


α-Synuclein Preformed Fibrils Bind to β-Neurexins and Impair β-Neurexin-Mediated Presynaptic Organization

 , , and
, , and 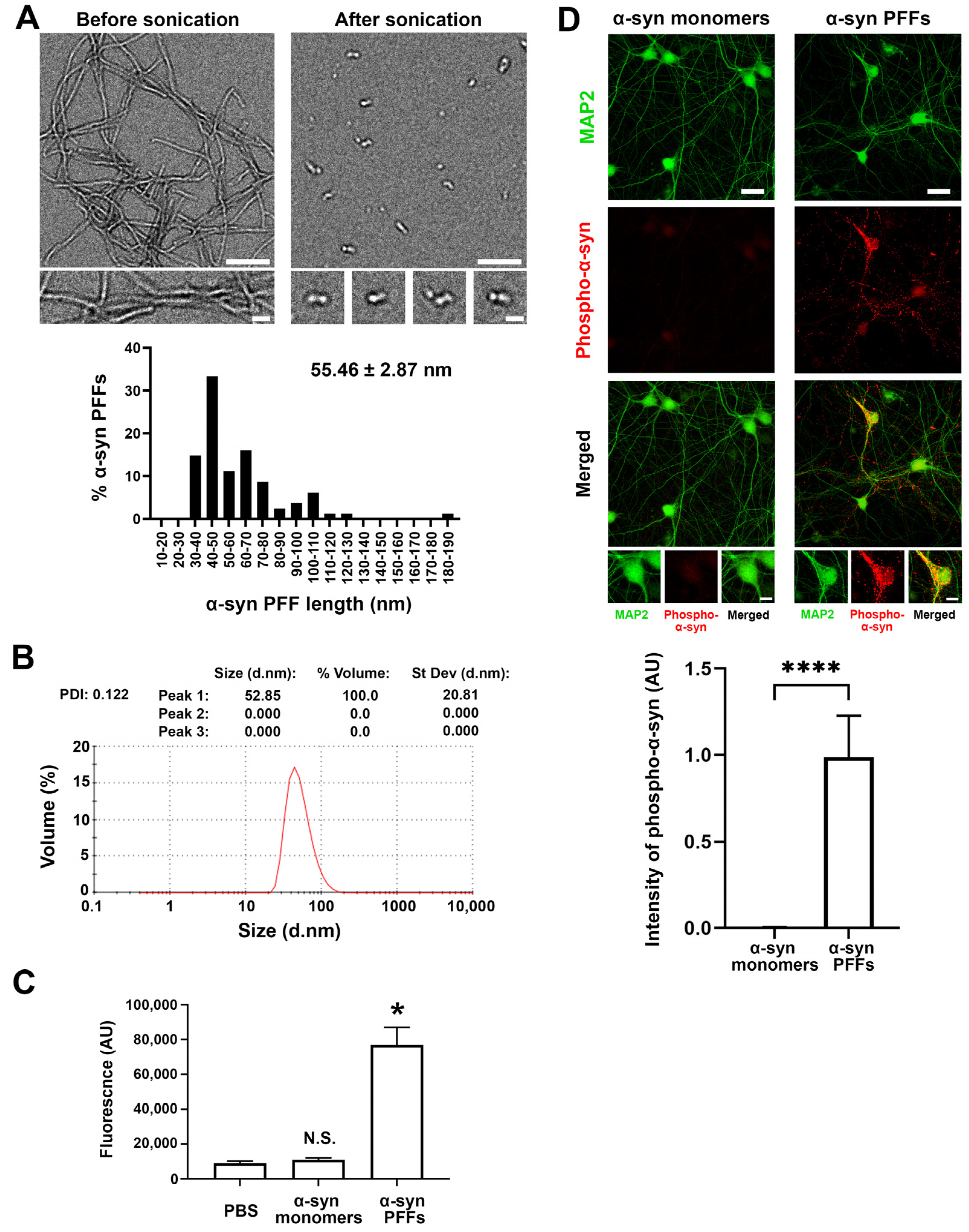{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Generation of α-Synuclein PFFs and Biotin Labelling
2.3. Validation of α-Synuclein PFFs Formation by Electron Microscopy, Dynamic Light Scattering, Thioflavin-T and Circular Dichroism Spectra Assays
2.4. Dot Blot
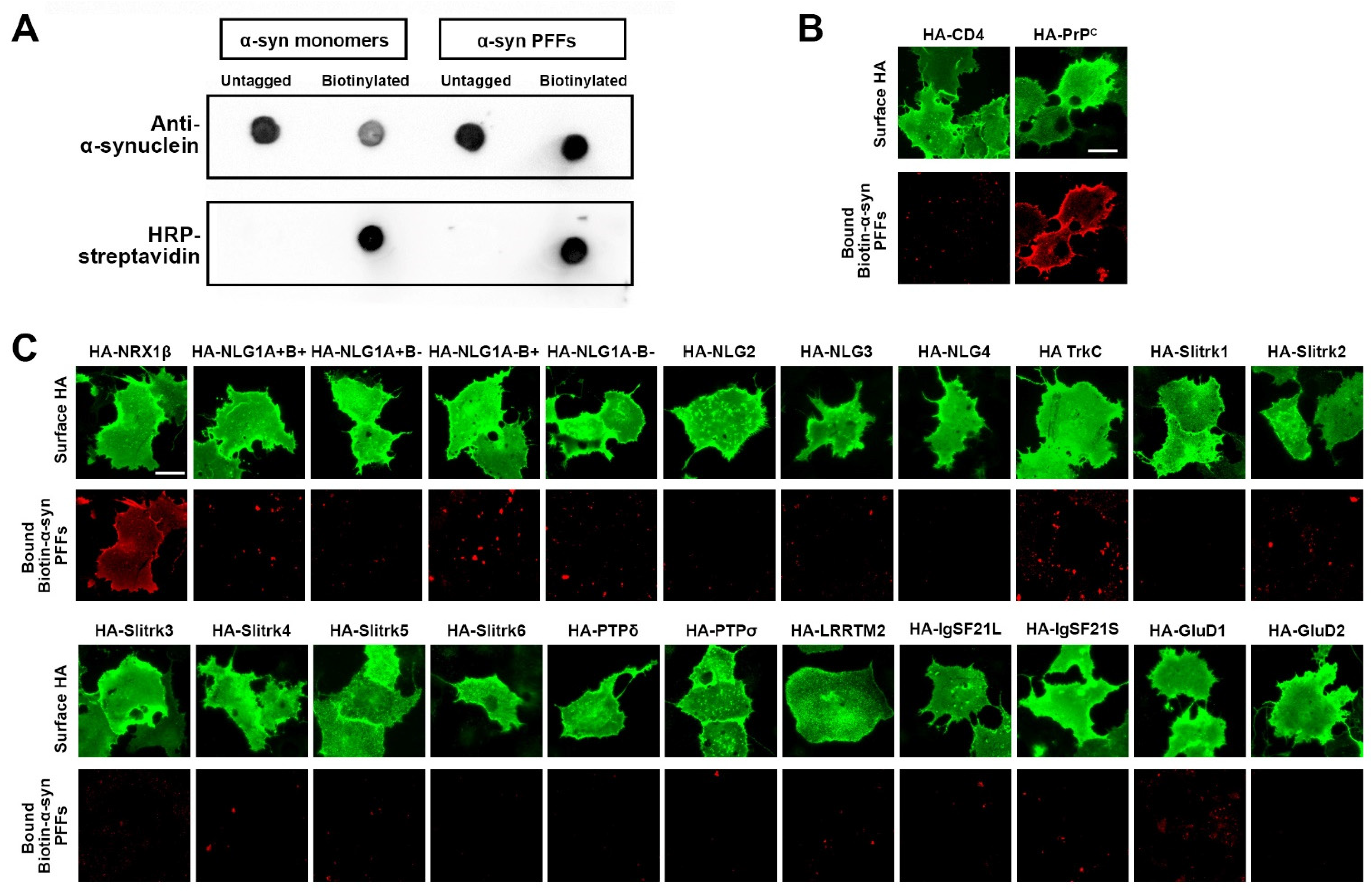
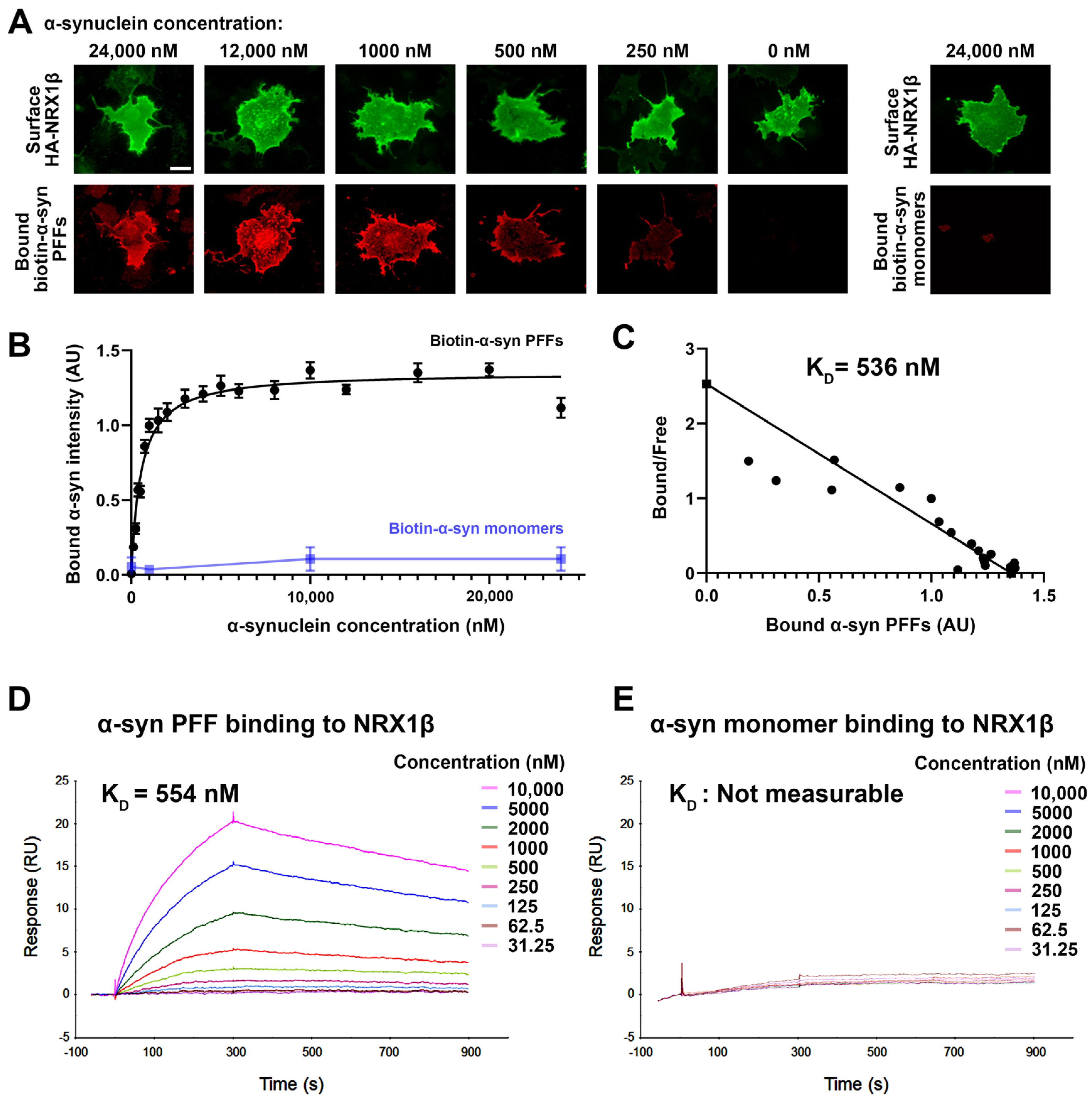
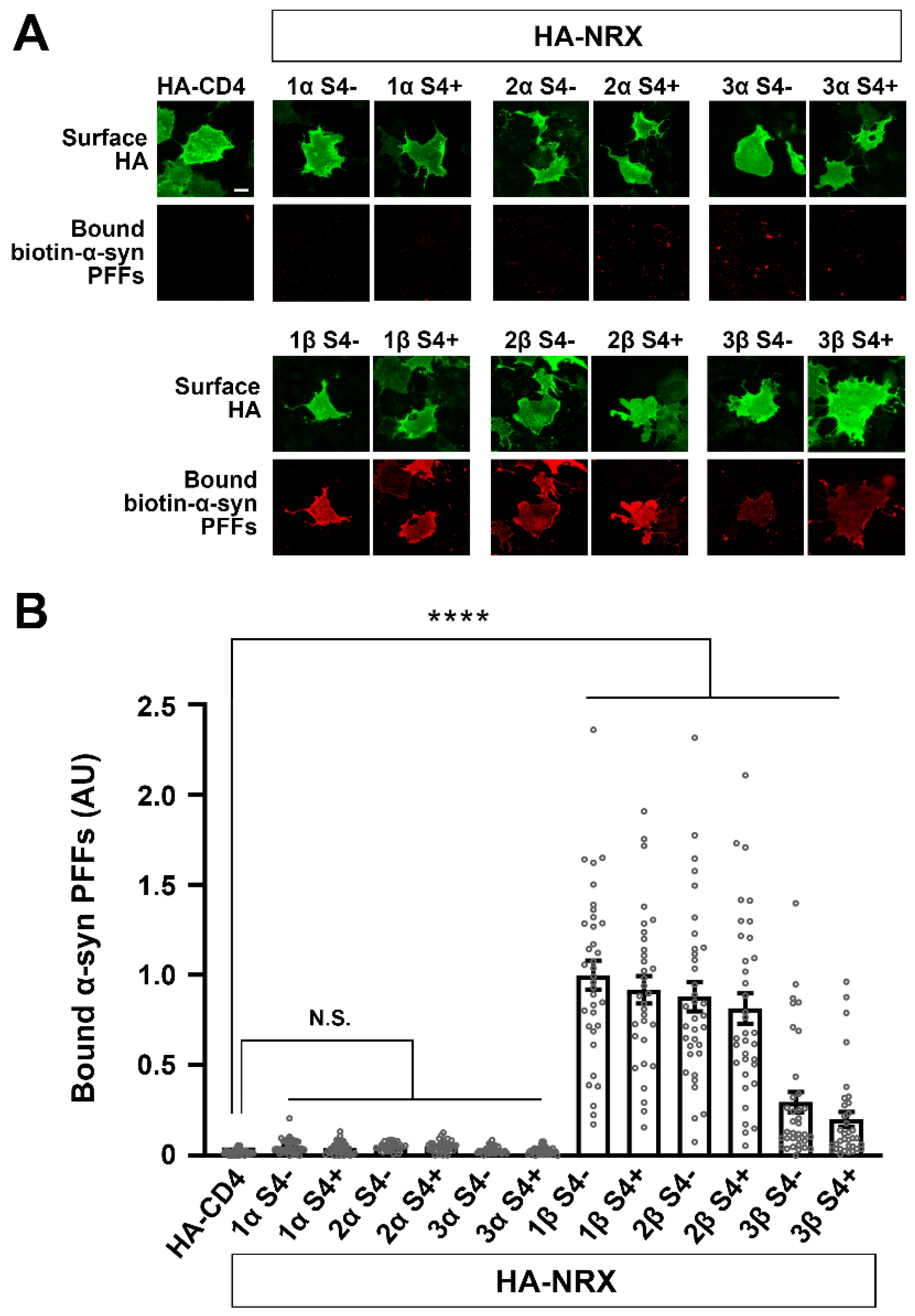
2.5. Cell Surface Binding Assays
2.6. Surface Plasmon Resonance
2.7. Neuron Culture, Transfection, and Neuronal Immunocytochemistry
2.8. Artificial Synapse Formation Assays
2.9. Imaging and Quantitative Fluorescence Analysis
2.10. Statistical Analysis
3. Results
3.1. Screening Synaptic Organizing Molecules for Interaction with α-Synuclein Preformed Fibrils Isolates Neurexin 1β as a Candidate α-Synuclein Binding Partner
3.2. α-Synuclein PFFs, but Not α-Synuclein Monomers, Bind Directly to NRX1β
3.3. α-Synuclein PFFs Specifically Bind to β-Isoforms of NRXs
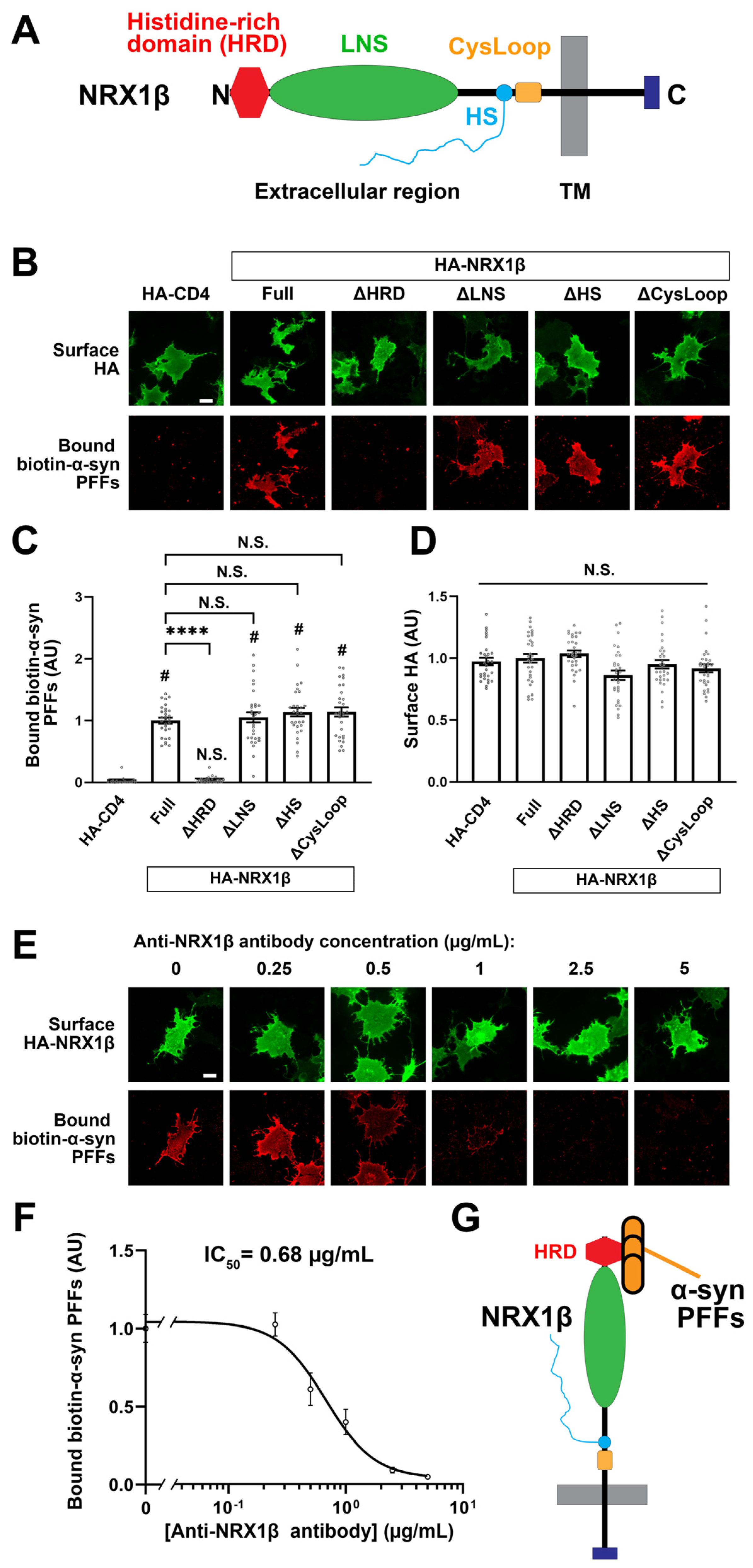
3.4. The N-Terminal Histidine-Rich Domain of β-NRXs Is Responsible for the Binding of α-Synuclein PFFs
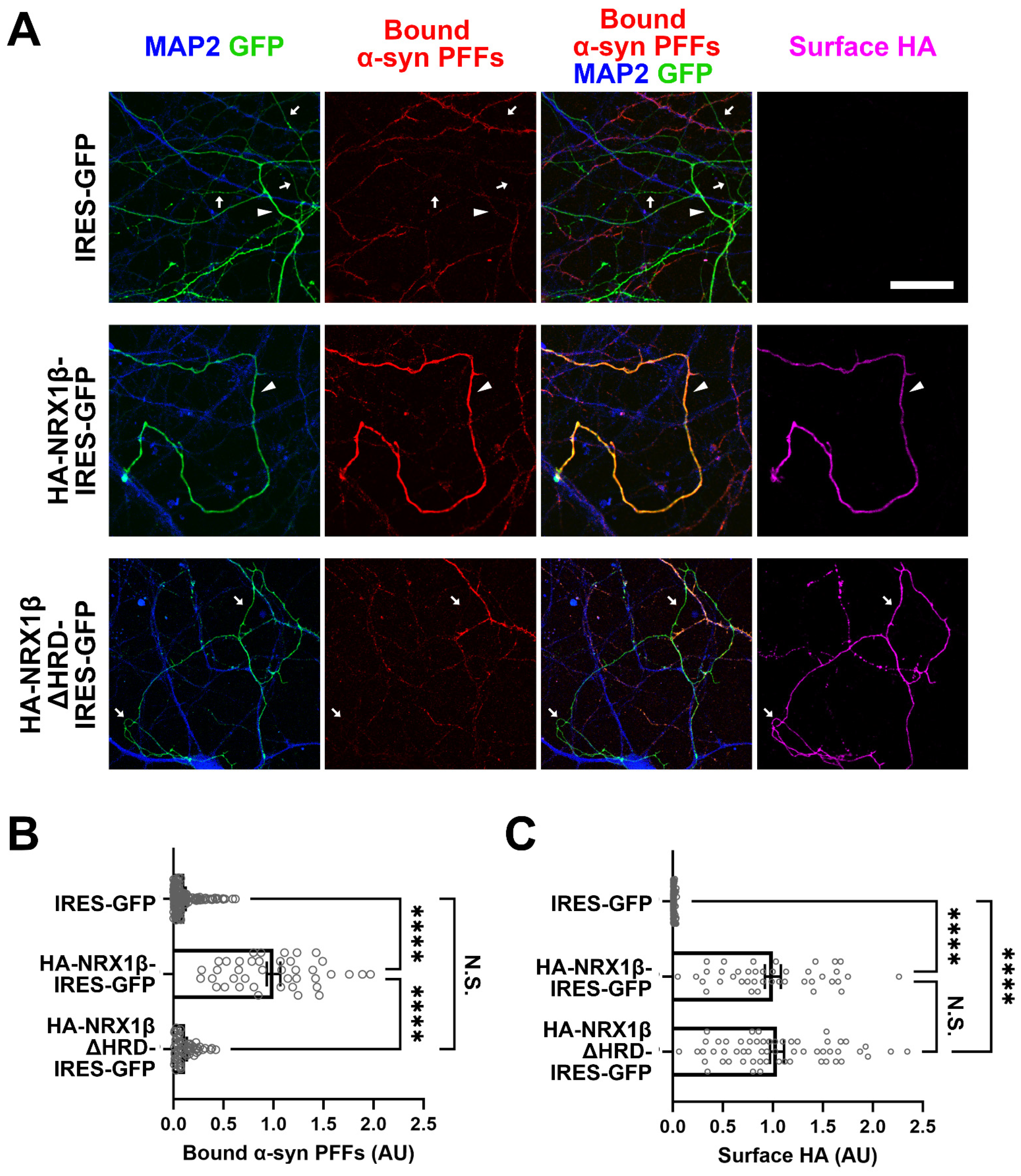
3.5. Neuronal Overexpression of NRX1β Enhances the Binding of α-Syn PFFs on the Axon Surface in an NRX1β HRD-Dependent Manner
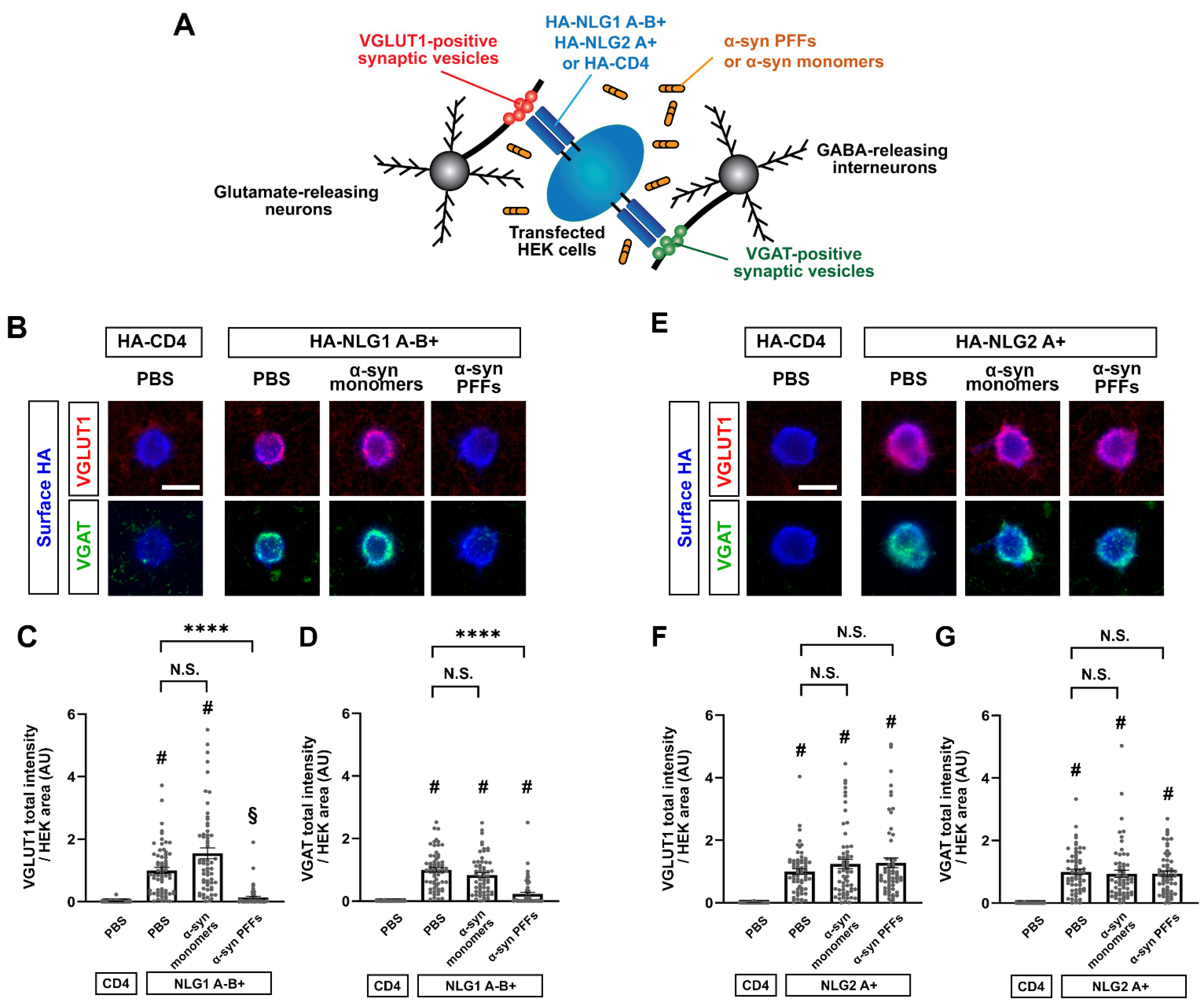
3.6. α-Syn PFF Treatment Diminishes β-NRX-Mediated Presynaptic Organization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dodel, R.; Csoti, I.; Ebersbach, G.; Fuchs, G.; Hahne, M.; Kuhn, W.; Oechsner, M.; Jost, W.; Reichmann, H.; Schulz, J.B. Lewy body dementia and Parkinson’s disease with dementia. J. Neurol. 2008, 255 (Suppl. S5), 39–47. [Google Scholar] [CrossRef] [PubMed]
- Galvin, J.E.; Lee, V.M.; Trojanowski, J.Q. Synucleinopathies: Clinical and pathological implications. Arch. Neurol. 2001, 58, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Gwinn-Hardy, K. Genetic classification of primary neurodegenerative disease. Science 1998, 282, 1075–1079. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. Alpha-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Desplats, P.; Sigurdson, C.; Tsigelny, I.; Masliah, E. Cell-to-cell transmission of non-prion protein aggregates. Nat. Rev. Neurol. 