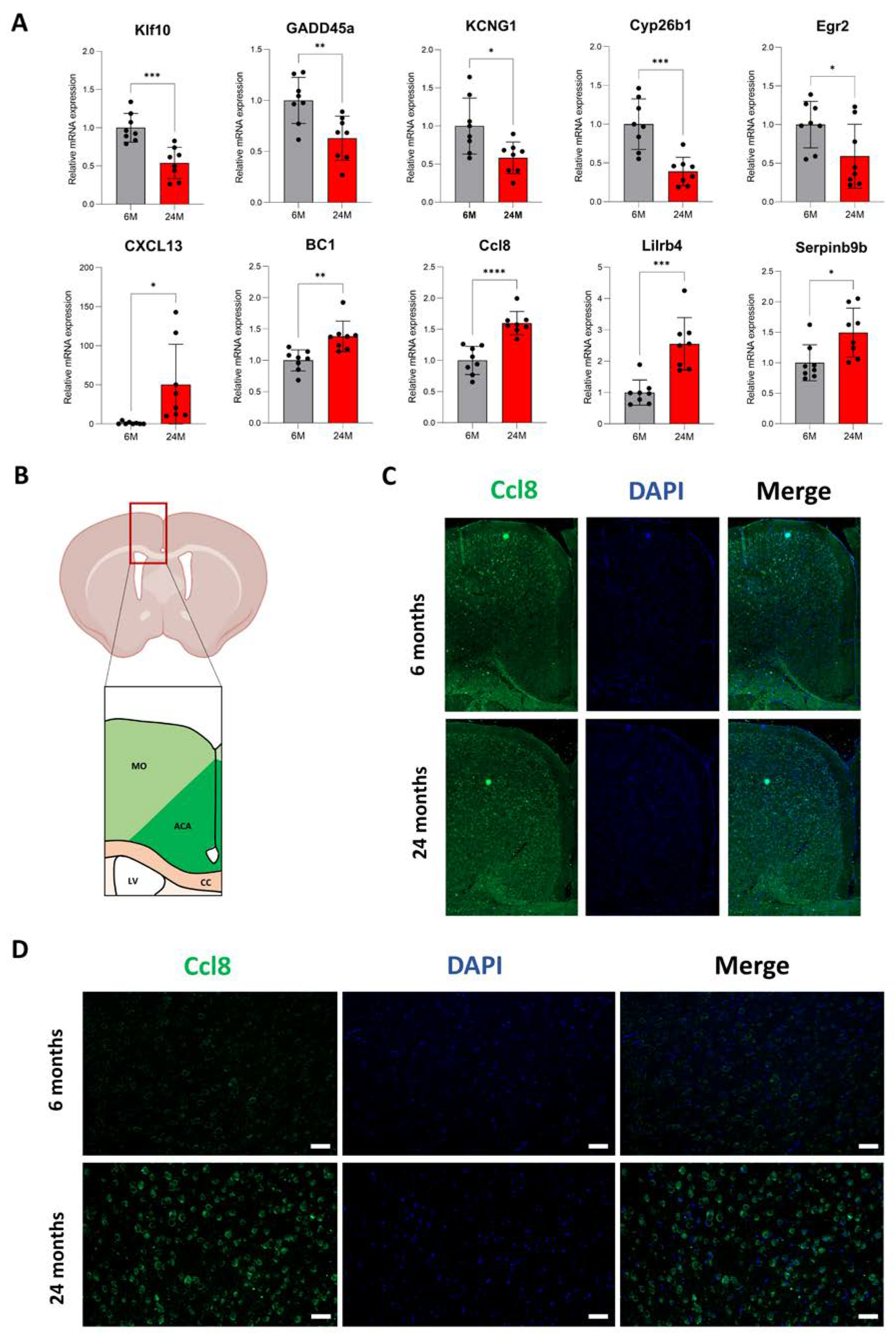
3.4. A Subset of Genes Depends on Translation Regulation in the Aging Cortex
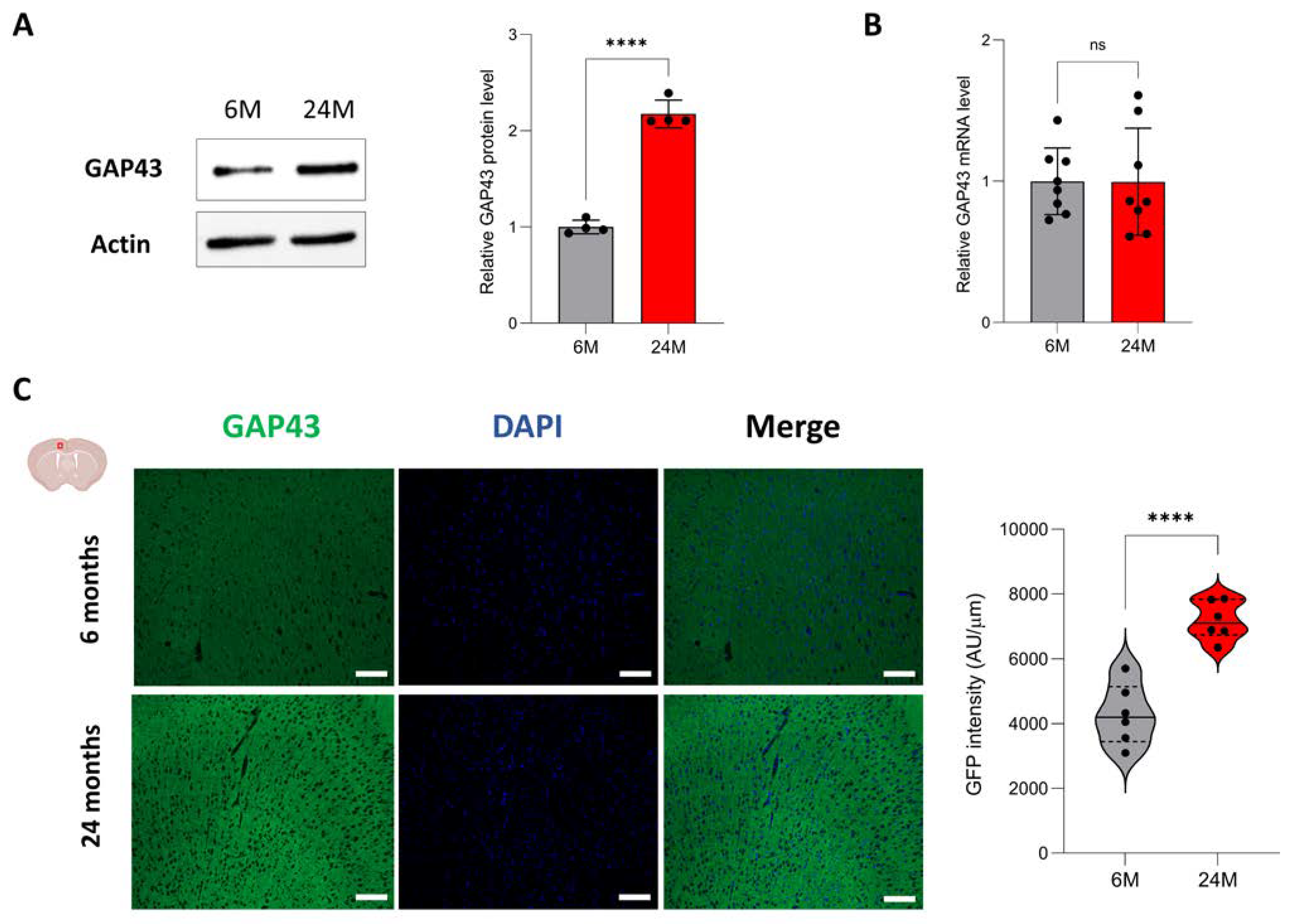
3.5. Aging Increases Protein Expression of Genes Associated with Neuroplasticity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moskalev, A.A.; Shaposhnikov, M.V.; Plyusnina, E.N.; Zhavoronkov, A.; Budovsky, A.; Yanai, H.; Fraifeld, V.E. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev. 2013, 12, 661–684. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet. 2007, 23, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Brunet, A. Histone methylation makes its mark on longevity. Trends Cell. Biol. 2012, 22, 42–49. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Taylor, R.C.; Dillin, A. Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 2011, 3, a004440. [Google Scholar] [CrossRef]
- Lemoine, M. The Evolution of the Hallmarks of Aging. Front. Genet. 2021, 12, 693071. [Google Scholar] [CrossRef] [PubMed]
- Dhondt, I.; Petyuk, V.A.; Bauer, S.; Brewer, H.M.; Smith, R.D.; Depuydt, G.; Braeckman, B.P. Changes of Protein Turnover in Aging Caenorhabditis elegans. Mol. Cell. Proteom. 2017, 16, 1621–1633. [Google Scholar] [CrossRef] [Green Version]
- Janssens, G.E.; Meinema, A.C.; Gonzalez, J.; Wolters, J.C.; Schmidt, A.; Guryev, V.; Bischoff, R.; Wit, E.C.; Veenhoff, L.M.; Heinemann, M. Protein biogenesis machinery is a driver of replicative aging in yeast. Elife 2015, 4, e08527. [Google Scholar] [CrossRef]
- Anisimova, A.S.; Meerson, M.B.; Gerashchenko, M.V.; Kulakovskiy, I.V.; Dmitriev, S.E.; Gladyshev, V.N. Multifaceted deregulation of gene expression and protein synthesis with age. Proc. Natl. Acad. Sci. USA 2020, 117, 15581–15590. [Google Scholar] [CrossRef]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- McManus, J.; Cheng, Z.; Vogel, C. Next-generation analysis of gene expression regulation--comparing the roles of synthesis and degradation. Mol. Biosyst. 2015, 11, 2680–2689. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Mayo, A.; Keren, L.; Alon, U. Central dogma rates and the trade-off between precision and economy in gene expression. Nat. Commun. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Rooney, M.S.; Mertins, P.; Przybylski, D.; Chevrier, N.; Satija, R.; Rodriguez, E.H.; Fields, A.P.; Schwartz, S.; Raychowdhury, R.; et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science 2015, 347, 1259038. [Google Scholar] [CrossRef]
