Are Proteinopathy and Oxidative Stress Two Sides of the Same Coin?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
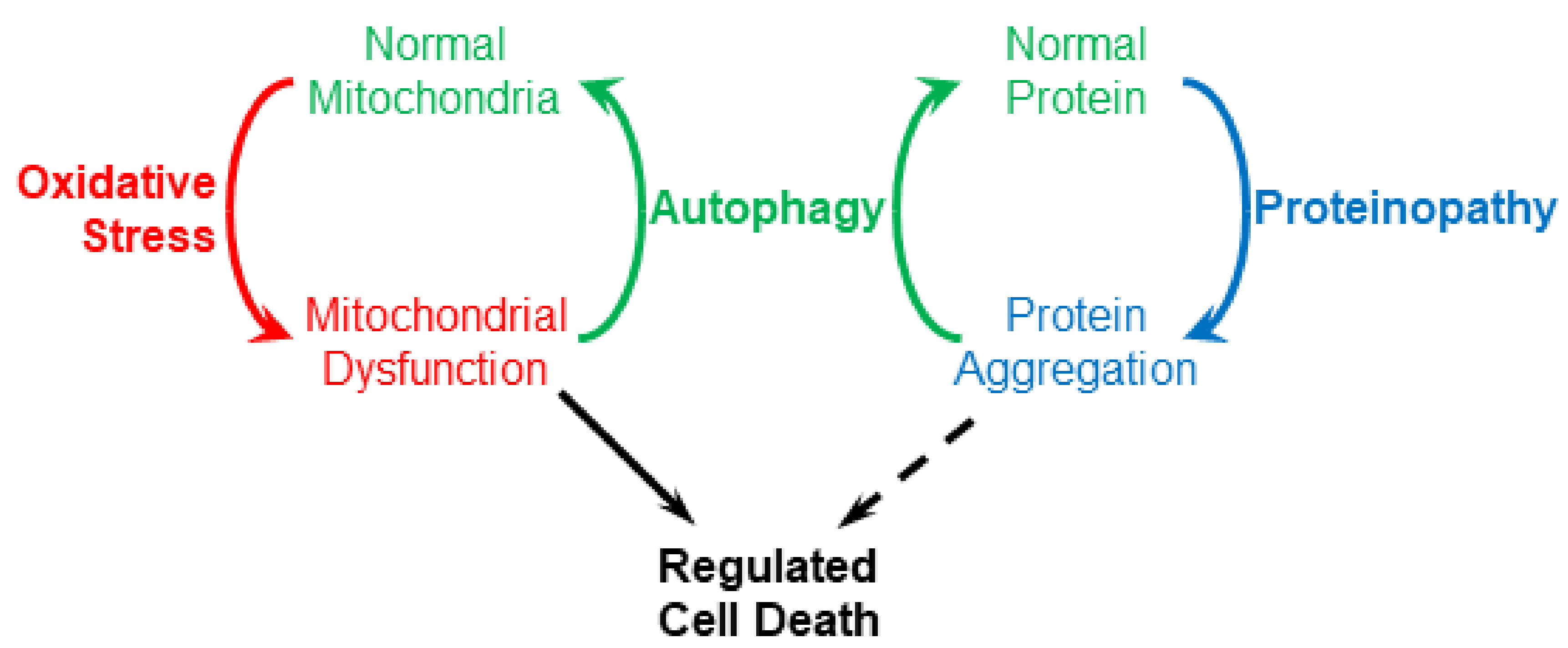
2. Proteinopathy and Autophagy
3. Mitochondrial Dysfunction and Mitophagy
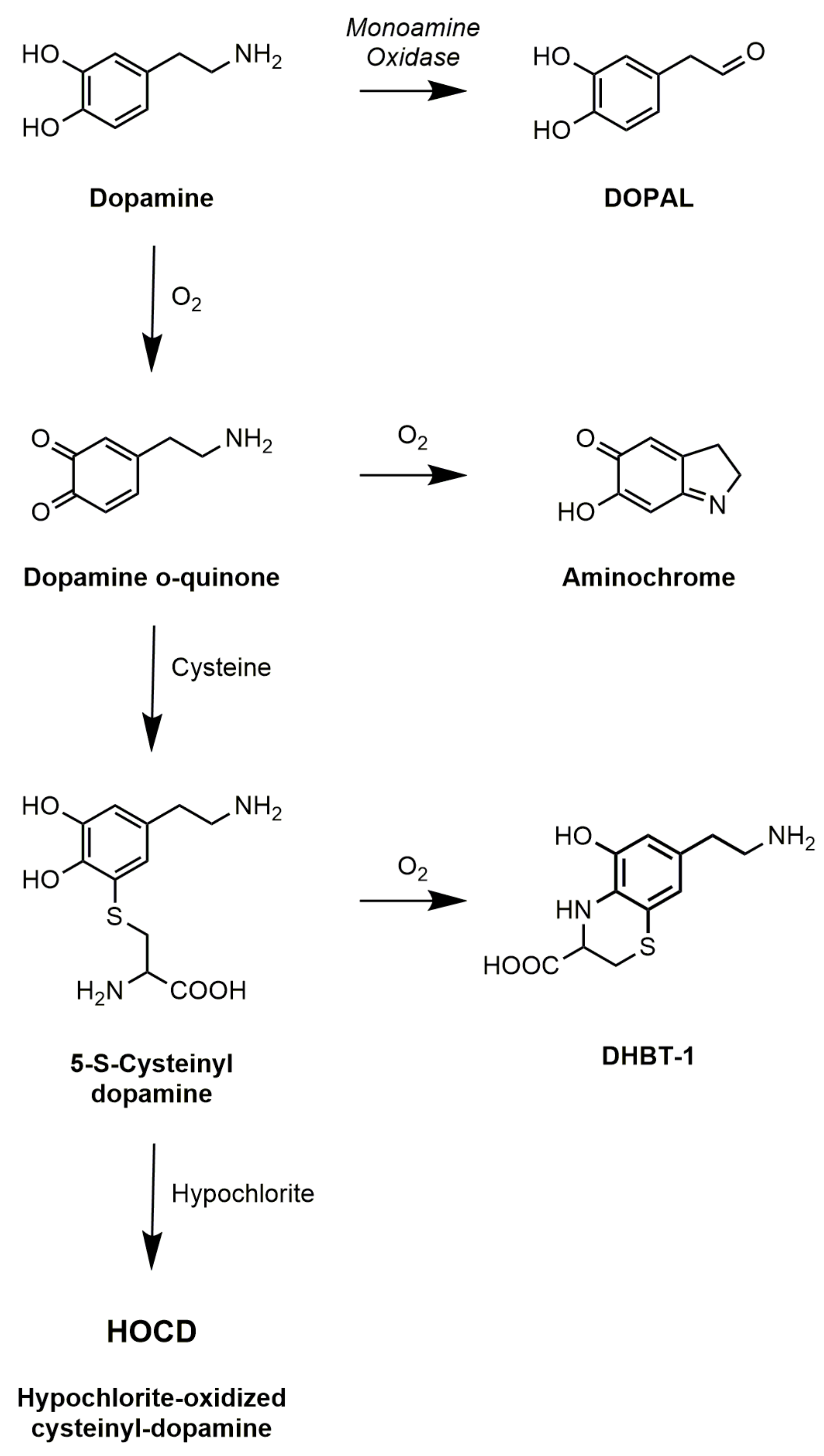
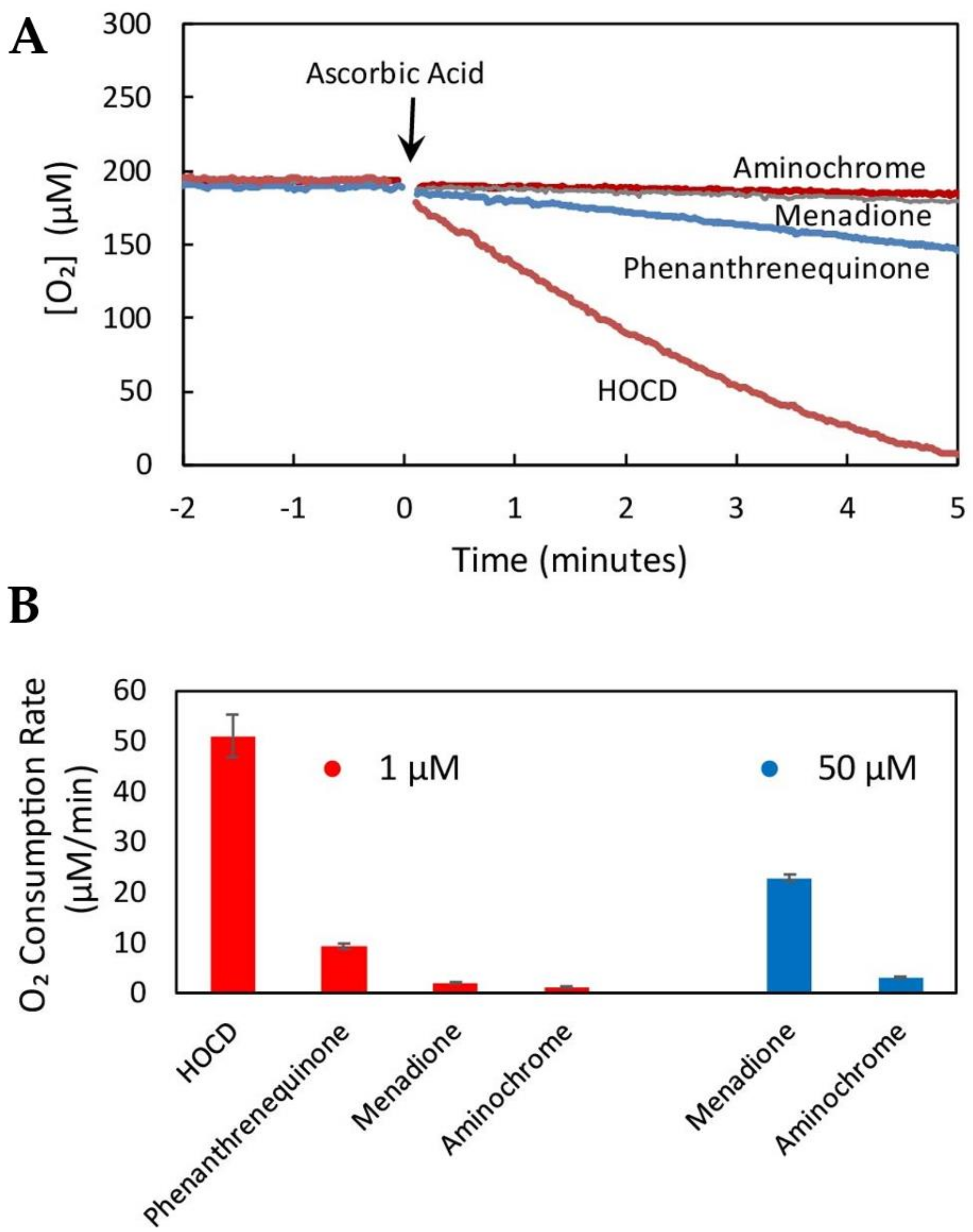
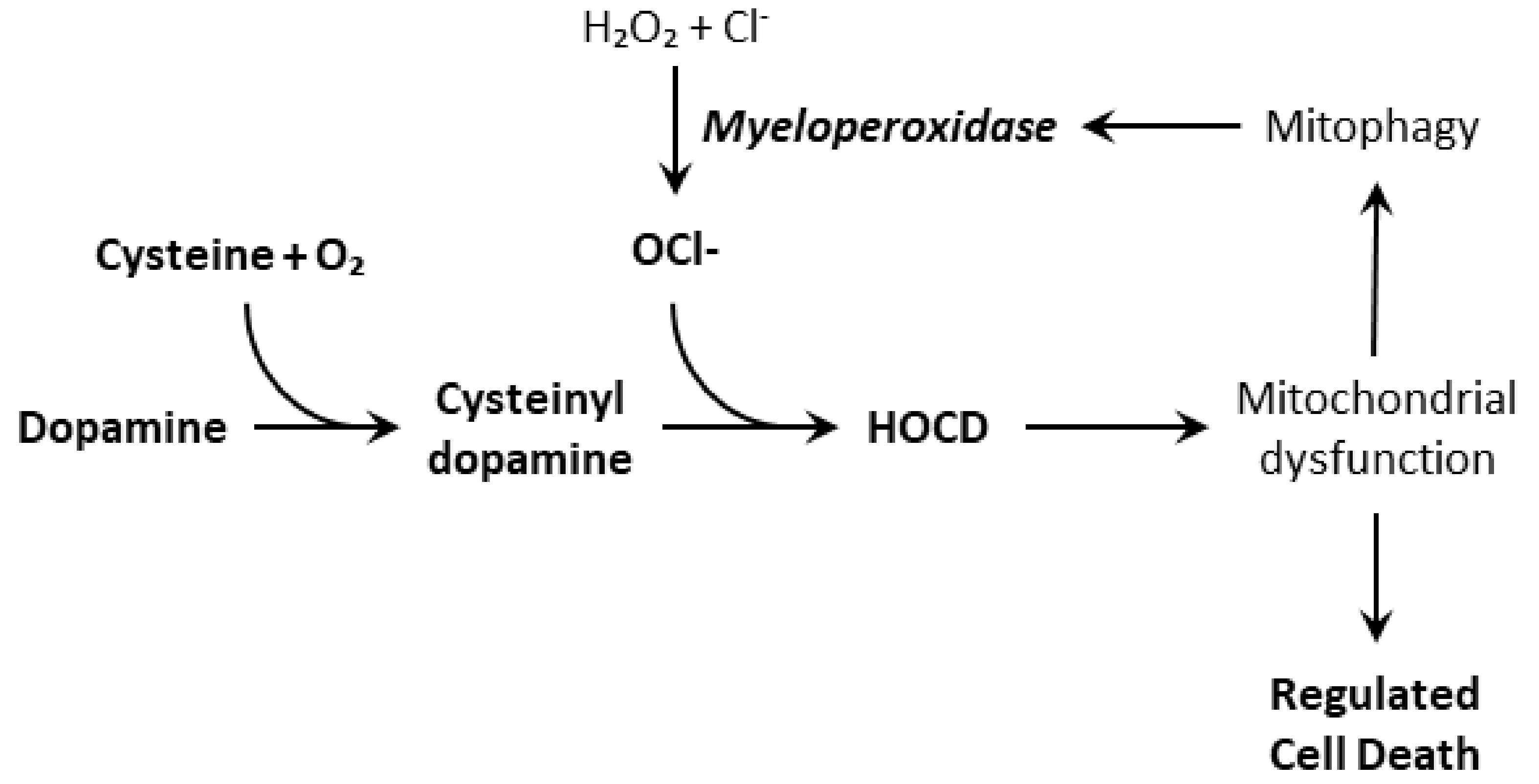
4. Dopamine Oxidation and HOCD
5. Conclusions
Conflicts of Interest
References
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.J.; Asmaro, K.; Hermiz, D.J.; Njus, M.M.; Saleh, A.H.; Beningo, K.A.; Njus, D. Hypochlorite converts cysteinyl-dopamine into a cytotoxic product: A possible factor in Parkinson’s Disease. Free Radic. Biol. Med. 2016, 101, 44–52. [Google Scholar] [CrossRef]
- Genova, M.L.; Abd-Elsalam, N.M.; Mahdy, E.S.M.E.; Bernacchia, A.; Lucarini, M.; Pedulli, G.F.; Lenaz, G. Redox cycling of adrenaline and adrenochrome catalyzed by mitochondrial Complex I. Arch. Biochem. Biophys. 2006, 447, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Mosca, L.; Tempera, I.; Lendaro, E.; Di Francesco, L.; d’Erme, M. Characterization of catechol-thioether-induced apoptosis in human SH-SY5Y neuroblastoma cells. J. Neurosci. Res. 2008, 86, 954–960. [Google Scholar] [CrossRef]