2010, 6, 702–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Stefanis, L. Alpha-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamguney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Rey, N.L.; Petit, G.H.; Bousset, L.; Melki, R.; Brundin, P. Transfer of human alpha-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol. 2013, 126, 555–573. [Google Scholar] [CrossRef] [Green Version]
- Okuzumi, A.; Kurosawa, M.; Hatano, T.; Takanashi, M.; Nojiri, S.; Fukuhara, T.; Yamanaka, T.; Miyazaki, H.; Yoshinaga, S.; Furukawa, Y.; et al. Rapid dissemination of alpha-synuclein seeds through neural circuits in an in-vivo prion-like seeding experiment. Acta Neuropathol. Commun. 2018, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic alpha-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Awa, S.; Suzuki, G.; Masuda-Suzukake, M.; Nonaka, T.; Saito, M.; Hasegawa, M. Phosphorylation of endogenous alpha-synuclein induced by extracellular seeds initiates at the pre-synaptic region and spreads to the cell body. Sci. Rep. 2022, 12, 1163. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, T.J.; Craig, A.M. Synaptic organizing complexes. Curr. Opin. Neurobiol. 2011, 21, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, A.M.; Graf, E.R.; Linhoff, M.W. How to build a central synapse: Clues from cell culture. Trends Neurosci. 2006, 29, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhof, T.C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Craig, A.M. Protein tyrosine phosphatases PTPdelta, PTPsigma, and LAR: Presynaptic hubs for synapse organization. Trends Neurosci. 2013, 36, 522–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Wit, J.; Ghosh, A. Control of neural circuit formation by leucine-rich repeat proteins. Trends Neurosci. 2014, 37, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Um, J.W.; Ko, J. LAR-RPTPs: Synaptic adhesion molecules that shape synapse development. Trends Cell Biol. 2013, 23, 465–475. [Google Scholar] [CrossRef]
- Sudhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, A.M.; Kang, Y. Neurexin-neuroligin signaling in synapse development. Curr. Opin. Neurobiol. 2007, 17, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bemben, M.A.; Shipman, S.L.; Nicoll, R.A.; Roche, K.W. The cellular and molecular landscape of neuroligins. Trends Neurosci. 