Peripheral Delivery of Neural Precursor Cells Ameliorates Parkinson’s Disease-Associated Pathology

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation of Neural Precursors for Cell Therapy
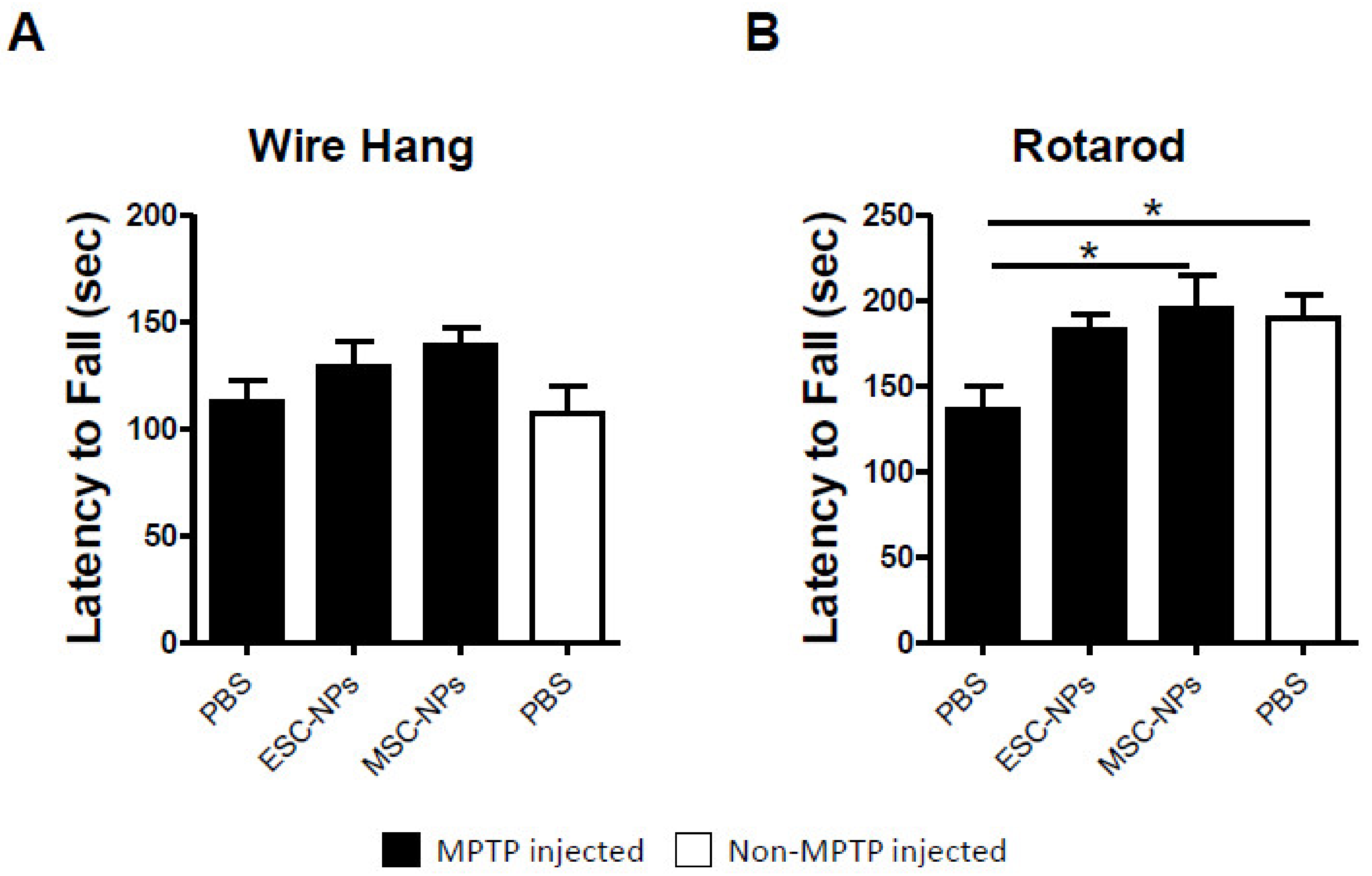
2.2. Short-Term Attenuation of PD-Induced Motor Deficits After Stem Cell Therapy
2.3. NP Therapy Does Not Increase Dopamine Levels but Modulates Neuroinflammation
2.4. Intravenous Treatment with NPs is Not Tumorigenic
3. Discussion
4. Materials and Methods
4.1. Preparation of Mouse NPs from ESC and MSCs
4.2. Immunocytochemistry
4.3. Induction of Parkinson’s Disease Phenotype
4.4. NP Treatment
4.5. Wire Hang
4.6. Rotarod
4.7. Immunohistochemistry
4.8. Quantitative Image Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease—The lancet. Lancet 2015, 86, 896–912. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 2003, 302, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H.V. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Cummings, J.L. Depression and parkinson’s disease: A review. Am. J. Psychiatry 1992, 149, 443–454. [Google Scholar] [PubMed]
- Fasano, A.; Visanji, N.P.; Liu, L.W.C.; Lang, A.E.; Pfeiffer, R.F. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol. 2015, 14, 625–639. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Liberatore, G.T.; Jackson-Lewis, V.; Vukosavic, S.; Mandir, A.S.; Vila, M.; Mcauliffe, W.G.; Dawson, V.L.; Dawson, T.M.; Przedborski, S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat. Med. 1999, 5, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Schwab, C.; Parent, A.; Doudet, D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann. Neurol. 2003, 54, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Ferreira, J. Current and future therapeutic strategies for Parkinson’s disease. Curr. Pharm. Des. 2009, 15, 3968–3976. [Google Scholar] [CrossRef]
- Bloem, B.R.; de Vries, N.M.; Ebersbach, G. Nonpharmacological treatments for patients with Parkinson’s disease. Mov. Disord. 2015, 30, 1504–1520. [Google Scholar] [CrossRef]
- Okun, M.S. Deep-brain stimulation for Parkinson’s disease the clinical problem. N. Engl. J. Med. 2012, 367, 1529–1538. [Google Scholar] [CrossRef]
- Olanow, C.W.; Kordower, J.H.; Lang, A.E.; Obeso, J.A. Dopaminergic transplantation for Parkinson’s disease: Current status and future prospects. Ann. Neurol. 2009, 66, 591–596. [Google Scholar] [CrossRef]
- Anisimov, S. V Cell-based therapeutic approaches for Parkinson’s disease: Progress and perspectives. Rev. Neurosci. 2009, 20, 347–382. [Google Scholar] [CrossRef]