- Hauser, D.N.; Dukes, A.A.; Mortimer, A.D.; Hastings, T.G. Dopamine quinone modifies and decreases the abundance of the mitochondrial selenoprotein glutathione peroxidase 4. Free Radic. Biol. Med. 2013, 65, 419–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vauzour, D.; Pinto, J.T.; Cooper, A.J.L.; Spencer, J.P.E. The neurotoxicity of 5-S -cysteinyl-dopamine is mediated by the early activation of ERK1/2 followed by the subsequent activation of ASK1/JNK1/2 pro-apoptotic signaling. Biochem. J. 2014, 463, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Arduini, I.; Soriano, M.E.; Giorgio, V.; Bernardi, P.; Bisaglia, M.; Bubacco, L. Dopamine oxidation products as mitochondrial endotoxins, a potential molecular mechanism for preferential neurodegeneration in Parkinson’s Disease. ACS Chem. Neurosci. 2018, 9, 2849–2858. [Google Scholar] [CrossRef]
- Herrera, A.; Muñoz, P.; Steinbusch, H.W.M.; Segura-Aguilar, J. Are dopamine oxidation metabolites involved in the loss of dopaminergic neurons in the Nigrostriatal system in Parkinson’s Disease? ACS Chem. Neurosci. 2017, 8, 702–711. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease—Locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.E.; Oddo, S. Autophagic/lysosomal dysfunction in Alzheimer’s Disease. Alzheimers Res. Ther. 2013, 5, 53. [Google Scholar] [CrossRef]
- Martin, D.D.O.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Shin, J.H.; Lee, J.E.; Choi, E.J. Role of autophagy in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2015, 1852, 2517–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Abraham, N.; Gao, G.; Yang, Q. Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Transl Neurodegener. 2016, 5, 19. [Google Scholar] [CrossRef]