- Locker, N.; Chamond, N.; Sargueil, B. A conserved structure within the HIV gag open reading frame that controls translation initiation directly recruits the 40S subunit and eIF3. Nucleic Acids Res. 2011, 39, 2367–2377. [Google Scholar] [CrossRef] [PubMed]
- Wethmar, K. The regulatory potential of upstream open reading frames in eukaryotic gene expression. Wiley Interdiscip. Rev. RNA 2014, 5, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Aebersold, R. The interdependence of transcript and protein abundance: New data--new complexities. Mol. Syst. Biol. 2016, 12, 856. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, A.R.; Gsponer, J.; Foster, L.J. Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol. Syst. Biol. 2013, 9, 689. [Google Scholar] [CrossRef] [PubMed]
- Li, G.W.; Burkhardt, D.; Gross, C.; Weissman, J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 2014, 157, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Teo, G.; Krueger, S.; Rock, T.M.; Koh, H.W.; Choi, H.; Vogel, C. Differential dynamics of the mammalian mRNA and protein expression response to misfolding stress. Mol. Syst. Biol. 2016, 12, 855. [Google Scholar] [CrossRef]
- Lackner, D.H.; Schmidt, M.W.; Wu, S.; Wolf, D.A.; Bahler, J. Regulation of transcriptome, translation, and proteome in response to environmental stress in fission yeast. Genome Biol. 2012, 13, R25. [Google Scholar] [CrossRef]
- Takemon, Y.; Chick, J.M.; Gerdes Gyuricza, I.; Skelly, D.A.; Devuyst, O.; Gygi, S.P.; Churchill, G.A.; Korstanje, R. Proteomic and transcriptomic profiling reveal different aspects of aging in the kidney. Elife 2021, 10, e62585. [Google Scholar] [CrossRef]
- Waldera-Lupa, D.M.; Kalfalah, F.; Florea, A.M.; Sass, S.; Kruse, F.; Rieder, V.; Tigges, J.; Fritsche, E.; Krutmann, J.; Busch, H.; et al. Proteome-wide analysis reveals an age-associated cellular phenotype of in situ aged human fibroblasts. Aging 2014, 6, 856–878. [Google Scholar] [CrossRef]
- Wei, Y.N.; Hu, H.Y.; Xie, G.C.; Fu, N.; Ning, Z.B.; Zeng, R.; Khaitovich, P. Transcript and protein expression decoupling reveals RNA binding proteins and miRNAs as potential modulators of human aging. Genome Biol. 2015, 16, 41. [Google Scholar] [CrossRef]