2015, 38, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Kasem, E.; Kurihara, T.; Tabuchi, K. Neurexins and neuropsychiatric disorders. Neurosci. Res. 2018, 127, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Tromp, A.; Mowry, B.; Giacomotto, J. Neurexins in autism and schizophrenia-a review of patient mutations, mouse models and potential future directions. Mol. Psychiatry 2021, 26, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Born, G.; Grayton, H.M.; Langhorst, H.; Dudanova, I.; Rohlmann, A.; Woodward, B.W.; Collier, D.A.; Fernandes, C.; Missler, M. Genetic targeting of NRXN2 in mice unveils role in excitatory cortical synapse function and social behaviors. Front. Synaptic Neurosci. 2015, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Dachtler, J.; Ivorra, J.L.; Rowland, T.E.; Lever, C.; Rodgers, R.J.; Clapcote, S.J. Heterozygous deletion of alpha-neurexin I or alpha-neurexin II results in behaviors relevant to autism and schizophrenia. Behav. Neurosci. 2015, 129, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Tanabe, Y.; Lee, A.K.; Hamel, E.; Takahashi, H. Amyloid-beta Oligomers Interact with Neurexin and Diminish Neurexin-mediated Excitatory Presynaptic Organization. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.K.; Khaled, H.; Chofflet, N.; Takahashi, H. Synaptic Organizers in Alzheimer’s Disease: A Classification Based on Amyloid-beta Sensitivity. Front. Cell Neurosci. 2020, 14, 281. [Google Scholar] [CrossRef]
- Domert, J.; Rao, S.B.; Agholme, L.; Brorsson, A.C.; Marcusson, J.; Hallbeck, M.; Nath, S. Spreading of amyloid-beta peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 2014, 65, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid beta-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Agholme, L.; Kurudenkandy, F.R.; Granseth, B.; Marcusson, J.; Hallbeck, M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J. Neurosci. 2012, 32, 8767–8777. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s disease: Synaptic dysfunction and Abeta. Mol. Neurodegener. 2009, 4, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, M.; Sabatini, B.L.; Sudhof, T.C. Synapses and Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2012, 4, a005777. [Google Scholar] [CrossRef] [Green Version]