- Correia, A.S.; Anisimov, S.V.; Li, J.Y.; Brundin, P. Stem cell-based therapy for Parkinson’s disease. Ann. Med. 2005, 37, 487–498. [Google Scholar] [CrossRef]
- Caplan, A.I.; Bruder, S.P. Mesenchymal stem cells: Building blocks for molecular medicine in the 21st century. Trends Mol. Med. 2001, 7, 259–264. [Google Scholar] [CrossRef]
- Yamanaka, K.T. Shinya induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar]
- Kim, S.U.; Lee, H.J.; Kim, Y.B. Neural stem cell-based treatment for neurodegenerative diseases. Neuropathology 2013, 33, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, J.; Wang, L.; Zhang, L.; Lu, M.; Chopp, M. Intracerebral transplantation of bone marrow stromal cells in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neurosci. Lett. 2001, 316, 67–70. [Google Scholar] [CrossRef]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef]
- Galindo, L.T.; Filippo, T.R.M.; Patricia, S.; Ariza, C.B.; Moreira, C.M.; Camara, N.O.S.; Porcionatto, M.A. Mesenchymal stem cell therapy modulates the inflammatory response in experimental traumatic brain injury. Neurol. Res. Int. 2011, 2011, 564089. [Google Scholar] [CrossRef]
- Drago, D.; Cossetti, C.; Iraci, N.; Gaude, E.; Musco, G.; Bachi, A.; Pluchino, S. The stem cell secretome and its role in brain repair. Biochimie 2013, 95, 2271–2285. [Google Scholar] [CrossRef] [Green Version]
- Joyce, N.; Annett, G.; Wirthlin, L.; Olson, S.; Bauer, G.; Nolta, A. Mesenchymal stem cells for the treatment of neurodegenerative disease. Regen. Med. 2010, 5, 933–946. [Google Scholar] [CrossRef] [Green Version]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef]
- Li, J.-Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Misra, V.; Lal, A.; El Khoury, R.; Chen, P.R.; Savitz, S.I. Intra-arterial delivery of cell therapies for stroke. Stem Cells Dev. 2012, 21, 1007–1015. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Tetzlaff, W.; Weiss, S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 1992, 12, 4565–4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, J.; Peterson, D.A.; Schinstine, M.; Gage, F.H. Proliferation, differentiation, and long-term culture of primary hippocampal neurons. Proc. Natl. Acad. Sci. USA 2006, 90, 3602–3606. [Google Scholar] [CrossRef] [PubMed]
- Balenci, L.; Inoue, T.; Karpowicz, P.; Willaime-Morawek, S.; DeVeale, B.; van der Kooy, D. E-Cadherin regulates neural stem cell self-renewal. J. Neurosci. 2009, 29, 3885–3896. [Google Scholar]