- Schaum, N.; Lehallier, B.; Hahn, O.; Palovics, R.; Hosseinzadeh, S.; Lee, S.E.; Sit, R.; Lee, D.P.; Losada, P.M.; Zardeneta, M.E.; et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 2020, 583, 596–602. [Google Scholar] [CrossRef]
- Ori, A.; Toyama, B.H.; Harris, M.S.; Bock, T.; Iskar, M.; Bork, P.; Ingolia, N.T.; Hetzer, M.W.; Beck, M. Integrated Transcriptome and Proteome Analyses Reveal Organ-Specific Proteome Deterioration in Old Rats. Cell Syst. 2015, 1, 224–237. [Google Scholar] [CrossRef] [Green Version]
- Ibrayeva, A.; Bay, M.; Pu, E.; Jorg, D.J.; Peng, L.; Jun, H.; Zhang, N.; Aaron, D.; Lin, C.; Resler, G.; et al. Early stem cell aging in the mature brain. Cell Stem Cell 2021, 28, 955–966.e7. [Google Scholar] [CrossRef]
- Bourgon, R.; Gentleman, R.; Huber, W. Independent filtering increases detection power for high-throughput experiments. Proc. Natl. Acad. Sci. USA 2010, 107, 9546–9551. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, S.; Combes, F.; Lazar, C.; Giai Gianetto, Q.; Gatto, L.; Dorffer, A.; Hesse, A.M.; Coute, Y.; Ferro, M.; Bruley, C.; et al. DAPAR & ProStaR: Software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 2017, 33, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef] [PubMed]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef] [PubMed]
- Hammelrath, L.; Skokic, S.; Khmelinskii, A.; Hess, A.; van der Knaap, N.; Staring, M.; Lelieveldt, B.P.F.; Wiedermann, D.; Hoehn, M. Morphological maturation of the mouse brain: An in vivo MRI and histology investigation. Neuroimage 2016, 125, 144–152. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef]
- Dutta, S.; Sengupta, P. Men and mice: Relating their ages. Life Sci. 2016, 152, 244–248. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.J.; Wu, C.C.; Phan, N.N.; Liu, Y.H.; Ta, H.D.K.; Anuraga, G.; Wu, Y.F.; Lee, K.H.; Chuang, J.Y.; Wang, C.Y. Prognoses and genomic analyses of proteasome 26S subunit, ATPase (PSMC) family genes in clinical breast cancer. Aging 2021, 13, 17970. [Google Scholar] [CrossRef] [PubMed]
- Xuan, D.T.M.; Wu, C.C.; Kao, T.J.; Ta, H.D.K.; Anuraga, G.; Andriani, V.; Athoillah, M.; Chiao, C.C.; Wu, Y.F.; Lee, K.H.; et al. Prognostic and immune infiltration signatures of proteasome 26S subunit, non-ATPase (PSMD) family genes in breast cancer patients. Aging 2021, 13, 24882–24913. [Google Scholar] [CrossRef] [PubMed]