- Takuma, H.; Tomiyama, T.; Kuida, K.; Mori, H. Amyloid beta peptide-induced cerebral neuronal loss is mediated by caspase-3 in vivo. J. Neuropathol. Exp. Neurol. 2004, 63, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Nguyen, L.N.; Kessels, H.W.; Hagiwara, H.; Sisodia, S.; Malinow, R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 2010, 13, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Kalaitzakis, M.E.; Graeber, M.B.; Gentleman, S.M.; Pearce, R.K. Striatal beta-amyloid deposition in Parkinson disease with dementia. J. Neuropathol. Exp. Neurol. 2008, 67, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Hepp, D.H.; Vergoossen, D.L.; Huisman, E.; Lemstra, A.W.; Netherlands Brain, B.; Berendse, H.W.; Rozemuller, A.J.; Foncke, E.M.; van de Berg, W.D. Distribution and Load of Amyloid-beta Pathology in Parkinson Disease and Dementia with Lewy Bodies. J. Neuropathol. Exp. Neurol. 2016, 75, 936–945. [Google Scholar] [CrossRef] [Green Version]
- Lippa, C.F.; Fujiwara, H.; Mann, D.M.; Giasson, B.; Baba, M.; Schmidt, M.L.; Nee, L.E.; O’Connell, B.; Pollen, D.A.; St George-Hyslop, P.; et al. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am. J. Pathol. 1998, 153, 1365–1370. [Google Scholar] [CrossRef]
- Arai, Y.; Yamazaki, M.; Mori, O.; Muramatsu, H.; Asano, G.; Katayama, Y. Alpha-synuclein-positive structures in cases with sporadic Alzheimer’s disease: Morphology and its relationship to tau aggregation. Brain Res. 2001, 888, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Sagara, Y.; Mallory, M.; Hashimoto, M.; Mucke, L. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 12245–12250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, G.R.; Aoto, J.; Tabuchi, K.; Foldy, C.; Covy, J.; Yee, A.X.; Wu, D.; Lee, S.J.; Chen, L.; Malenka, R.C.; et al. beta-Neurexins Control Neural Circuits by Regulating Synaptic Endocannabinoid Signaling. Cell 2015, 162, 593–606. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.K.; Yi, N.; Khaled, H.; Feller, B.; Takahashi, H. SorCS1 inhibits amyloid-beta binding to neurexin and rescues amyloid-beta-induced synaptic pathology. Life Sci. Alliance 2023, 6, e202201681. [Google Scholar] [CrossRef]
- Takahashi, H.; Arstikaitis, P.; Prasad, T.; Bartlett, T.E.; Wang, Y.T.; Murphy, T.H.; Craig, A.M. Postsynaptic TrkC and presynaptic PTPsigma function as a bidirectional excitatory synaptic organizing complex. Neuron 2011, 69, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Katayama, K.; Sohya, K.; Miyamoto, H.; Prasad, T.; Matsumoto, Y.; Ota, M.; Yasuda, H.; Tsumoto, T.; Aruga, J.; et al. Selective control of inhibitory synapse development by Slitrk3-PTPdelta trans-synaptic interaction. Nat. Neurosci. 2012, 15, 389–398. [Google Scholar] [CrossRef]
