
A Novel CD206 Targeting Peptide Inhibits Bleomycin-Induced Pulmonary Fibrosis in Mice

 ,
,  , and
, and
Abstract
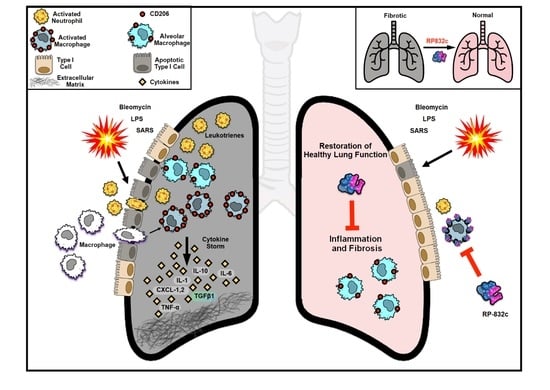
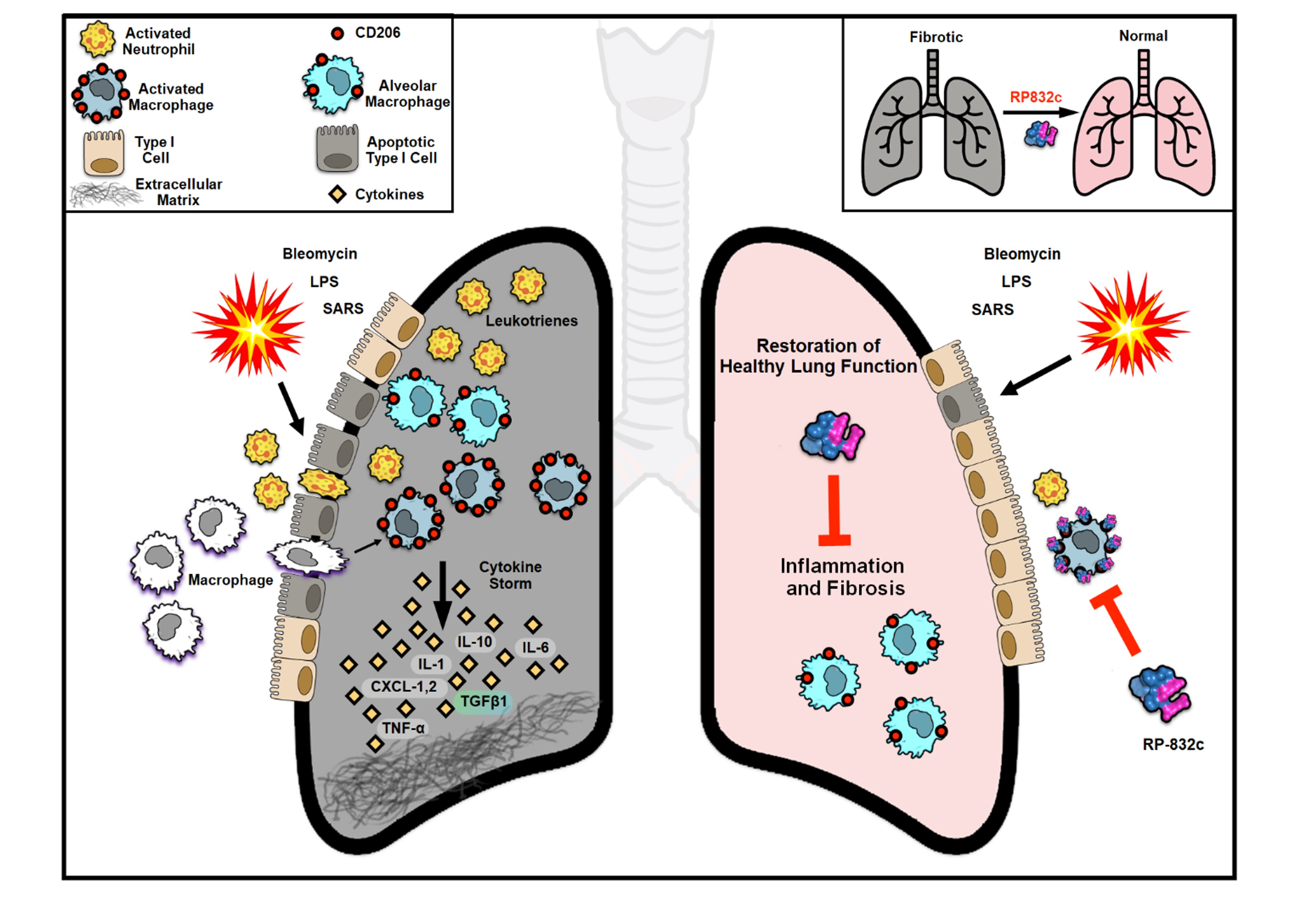
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture of Primary Cells
2.2. Cell Viability Assay
2.3. Animal Experiments
2.4. Histological and Immunostaining Evaluation
2.5. Immunofluorescence
2.6. Quantitative Real-Time PCR
2.7. Statistical Analysis
3. Results
3.1. RP-832c Targets CD206 Positive Macrophages
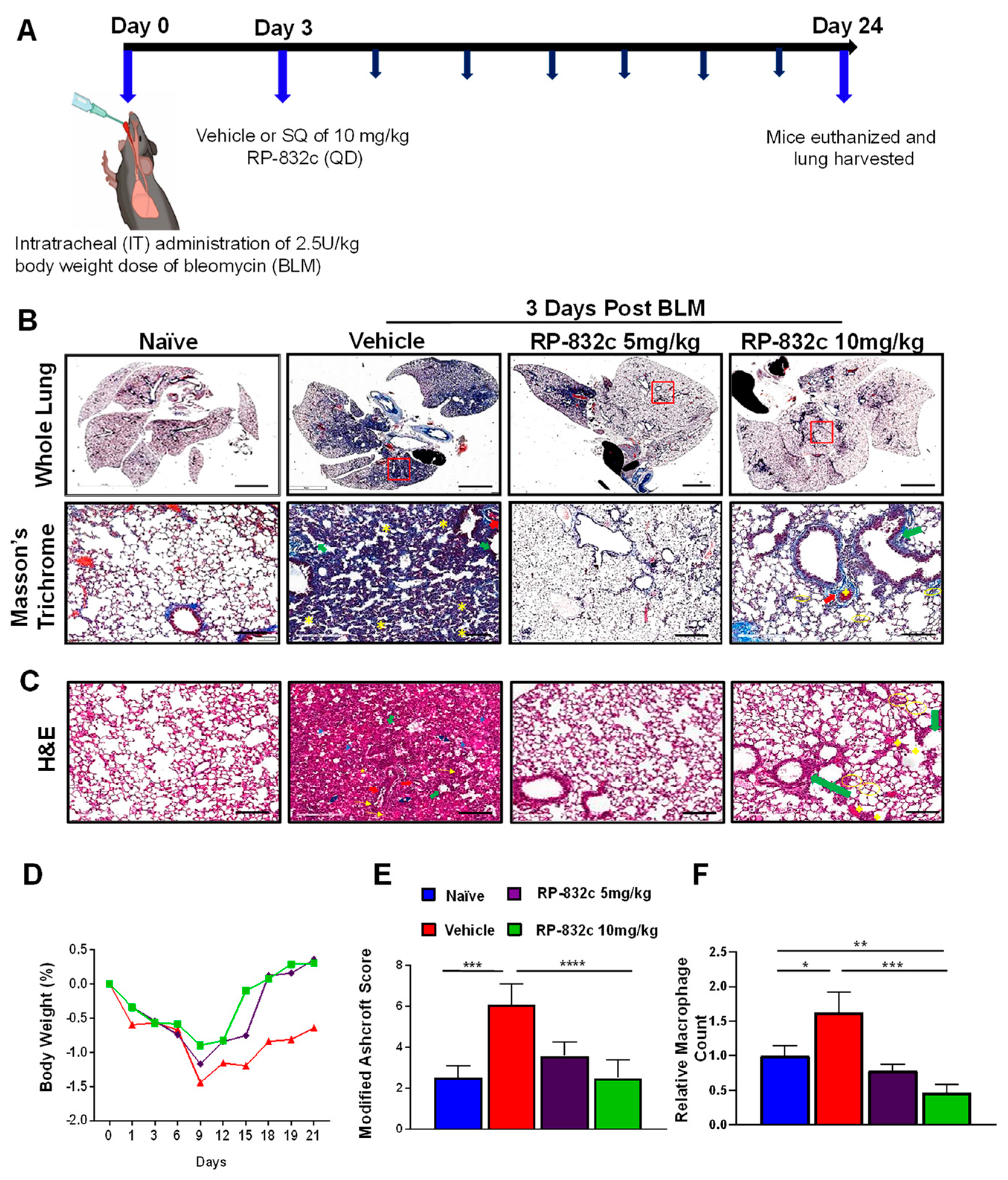
3.2. RP-832c Prevented Fibrosis in an Early Model of BLM-Induced Lung Fibrosis
3.3. RP-832c Peptide Treated Firosis and Reduced Expression of CD206 and Fibrosis Markers in a Late Model of BLM-Induced Lung Fibrosis Model
3.4. RP-832c Lacks Significant Toxicity
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirani, N.; Antonicelli, F.; Strieter, R.M.; Wiesener, M.S.; Ratcliffe, P.J.; Haslett, C.; Donnelly, S.C. The regulation of interleukin-8 by hypoxia in human macrophages--a potential role in the pathogenesis of acute respiratory distress syndrome (ARDS). Mol. Med. 2001, 7, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Carrillo, G.; Estrada, A.; Mejia, M.; Becerril, C.; Cisneros, J.; Gaxiola, M.; Pérez-Padilla, R.; Navarro, C.; Richards, T.; et al. Accelerated variant of idiopathic pulmonary fibrosis: Clinical behavior and gene expression pattern. PLoS ONE 2007, 2, e482. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.; Bailey, N.W.; Yang, J.; Steel, M.P.; Groshong, S.; Kervitsky, L.; Brown, K.K.; Schwarz, M.I.; Schwartz, D.A. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF). PLoS ONE 2009, 4, e5134. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology, and regulation. Mucosal Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, J.C.; Kropski, J.A.; Blackwell, T.S. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018, 71–72, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Geng, Y.; Li, L.; Li, X.; Yan, X.; Fang, Y.; Li, X.; Dong, S.; Liu, X.; Yang, X.; et al. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J. Exp. Med. 2015, 212, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- McCubbrey, A.L.; Barthel, L.; Mohning, M.P.; Redente, E.F.; Mould, K.J.; Thomas, S.M.; Leach, S.M.; Danhorn, T.; Gibbings, S.L.; Jakubzick, C.V.; et al. Deletion of c-FLIP from CD11b(hi) Macrophages Prevents the Development of Bleomycin-induced Lung Fibrosis. Am. J. Respir. Cell. Mol. Biol. 2018, 58, 66–78. [Google Scholar] [CrossRef]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at the single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; López-Giráldez, F.; Dash, B.C.; Muñoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362, eaar2971. [Google Scholar] [CrossRef] [PubMed]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Mackinnon, A.C.; Farnworth, S.L.; Kipari, T.; Haslett, C.; Iredale, J.P.; Liu, F.-T.; Hughes, J.; Sethi, T. Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am. J. Pathol. 2008, 172, 288–298. [Google Scholar] [CrossRef]
- Zhou, X.; Franklin, R.A.; Adler, M.; Jacox, J.B.; Bailis, W.; Shyer, J.A.; Flavell, R.A.; Mayo, A.; Alon, U.; Medzhitov, R. Circuit design features of a stable two-cell system. Cell 2018, 172, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Lodyga, M.; Cambridge, E.; Karvonen, H.M.; Pakshir, P.; Wu, B.; Boo, S.; Kiebalo, M.; Kaarteenaho, R.; Glogauer, M.; Kapoor, M.; et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGFβ. Sci. Signal. 2019, 12, eaao3469. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Rosada, R.; Moreira, A.P.; Joshi, A.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Mathai, S.; Gulati, M.; Herzog, E.L.; et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS ONE 2010, 5, e9683. [Google Scholar] [CrossRef]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Leicester, K.L.; Olynyk, J.K.; Brunt, E.M.; Britton, R.S.; Bacon, B.R. CD14-positive hepatic monocytes/macrophages increase in hereditary hemochromatosis. Liver Int. 2004, 24, 446–451. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1 (LPS+) vs. Classically and M2 (LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Minutti, C.M.; Modak, R.V.; Macdonald, F.; Li, F.; Smyth, D.J.; Dorward, D.A.; Blair, N.; Husovsky, C.; Muir, A.; Giampazolias, E.; et al. A macrophage-pericyte axis directs tissue restoration via amphiregulin-induced transforming growth factor beta activation. Immunity 2019, 50, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.L.; Koh, T.J. Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 2013, 93, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Apte, S.H.; Groves, P.; Olver, S.; Baz, A.; Doolan, D.L.; Kelso, A.; Kienzle, N. IFN-gamma inhibits IL-4-induced type 2 cytokine expression by CD8 T cells in vivo and modulates the anti-tumor response. J. Immunol. 2010, 185, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.T.; Benveniste, E.N. IL-4-activated STAT-6 inhibits IFN-gamma-induced CD40 gene expression in macrophages/microglia. J. Immunol. 2000, 165, 6235–6243. [Google Scholar] [CrossRef] [PubMed]