- Casoli, T.; Spagna, C.; Fattoretti, P.; Gesuita, R.; Bertoni-Freddari, C. Neuronal plasticity in aging: A quantitative immunohistochemical study of GAP-43 distribution in discrete regions of the rat brain. Brain Res. 1996, 714, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Jeanne, M.; Demory, H.; Moutal, A.; Vuillaume, M.L.; Blesson, S.; Thepault, R.A.; Marouillat, S.; Halewa, J.; Maas, S.M.; Motazacker, M.M.; et al. Missense variants in DPYSL5 cause a neurodevelopmental disorder with corpus callosum agenesis and cerebellar abnormalities. Am. J. Hum. Genet. 2021, 108, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.J.; Proietti Onori, M.; Borgesius, N.Z.; van de Bree, J.E.; Elgersma-Hooisma, M.; Nio, E.; Bezstarosti, K.; Buitendijk, G.H.S.; Aghadavoud Jolfaei, M.; Demmers, J.A.A.; et al. CAMK2-Dependent Signaling in Neurons Is Essential for Survival. J. Neurosci. 2019, 39, 5424–5439. [Google Scholar] [CrossRef]
- Li, L.; Cao, X.H.; Chen, S.R.; Han, H.D.; Lopez-Berestein, G.; Sood, A.K.; Pan, H.L. Up-regulation of Cavbeta3 subunit in primary sensory neurons increases voltage-activated Ca2+ channel activity and nociceptive input in neuropathic pain. J. Biol. Chem. 2012, 287, 6002–6013. [Google Scholar] [CrossRef]
- Rasmussen, A.H.; Rasmussen, H.B.; Silahtaroglu, A. The DLGAP family: Neuronal expression, function and role in brain disorders. Mol. Brain. 2017, 10, 43. [Google Scholar] [CrossRef]
- Pallotto, M.; Deprez, F. Regulation of adult neurogenesis by GABAergic transmission: Signaling beyond GABAA-receptors. Front Cell Neurosci. 2014, 8, 166. [Google Scholar] [CrossRef]
- Ramanathan, D.; Conner, J.M.; Tuszynski, M.H. A form of motor cortical plasticity that correlates with recovery of function after brain injury. Proc. Natl. Acad. Sci. USA 2006, 103, 11370–11375. [Google Scholar] [CrossRef] [Green Version]
- Rosskothen-Kuhl, N.; Illing, R.B. Gap43 transcription modulation in the adult brain depends on sensory activity and synaptic cooperation. PLoS ONE 2014, 9, e92624. [Google Scholar] [CrossRef]
- Svennerholm, L.; Bostrom, K.; Jungbjer, B. Changes in weight and compositions of major membrane components of human brain during the span of adult human life of Swedes. Acta Neuropathol. 1997, 94, 345–352. [Google Scholar] [CrossRef]