- Tanabe, Y.; Naito, Y.; Vasuta, C.; Lee, A.K.; Soumounou, Y.; Linhoff, M.W.; Takahashi, H. IgSF21 promotes differentiation of inhibitory synapses via binding to neurexin2α. Nat. Commun. 2017, 8, 408. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.R.; Polinski, N.K.; Duffy, M.F.; Kemp, C.J.; Luk, K.C.; Volpicelli-Daley, L.A.; Kanaan, N.M.; Sortwell, C.E. Generation of Alpha-Synuclein Preformed Fibrils from Monomers and Use In Vivo. J. Vis. Exp. 2019, 148, e59758. [Google Scholar] [CrossRef] [Green Version]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014, 9, 2135–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panchal, J.; Kotarek, J.; Marszal, E.; Topp, E.M. Analyzing subvisible particles in protein drug products: A comparison of dynamic light scattering (DLS) and resonant mass measurement (RMM). AAPS J. 2014, 16, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Urrea, L.; Segura-Feliu, M.; Masuda-Suzukake, M.; Hervera, A.; Pedraz, L.; Garcia Aznar, J.M.; Vila, M.; Samitier, J.; Torrents, E.; Ferrer, I.; et al. Involvement of Cellular Prion Protein in alpha-Synuclein Transport in Neurons. Mol. Neurobiol. 2018, 55, 1847–1860. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, D.G.; Temido-Ferreira, M.; Vicente Miranda, H.; Batalha, V.L.; Coelho, J.E.; Szego, E.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. Alpha-synuclein interacts with PrP (C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Domingues, R.; Sant’Anna, R.; da Fonseca, A.C.C.; Robbs, B.K.; Foguel, D.; Outeiro, T.F. Extracellular alpha-synuclein: Sensors, receptors, and responses. Neurobiol. Dis. 2022, 168, 105696. [Google Scholar] [CrossRef]
- Reissner, C.; Runkel, F.; Missler, M. Neurexins. Genome Biol. 2013, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, H.; Gollan, L.; Scholl, F.G.; Mahadomrongkul, V.; Dobler, E.; Limthong, N.; Peck, M.; Aoki, C.; Scheiffele, P. Silencing of neuroligin function by postsynaptic neurexins. J. Neurosci. 2007, 27, 2815–2824. [Google Scholar] [CrossRef] [Green Version]
- Futai, K.; Doty, C.D.; Baek, B.; Ryu, J.; Sheng, M. Specific trans-synaptic interaction with inhibitory interneuronal neurexin underlies differential ability of neuroligins to induce functional inhibitory synapses. J. Neurosci. 2013, 33, 3612–3623. [Google Scholar] [CrossRef] [Green Version]