- Moors, T.E.; Hoozemans, J.J.M.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; van de Berg, W.D.J. Therapeutic potential of autophagy enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Al Mamun, A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s disease: From molecular mechanisms to therapeutic implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Pircs, K.; Petri, R.; Madsen, S.; Brattas, P.L.; Vuono, R.; Ottosson, D.R.; St-Amour, I.; Hersbach, B.A.; Matusiak-Bruckner, M.; Lundh, S.H.; et al. Huntingtin Aggregation Impairs Autophagy, Leading to Argonaute-2 Accumulation and Global MicroRNA Dysregulation. Cell Rep. 2018, 24, 1397–1406. [Google Scholar] [CrossRef]
- Nguyen, D.K.H.; Thombre, R.; Wang, J. Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 2018. [Google Scholar] [CrossRef]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Uchihara, T.; Fukuda, T.; Noda, S.; Kondo, H.; Saiki, S.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 2018, 8, 2813. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H.V. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson disease. Hum. Mol. Gen. 2016, 25, 3432–3445. [Google Scholar] [CrossRef]
- Manzoni, C. The LRRK2-macroautophagy axis and its relevance to Parkinson’s disease. Biochem. Soc. Trans. 2017, 45, 155–162. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, 557. [Google Scholar] [CrossRef] [PubMed]