- Salat, D.H.; Buckner, R.L.; Snyder, A.Z.; Greve, D.N.; Desikan, R.S.; Busa, E.; Morris, J.C.; Dale, A.M.; Fischl, B. Thinning of the cerebral cortex in aging. Cereb. Cortex 2004, 14, 721–730. [Google Scholar] [CrossRef]
- Dotson, V.M.; Szymkowicz, S.M.; Sozda, C.N.; Kirton, J.W.; Green, M.L.; O’Shea, A.; McLaren, M.E.; Anton, S.D.; Manini, T.M.; Woods, A.J. Age Differences in Prefrontal Surface Area and Thickness in Middle Aged to Older Adults. Front. Aging Neurosci. 2015, 7, 250. [Google Scholar] [CrossRef] [PubMed]
- Fjell, A.M.; Westlye, L.T.; Grydeland, H.; Amlien, I.; Espeseth, T.; Reinvang, I.; Raz, N.; Dale, A.M.; Walhovd, K.B.; Alzheimer Disease Neuroimaging, I. Accelerating cortical thinning: Unique to dementia or universal in aging? Cereb. Cortex 2014, 24, 919–934. [Google Scholar] [CrossRef]
- Draganski, B.; Gaser, C.; Kempermann, G.; Kuhn, H.G.; Winkler, J.; Buchel, C.; May, A. Temporal and spatial dynamics of brain structure changes during extensive learning. J. Neurosci. 2006, 26, 6314–6317. [Google Scholar] [CrossRef]
- Engvig, A.; Fjell, A.M.; Westlye, L.T.; Moberget, T.; Sundseth, O.; Larsen, V.A.; Walhovd, K.B. Effects of memory training on cortical thickness in the elderly. Neuroimage 2010, 52, 1667–1676. [Google Scholar] [CrossRef]
- Pauwels, L.; Chalavi, S.; Swinnen, S.P. Aging and brain plasticity. Aging 2018, 10, 1789–1790. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, D.A.; Arfanakis, K.; Kelly, J.F.; Rajendran, N.; Buchman, A.S.; Morris, M.C.; Barnes, L.L.; Bennett, D.A. Regional brain cortical thinning and systemic inflammation in older persons without dementia. J. Am. Geriatr. Soc. 2010, 58, 1823–1825. [Google Scholar] [CrossRef]
- Krishnadas, R.; McLean, J.; Batty, G.D.; Burns, H.; Deans, K.A.; Ford, I.; McConnachie, A.; McGinty, A.; McLean, J.S.; Millar, K.; et al. Cardio-metabolic risk factors and cortical thickness in a neurologically healthy male population: Results from the psychological, social and biological determinants of ill health (pSoBid) study. Neuroimage Clin. 2013, 2, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Poletti, S.; Leone, G.; Hoogenboezem, T.A.; Ghiglino, D.; Vai, B.; de Wit, H.; Wijkhuijs, A.J.M.; Locatelli, C.; Colombo, C.; Drexhage, H.A.; et al. Markers of neuroinflammation influence measures of cortical thickness in bipolar depression. Psychiatry Res. Neuroimaging 2019, 285, 64–66. [Google Scholar] [CrossRef]