- Klatt, O.; Repetto, D.; Brockhaus, J.; Reissner, C.; El Khallouqi, A.; Rohlmann, A.; Heine, M.; Missler, M. Endogenous beta-neurexins on axons and within synapses show regulated dynamic behavior. Cell Rep. 2021, 35, 109266. [Google Scholar] [CrossRef]
- Scheiffele, P.; Fan, J.; Choih, J.; Fetter, R.; Serafini, T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 2000, 101, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Boucard, A.A.; Chubykin, A.A.; Comoletti, D.; Taylor, P.; Sudhof, T.C. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron 2005, 48, 229–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chih, B.; Gollan, L.; Scheiffele, P. Alternative splicing controls selective trans-synaptic interactions of the neuroligin-neurexin complex. Neuron 2006, 51, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli-Daley, L.A.; Gamble, K.L.; Schultheiss, C.E.; Riddle, D.M.; West, A.B.; Lee, V.M. Formation of alpha-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 2014, 25, 4010–4023. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schutz, O.; et al. Alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef] [Green Version]
- Colom-Cadena, M.; Pegueroles, J.; Herrmann, A.G.; Henstridge, C.M.; Munoz, L.; Querol-Vilaseca, M.; Martin-Paniello, C.S.; Luque-Cabecerans, J.; Clarimon, J.; Belbin, O.; et al. Synaptic phosphorylated alpha-synuclein in dementia with Lewy bodies. Brain 2017, 140, 3204–3214. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353, aah3374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.R.; Cha, S.H.; Kang, S.J.; Kim, J.B.; Jou, I.; Park, S.M. Prion-like Propagation of alpha-Synuclein Is Regulated by the FcgammaRIIB-SHP-1/2 Signaling Pathway in Neurons. Cell Rep. 2018, 22, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, L.; Marano, M.M.; Tandon, A. Import and Export of Misfolded alpha-Synuclein. Front. Neurosci. 2018, 12, 344. [Google Scholar] [CrossRef] [Green Version]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, U.; Kayed, R. Amyloid beta, Tau, and alpha-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog. Neurobiol. 2022, 214, 102270. [Google Scholar] [CrossRef]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudenko, G.; Nguyen, T.; Chelliah, Y.; Sudhof, T.C.; Deisenhofer, J. The structure of the ligand-binding domain of neurexin Ibeta: Regulation of LNS domain function by alternative splicing. Cell 1999, 99, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gokce, O.; Sudhof, T.C. Membrane-tethered monomeric neurexin LNS-domain triggers synapse formation. J. Neurosci. 