- Martire, S.; Mosca, L.; d’Erme, M. PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 2015, 146, 53–64. [Google Scholar] [CrossRef]
- Mandir, A.S.; Przedborski, S.; Jackson-Lewis, V.; Wang, Z.Q.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Hoffman, B.E.; Guastella, D.B.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc. Natl. Acad. Sci. USA 1999, 96, 5774–5779. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Cho, H.M.; Choi, S.Y.; Suguira, Y.; Hayasaka, T.; Setou, M.; Koh, H.C.; Mi Hwang, E.; Park, J.Y.; Kang, S.J.; et al. (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Cell Death Dis. 2013, 4, e919. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.J. Understanding the mechanism of oxidative stress generation by dopamine oxidized metabolites: Implications in Parkinson’s disease. Ph.D. Thesis, Wayne State University, Detroit, MI, USA, 2017. [Google Scholar]
- Keane, P.C.; Kurzawa, M.; Blain, P.G.; Morris, C.M. Mitochondrial dysfunction in Parkinson’s disease. Parkinsons Dis. 2011, 2011, 716871. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef]
- Hroudová, J.; Singh, N.; Fišar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 9. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Ali, R.A.; Ibrahim, N.M.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta 2009, 1787, 384–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Gen. 2011, 20, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Majcher, V.; Goode, A.; James, V.; Layfield, R. Autophagy receptor defects and ALS-FTLD. Mol. Cell Neurosci. 