- Sörös, P. Increased thickness of the orbitofrontal and anterior cingulate cortex in healthy aging. In Proceedings of the Conference Abstract: The 20th Annual Rotman Research Institute Conference, The frontal lobes, Toronto, Canada, 22–26 March 2010. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, M.; Lin, S.; Jian, R.; Li, X.; Chan, J.; Dong, G.; Fang, H.; Robinson, A.E.; Consortium, G.T.; et al. A Quantitative Proteome Map of the Human Body. Cell 2020, 183, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Velasco, O.; Papy-Garcia, D.; Le Douaron, G.; Sanchez-Santos, J.M.; De Las Rivas, J. Transcriptomic landscape, gene signatures and regulatory profile of aging in the human brain. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194491. [Google Scholar] [CrossRef]
- Erraji-Benchekroun, L.; Underwood, M.D.; Arango, V.; Galfalvy, H.; Pavlidis, P.; Smyrniotopoulos, P.; Mann, J.J.; Sibille, E. Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biol. Psychiatry 2005, 57, 549–558. [Google Scholar] [CrossRef] [PubMed]
- French, L.; Ma, T.; Oh, H.; Tseng, G.C.; Sibille, E. Age-Related Gene Expression in the Frontal Cortex Suggests Synaptic Function Changes in Specific Inhibitory Neuron Subtypes. Front. Aging Neurosci. 2017, 9, 162. [Google Scholar] [CrossRef] [PubMed]
- Carrard, G.; Bulteau, A.L.; Petropoulos, I.; Friguet, B. Impairment of proteasome structure and function in aging. Int. J. Biochem. Cell. Biol. 2002, 34, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Gee, J.; Ding, Q. The proteasome in brain aging. Ageing Res. Rev. 2002, 1, 279–293. [Google Scholar] [CrossRef]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Gonskikh, Y.; Polacek, N. Alterations of the translation apparatus during aging and stress response. Mech. Ageing. Dev. 2017, 168, 30–36. [Google Scholar] [CrossRef]
- Skariah, G.; Todd, P.K. Translational control in aging and neurodegeneration. Wiley Interdiscip. Rev. RNA 2021, 12, e1628. [Google Scholar] [CrossRef]
- Shcherbakov, D.; Nigri, M.; Akbergenov, R.; Brilkova, M.; Mantovani, M.; Petit, P.I.; Grimm, A.; Karol, A.A.; Teo, Y.; Sanchon, A.C.; et al. Premature aging in mice with error-prone protein synthesis. Sci. Adv. 2022, 8, eabl9051. [Google Scholar] [CrossRef] [PubMed]