2013, 33, 14617–14628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roppongi, R.T.; Dhume, S.H.; Padmanabhan, N.; Silwal, P.; Zahra, N.; Karimi, B.; Bomkamp, C.; Patil, C.S.; Champagne-Jorgensen, K.; Twilley, R.E.; et al. LRRTMs Organize Synapses through Differential Engagement of Neurexin and PTPsigma. Neuron 2020, 106, 108–125. [Google Scholar] [CrossRef]
- Han, K.A.; Kim, Y.J.; Yoon, T.H.; Kim, H.; Bae, S.; Um, J.W.; Choi, S.Y.; Ko, J. LAR-RPTPs Directly Interact with Neurexins to Coordinate Bidirectional Assembly of Molecular Machineries. J. Neurosci. 2020, 40, 8438–8462. [Google Scholar] [CrossRef] [PubMed]
- Savas, J.N.; Ribeiro, L.F.; Wierda, K.D.; Wright, R.; DeNardo-Wilke, L.A.; Rice, H.C.; Chamma, I.; Wang, Y.Z.; Zemla, R.; Lavallee-Adam, M.; et al. The Sorting Receptor SorCS1 Regulates Trafficking of Neurexin and AMPA Receptors. Neuron 2015, 87, 764–780. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M. Alpha-Synuclein (alphaSyn) Preformed Fibrils Induce Endogenous alphaSyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. J. Neurosci. 2019, 39, 5080–5094. [Google Scholar] [CrossRef] [Green Version]
- Covey, D.P.; Mateo, Y.; Sulzer, D.; Cheer, J.F.; Lovinger, D.M. Endocannabinoid modulation of dopamine neurotransmission. Neuropharmacology 2017, 124, 52–61. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, J.; Moreno-Martet, M.; Rodriguez-Cueto, C.; Palomo-Garo, C.; Gomez-Canas, M.; Valdeolivas, S.; Guaza, C.; Romero, J.; Guzman, M.; Mechoulam, R.; et al. Prospects for cannabinoid therapies in basal ganglia disorders. Br. J. Pharmacol. 2011, 163, 1365–1378. [Google Scholar] [CrossRef] [Green Version]
- Glass, M.; Felder, C.C. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: Evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997, 17, 5327–5333. [Google Scholar] [CrossRef] [Green Version]