- Gurujeyalakshmi, G.; Giri, S.N. Molecular mechanisms of antifibrotic effect of interferon-gamma in bleomycin-mouse model of lung fibrosis: Downregulation of TGF-beta and procollagen I and III gene expression. Exp. Lung Res. 1995, 21, 791–808. [Google Scholar] [CrossRef]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defense peptides: Beyond antimicrobial activity. Nat. Rev. Immunol. 2016, 16, 321–334. [Google Scholar] [CrossRef]
- Steinstraesser, L.; Kraneburg, U.M.; Hirsch, T.; Kesting, M.; Steinau, H.U.; Jacobsen, F.; Al-Benna, S. Host defense peptides as effector molecules of the innate immune response: A sledgehammer for drug resistance? Int. J. Mol. Sci. 2009, 10, 3951–3970. [Google Scholar] [CrossRef]
- Jaynes, J.M.; Sable, R.; Ronzetti, M.; Bautista, W.; Knotts, Z.; Abisoye-Ogunniyan, A.; Li, D.; Calvo, R.; Dashnyam, M.; Singh, A.; et al. Mannose receptor (CD206) activation in tumor-associated macrophages enhances adaptive and innate antitumor immune responses. Sci. Transl. Med. 2020, 12, eaax6337. [Google Scholar] [CrossRef]
- Kim, T.H.; Lee, Y.H.; Kim, K.H.; Lee, S.H.; Cha, J.Y.; Shin, E.K.; Jung, S.; Jang, A.S.; Park, S.W.; Uh, S.T.; et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. Investig. 2007, 117, 3786–3799. [Google Scholar]
- Kim, T.H.; Lee, Y.H.; Kim, K.H.; Lee, S.H.; Cha, J.Y.; Shin, E.K.; Jung, S.; Jang, A.S.; Park, S.W.; Uh, S.T.; et al. Role of Lung Apolipoprotein A-I in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Francipane, M.G.; Medema, J.P.; Stassi, G. Colon cancer stem cells: Promise of targeted therapy. Gastro 2010, 138, 2151–2162. [Google Scholar] [CrossRef] [PubMed]
- Lierova, A.; Jelicova, M.; Nemcova, M.; Proksova, M.; Pejchal, J.; Zarybnicka, L.; Sinkorova, Z. Cytokines and radiation-induced pulmonary injuries. J. Radiat. Res. 2018, 59, 709–753. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Frederick, E.; Hausburg, M.; Goldberg, L.; Hoke, M.; Roshon, M.; Mains, C.; Bar-Or, D. The novel immunomodulatory biologic LMWF5A for pharmacological attenuation of the “cytokine storm” in COVID-19 patients: A hypothesis. Patient Saf. Surg. 2020, 14, 21. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Obstacles and opportunities for understanding macrophage polarization. J. Leukoc. Biol. 2011, 89, 557–563. [Google Scholar] [CrossRef]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef]