- Brilkova, M.; Nigri, M.; Kumar, H.S.; Moore, J.; Mantovani, M.; Keller, C.; Grimm, A.; Eckert, A.; Shcherbakov, D.; Akbergenov, R.; et al. Error-prone protein synthesis recapitulates early symptoms of Alzheimer disease in aging mice. Cell. Rep. 2022, 40, 111433. [Google Scholar] [CrossRef]
- Tasaki, S.; Xu, J.; Avey, D.R.; Johnson, L.; Petyuk, V.A.; Dawe, R.J.; Bennett, D.A.; Wang, Y.; Gaiteri, C. Inferring protein expression changes from mRNA in Alzheimer’s dementia using deep neural networks. Nat. Commun. 2022, 13, 655. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Cat n° | Dilution |
|---|---|---|---|
| Anti-Actin | Sigma Aldrich | A5441 | 1:20,000 |
| Anti-GAP43 | Abclonal | A19055 | 1:1000 |
| Anti-mouse HRP | Biorad | 170-6516 | 1:5000 |
| Anti-rabbit HRP | Biorad | 170-6515 | 1:5000 |
| Antibody | Company | Cat n° | Dilution |
|---|---|---|---|
| Anti-GAP43 | Abclonal | A19055 | 1:50 |
| Anti-CCL8 | Abclonal | A6977 | 1:100 |
| Alexa Fluor 488 anti-rabbit | Invitrogen | A-11017 | 1:1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khatir, I.; Brunet, M.A.; Meller, A.; Amiot, F.; Patel, T.; Lapointe, X.; Avila Lopez, J.; Guilloy, N.; Castonguay, A.; Husain, M.A.; et al. Decoupling of mRNA and Protein Expression in Aging Brains Reveals the Age-Dependent Adaptation of Specific Gene Subsets. Cells 2023, 12, 615. https://doi.org/10.3390/cells12040615
Khatir I, Brunet MA, Meller A, Amiot F, Patel T, Lapointe X, Avila Lopez J, Guilloy N, Castonguay A, Husain MA, et al. Decoupling of mRNA and Protein Expression in Aging Brains Reveals the Age-Dependent Adaptation of Specific Gene Subsets. Cells. 2023; 12(4):615. https://doi.org/10.3390/cells12040615
Chicago/Turabian StyleKhatir, Inès, Marie A. Brunet, Anna Meller, Florent Amiot, Tushar Patel, Xavier Lapointe, Jessica Avila Lopez, Noé Guilloy, Anne Castonguay, Mohammed Amir Husain, and et al. 2023. "Decoupling of mRNA and Protein Expression in Aging Brains Reveals the Age-Dependent Adaptation of Specific Gene Subsets" Cells 12, no. 4: 615. https://doi.org/10.3390/cells12040615
APA StyleKhatir, I., Brunet, M. A., Meller, A., Amiot, F., Patel, T., Lapointe, X., Avila Lopez, J., Guilloy, N., Castonguay, A., Husain, M. A., Germain, J. S., Boisvert, F. -M., Plourde, M., Roucou, X., & Laurent, B. (2023). Decoupling of mRNA and Protein Expression in Aging Brains Reveals the Age-Dependent Adaptation of Specific Gene Subsets. Cells, 12(4), 615. https://doi.org/10.3390/cells12040615