- Götz, M.; Sirko, S.; Beckers, J.; Irmler, M. Reactive astrocytes as neural stem or progenitor cells: In vivo lineage, In vitro potential, and Genome-wide expression analysis. Glia 2015, 63, 1452–1468. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.-K.; Ji, K.; Min, K.; Joe, E.-H. Brain inflammation and microglia: Facts and misconceptions. Exp. Neurobiol. 2013, 22, 59–67. [Google Scholar] [CrossRef]
- Lotia, M.; Jankovic, J. New and emerging medical therapies in Parkinson’s disease. Expert Opin. Pharmacother. 2016, 17, 895–909. [Google Scholar] [CrossRef]
- Georgiev, D.; Hamberg, K.; Hariz, M.; Forsgren, L.; Hariz, G.M. Gender differences in Parkinson’s disease: A clinical perspective. Acta Neurol. Scand. 2017, 136, 570–584. [Google Scholar] [CrossRef]
- Chao, Y.X.; He, B.P.; Tay, S.S.W. Mesenchymal stem cell transplantation attenuates blood brain barrier damage and neuroinflammation and protects dopaminergic neurons against MPTP toxicity in the substantia nigra in a model of Parkinson’s disease. J. Neuroimmunol. 2009, 216, 39–50. [Google Scholar] [CrossRef]
- Kesslak, J.P. Transplantation of embryonic dopamine neurons for severe parkinson’s disease. N Engl J Med 2001, 344, 710–719. [Google Scholar] [CrossRef]
- Chen, D.; Fu, W.; Zhuang, W.; Lv, C.; Li, F.; Wang, X. Therapeutic effects of intranigral transplantation of mesenchymal stem cells in rat models of Parkinson’s disease. J. Neurosci. Res. 2017, 95, 907–917. [Google Scholar] [CrossRef]
- Zawada, W.M.; Banninger, G.P.; Thornton, J.; Marriott, B.; Cantu, D.; Rachubinski, A.L.; Das, M.; Griffin, W.S.T.; Jones, S.M. Generation of reactive oxygen species in 1-methyl-4-phenylpyridinium (MPP+) treated dopaminergic neurons occurs as an NADPH oxidase-dependent two-wave cascade. J. Neuroinflammation 2011, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, J.; Sheng, R.; Li, M.; Wang, Y.; Han, R.; Han, F.; Chen, Z.; Qin, Z.H. Reduced Nicotinamide Adenine Dinucleotide Phosphate inhibits MPTP-induced neuroinflammation and neurotoxicity. Neuroscience 2018, 391, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Emborg, M.E.; Bloch, J.; Ma, S.Y.; Chu, Y.; Leventhal, L.; McBride, J.; Chen, E.Y.; Palfi, S.; Roitberg, B.Z.; et al. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 2000, 290, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.K.; Bunnage, M.; Plaha, P.; Svendsen, C.N.; Heywood, P.; Gill, S.S. Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: A two-year outcome study. Ann. Neurol. 2005, 57, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.; Camnasio, S.; Binetti, M.; Jung, C.B.; Moretti, A.; Cattaneo, E. Neuropotent self-renewing neural stem (NS) cells derived from mouse induced pluripotent stem (iPS) cells. Mol. Cell. Neurosci. 2010, 43, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Hirose, Y.; Ohara, S.; Ono, T.; Watanabe, Y. A simple quantitative bradykinesia test in MPTP-treated mice. Res. Commun. Chem. Pathol. Pharmacol. 1985, 50, 435–441. [Google Scholar] [PubMed]