- Kaku, Y.; Imaoka, H.; Morimatsu, Y.; Komohara, Y.; Ohnishi, K.; Oda, H.; Takenaka, S.; Matsuoka, M.; Kawayama, T.; Takeya, M.; et al. Overexpression of CD163, CD204, and CD206 on alveolar macrophages in the lungs of patients with severe chronic obstructive pulmonary disease. PLoS ONE 2014, 9, e87400. [Google Scholar] [CrossRef]
- Moore, B.B.; Paine, R., 3rd; Christensen, P.J.; Moore, T.A.; Sitterding, S.; Ngan, R.; Wilke, C.A.; Kuziel, W.A.; Toews, G.B. Protection from pulmonary fibrosis in the absence of CCR2 signaling. J. Immunol. 2001, 167, 4368–4377. [Google Scholar] [CrossRef]
- Xu, J.; Flaczyk, A.; Neal, L.M.; Fa, Z.; Cheng, D.; Ivey, M.; Moore, B.B.; Curtis, J.L.; Osterholzer, J.J.; Olszewski, M.A. Exploitation of Scavenger Receptor, Macrophage Receptor with Collagenous Structure, by Cryptococcus neoformans Promotes Alternative Activation of Pulmonary Lymph Node CD11b (+) Conventional Dendritic Cells and Non-Protective Th2 Bias. Front. Immunol. 2017, 8, 1231. [Google Scholar] [CrossRef] [PubMed]
- Salazar, F.; Hall, L.; Negm, O.H.; Awuah, D.; Tighe, P.J.; Shakib, F.; Ghaemmaghami, A.M. The mannose receptor negatively modulates the Toll-like receptor 4-aryl hydrocarbon receptor-indoleamine 2,3-dioxygenase axis in dendritic cells affecting T helper cell polarization. J. Allergy Clin. Immunol. 2016, 137, 1841–1851.e1842. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.N.; Hyde, D.M.; A Hollinger, M. Effect of antibody to transforming growth factor beta on bleomycin-induced accumulation of lung collagen in mice. Thorax 1993, 48, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Madsen, D.H.; Leonard, D.; Masedunskas, A.; Moyer, A.; Jürgensen, H.J.; Peters, D.E.; Amornphimoltham, P.; Selvaraj, A.; Yamada, S.S.; Brenner, D.A.; et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell. Biol. 2013, 202, 951–966. [Google Scholar] [CrossRef]
- Paracuellos, P.; Briggs, D.C.; Carafoli, F.; Lončar, T.; Hohenester, E. Insights into Collagen Uptake by C-type Mannose Receptors from the Crystal Structure of Endo180 Domains 1-4. Structure 2015, 23, 2133–2142. [Google Scholar] [CrossRef]