- Roppongi, R.T.; Karimi, B.; Siddiqui, T.J. Role of LRRTMs in synapse development and plasticity. Neurosci. Res. 2017, 116, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Yuzaki, M. The C1q complement family of synaptic organizers: Not just complementary. Curr. Opin. Neurobiol. 2017, 45, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Soler-Llavina, G.J.; Fuccillo, M.V.; Ko, J.; Sudhof, T.C.; Malenka, R.C. The neurexin ligands, neuroligins and leucine-rich repeat transmembrane proteins, perform convergent and divergent synaptic functions in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 16502–16509. [Google Scholar] [CrossRef] [Green Version]
- Dhume, S.H.; Connor, S.A.; Mills, F.; Tari, P.K.; Au-Yeung, S.H.M.; Karimi, B.; Oku, S.; Roppongi, R.T.; Kawabe, H.; Bamji, S.X.; et al. Distinct but overlapping roles of LRRTM1 and LRRTM2 in developing and mature hippocampal circuits. eLife 2022, 11, e64742. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Patzke, C.; Liakath-Ali, K.; Seigneur, E.; Sudhof, T.C. GluD1 is a signal transduction device disguised as an ionotropic receptor. Nature 2021, 595, 261–265. [Google Scholar] [CrossRef]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.M.; Price, N.C. The use of circular dichroism in the investigation of protein structure and function. Curr. Protein. Pept. Sci. 2000, 1, 349–384. [Google Scholar] [CrossRef] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feller, B.; Fallon, A.; Luo, W.; Nguyen, P.T.; Shlaifer, I.; Lee, A.K.; Chofflet, N.; Yi, N.; Khaled, H.; Karkout, S.; et al. α-Synuclein Preformed Fibrils Bind to β-Neurexins and Impair β-Neurexin-Mediated Presynaptic Organization. Cells 2023, 12, 1083. https://doi.org/10.3390/cells12071083
Feller B, Fallon A, Luo W, Nguyen PT, Shlaifer I, Lee AK, Chofflet N, Yi N, Khaled H, Karkout S, et al. α-Synuclein Preformed Fibrils Bind to β-Neurexins and Impair β-Neurexin-Mediated Presynaptic Organization. Cells. 2023; 12(7):1083. https://doi.org/10.3390/cells12071083
Chicago/Turabian StyleFeller, Benjamin, Aurélie Fallon, Wen Luo, Phuong Trang Nguyen, Irina Shlaifer, Alfred Kihoon Lee, Nicolas Chofflet, Nayoung Yi, Husam Khaled, Samer Karkout, and et al. 2023. "α-Synuclein Preformed Fibrils Bind to β-Neurexins and Impair β-Neurexin-Mediated Presynaptic Organization" Cells 12, no. 7: 1083. https://doi.org/10.3390/cells12071083
APA StyleFeller, B., Fallon, A., Luo, W., Nguyen, P. T., Shlaifer, I., Lee, A. K., Chofflet, N., Yi, N., Khaled, H., Karkout, S., Bourgault, S., Durcan, T. M., & Takahashi, H. (2023). α-Synuclein Preformed Fibrils Bind to β-Neurexins and Impair β-Neurexin-Mediated Presynaptic Organization. Cells, 12(7), 1083. https://doi.org/10.3390/cells12071083