- van Putten, M.; Aartsma-Rus, A.; Louvain-la-Neuve, L. The use of hanging wire tests to monitor muscle strength and condition over time. Treat. NMD Neuromuscul. Netw. 2012, 4, 1–12. [Google Scholar]
- Colotla, V.A.; Flores, E.; Oscos, A.; Meneses, A.; Tapia, R. Effects of MPTP on locomotor activity in mice. Neurotoxicol. Teratol. 1990, 12, 405–407. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, I.; Baglietto-Vargas, D.; Sanchez-Varo, R.; Jimenez, S.; Trujillo-Estrada, L.; Sanchez-Mejias, E.; Del Rio, J.C.; Torres, M.; Romero-Acebal, M.; Ruano, D.; et al. Extracellular amyloid-β and cytotoxic glial activation induce significant entorhinal neuron loss in young PS1M146L/APP751SL mice. J. Alzheimer’s Dis. 2009, 18, 755–776. [Google Scholar] [CrossRef]
- Jimenez, S.; Baglietto-Vargas, D.; Caballero, C.; Moreno-Gonzalez, I.; Torres, M.; Sanchez-Varo, R.; Ruano, D.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Inflammatory response in the Hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: Age-dependent switch in the microglial phenotype from alternative to classic. J. Neurosci. 2008, 28, 11650–11661. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edwards, G., III; Gamez, N.; Armijo, E.; Kramm, C.; Morales, R.; Taylor-Presse, K.; Schulz, P.E.; Soto, C.; Moreno-Gonzalez, I. Peripheral Delivery of Neural Precursor Cells Ameliorates Parkinson’s Disease-Associated Pathology. Cells 2019, 8, 1359. https://doi.org/10.3390/cells8111359
Edwards G III, Gamez N, Armijo E, Kramm C, Morales R, Taylor-Presse K, Schulz PE, Soto C, Moreno-Gonzalez I. Peripheral Delivery of Neural Precursor Cells Ameliorates Parkinson’s Disease-Associated Pathology. Cells. 2019; 8(11):1359. https://doi.org/10.3390/cells8111359
Chicago/Turabian StyleEdwards, George, III, Nazaret Gamez, Enrique Armijo, Carlos Kramm, Rodrigo Morales, Kathleen Taylor-Presse, Paul E. Schulz, Claudio Soto, and Ines Moreno-Gonzalez. 2019. "Peripheral Delivery of Neural Precursor Cells Ameliorates Parkinson’s Disease-Associated Pathology" Cells 8, no. 11: 1359. https://doi.org/10.3390/cells8111359
APA StyleEdwards, G., III, Gamez, N., Armijo, E., Kramm, C., Morales, R., Taylor-Presse, K., Schulz, P. E., Soto, C., & Moreno-Gonzalez, I. (2019). Peripheral Delivery of Neural Precursor Cells Ameliorates Parkinson’s Disease-Associated Pathology. Cells, 8(11), 1359. https://doi.org/10.3390/cells8111359