2015, 66, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.Y.; Choi, D.K.; Lee, D.K.; Hong, Y.J.; Park, E.J. Resveratrol confers protection against rotenone-induced neurotoxicity by modulating myeloperoxidase levels in glial cells. PLoS ONE 2013, 8, e60654. [Google Scholar] [CrossRef]
- Jinsmaa, Y.; Sharabi, Y.; Sullivan, P.; Isonaka, R.; Goldstein, D.S. 3,4-Dihydroxyphenyl acetaldehyde-induced protein modifications and their mitigation by N-acetylcysteine. J. Pharmacol. Exp. Ther. 2018, 366, 113–124. [Google Scholar] [CrossRef]
- Fitzmaurice, A.G.; Rhodes, S.L.; Lulla, A.; Murphy, N.P.; Lam, H.A.; O’Donnell, K.C.; Barnhill, L.; Casida, J.E.; Cockburn, M.; Sagasti, A.; et al. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc. Natl. Acad. Sci. USA 2013, 110, 636–641. [Google Scholar] [CrossRef]
- Rosengren, E.; Linder-Eliasson, E.; Carlsson, A. Detection of 5-S-cysteinyldopamine in human brain. J. Neural Transm. 1985, 63, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.M.; Dryhurst, G. Further insights into the influence of L-cysteine on the oxidation chemistry of dopamine: Reaction pathways of potential relevance to Parkinson’s Disease. Chem. Res. Toxicol. 1996, 9, 751–763. [Google Scholar] [CrossRef]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Munoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine, and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef] [PubMed]