- Chang, C.H.; Juan, Y.H.; Hu, H.C.; Kao, K.C.; Lee, C.S. Reversal of lung fibrosis: An unexpected finding in survivor of acute respiratory distress syndrome. QJM Int. J. Med. 2018, 111, 47–48. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Saline | RP-832c 50 mg/kg | |||
|---|---|---|---|---|
| Parameter | n = 5 | n = 10 | Mean Difference | p Value |
| RBC/mm3 | 8.3 ± 0.6 | 8.5 ± 0.7 | 0.2 | 0.7295 |
| WBC/μL | 5.7 ± 1.8 | 7.7 ± 3.2 | 2.0 | 0.2163 |
| Total Protein (g/dL) | 5.6 ± 0.8 | 5.9 ± 0.5 | 0.3 | 0.5797 |
| AST (mg/dL) | 140.0 ± 75.5 | 91.2 ± 21.7 | −48.8 | 0.1237 |
| Phosphorus (mg/dL) | 8.8 ± 2.4 | 7.7 ± 0.5 | −1.1 | 0.2623 |
| Creatinine (mg/dL) | 0.2 ± 0 | 0.2 ± 0 | 0.0 | - |
| ALT (mg/dL) | 35.8 ± 18.9 | 27.2 ± 10 | −8.6 | 0.3808 |
| Albumin (g/dL) | 2.9 ± 0.3 | 3.1± 0.3 | 0.2 | 0.3982 |
| BUN (mg/dL) | 22.4 ± 3.4 | 24.0 ± 4.3 | 1.6 | 0.3087 |
| Globulin (g/dL) | 2.6 ± 0.5 | 2.7 ± 0.2 | 0.1 | 0.4413 |
| Alb/Globulin Ratio | 1.1 ± 0.2 | 1.2 ± 0.1 | 0.1 | 0.3295 |
| BUN/Creatinine Ratio | 111.9 ± 17 | 120.1 ± 21.6 | 8.2 | 0.3087 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghebremedhin, A.; Salam, A.B.; Adu-Addai, B.; Noonan, S.; Stratton, R.; Ahmed, M.S.U.; Khantwal, C.; Martin, G.R.; Lin, H.; Andrews, C.; et al. A Novel CD206 Targeting Peptide Inhibits Bleomycin-Induced Pulmonary Fibrosis in Mice. Cells 2023, 12, 1254. https://doi.org/10.3390/cells12091254
Ghebremedhin A, Salam AB, Adu-Addai B, Noonan S, Stratton R, Ahmed MSU, Khantwal C, Martin GR, Lin H, Andrews C, et al. A Novel CD206 Targeting Peptide Inhibits Bleomycin-Induced Pulmonary Fibrosis in Mice. Cells. 2023; 12(9):1254. https://doi.org/10.3390/cells12091254
Chicago/Turabian StyleGhebremedhin, Anghesom, Ahmad Bin Salam, Benjamin Adu-Addai, Steve Noonan, Richard Stratton, Md Shakir Uddin Ahmed, Chandra Khantwal, George R. Martin, Huixian Lin, Chris Andrews, and et al. 2023. "A Novel CD206 Targeting Peptide Inhibits Bleomycin-Induced Pulmonary Fibrosis in Mice" Cells 12, no. 9: 1254. https://doi.org/10.3390/cells12091254
APA StyleGhebremedhin, A., Salam, A. B., Adu-Addai, B., Noonan, S., Stratton, R., Ahmed, M. S. U., Khantwal, C., Martin, G. R., Lin, H., Andrews, C., Karanam, B., Rudloff, U., Lopez, H., Jaynes, J., & Yates, C. (2023). A Novel CD206 Targeting Peptide Inhibits Bleomycin-Induced Pulmonary Fibrosis in Mice. Cells, 12(9), 1254. https://doi.org/10.3390/cells12091254