- Gellhaar, S.; Sunnemark, D.; Eriksson, H.; Olson, L.; Galter, D. Myeloperoxidase-immunoreactive cells are significantly increased in brain areas affected by neurodegeneration in Parkinson’s and Alzheimer’s disease. Cell Tissue Res. 2017, 369, 445–454. [Google Scholar] [CrossRef]
- Scudamore, O.; Ciossek, T. Increased oxidative stress exacerbates alpha-Synuclein aggregation in vivo. J. Neuropath. Exp. Neurol. 2018, 77, 443–453. [Google Scholar] [CrossRef]
- Xiang, W.; Schlachetzki, J.C.M.; Helling, S.; Bussmann, J.C.; Berlinghof, M.; Schäffer, T.E.; Marcus, K.; Winkler, J.; Klucken, J.; Becker, C.-M. Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 2013, 54, 71–83. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehta, N.J.; Marwah, P.K.; Njus, D. Are Proteinopathy and Oxidative Stress Two Sides of the Same Coin? Cells 2019, 8, 59. https://doi.org/10.3390/cells8010059
Mehta NJ, Marwah PK, Njus D. Are Proteinopathy and Oxidative Stress Two Sides of the Same Coin? Cells. 2019; 8(1):59. https://doi.org/10.3390/cells8010059
Chicago/Turabian StyleMehta, Nihar J., Praneet Kaur Marwah, and David Njus. 2019. "Are Proteinopathy and Oxidative Stress Two Sides of the Same Coin?" Cells 8, no. 1: 59. https://doi.org/10.3390/cells8010059
APA StyleMehta, N. J., Marwah, P. K., & Njus, D. (2019). Are Proteinopathy and Oxidative Stress Two Sides of the Same Coin? Cells, 8(1), 59. https://doi.org/10.3390/cells8010059