Targeting Hedgehog Signalling through the Ubiquitylation Process: The Multiple Roles of the HECT-E3 Ligase Itch

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
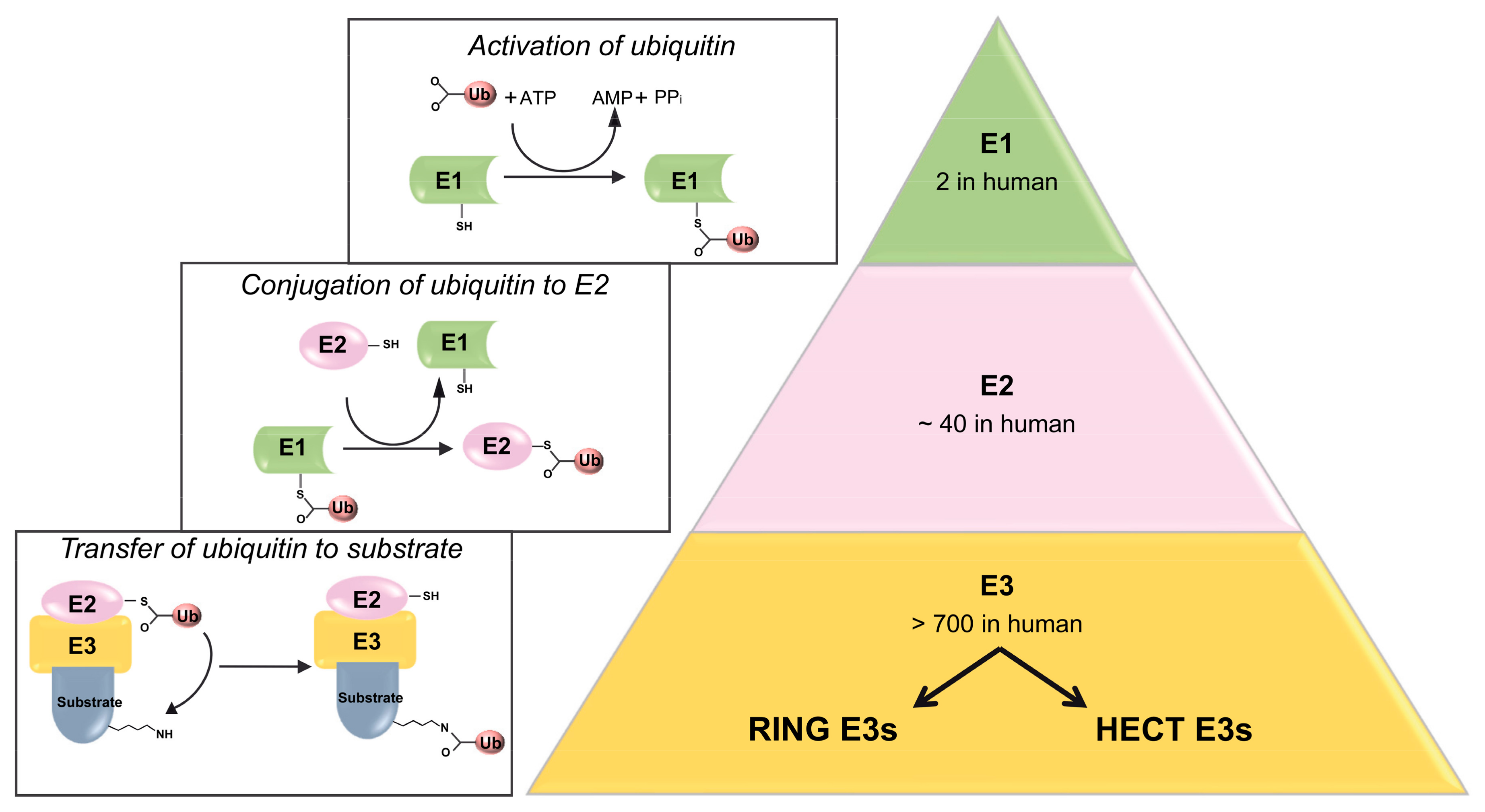
2. Ubiquitylation Processes: Not Only a Degradative Pathway
3. The HECT E3 Ubiquitin Ligase Itch: An Overview
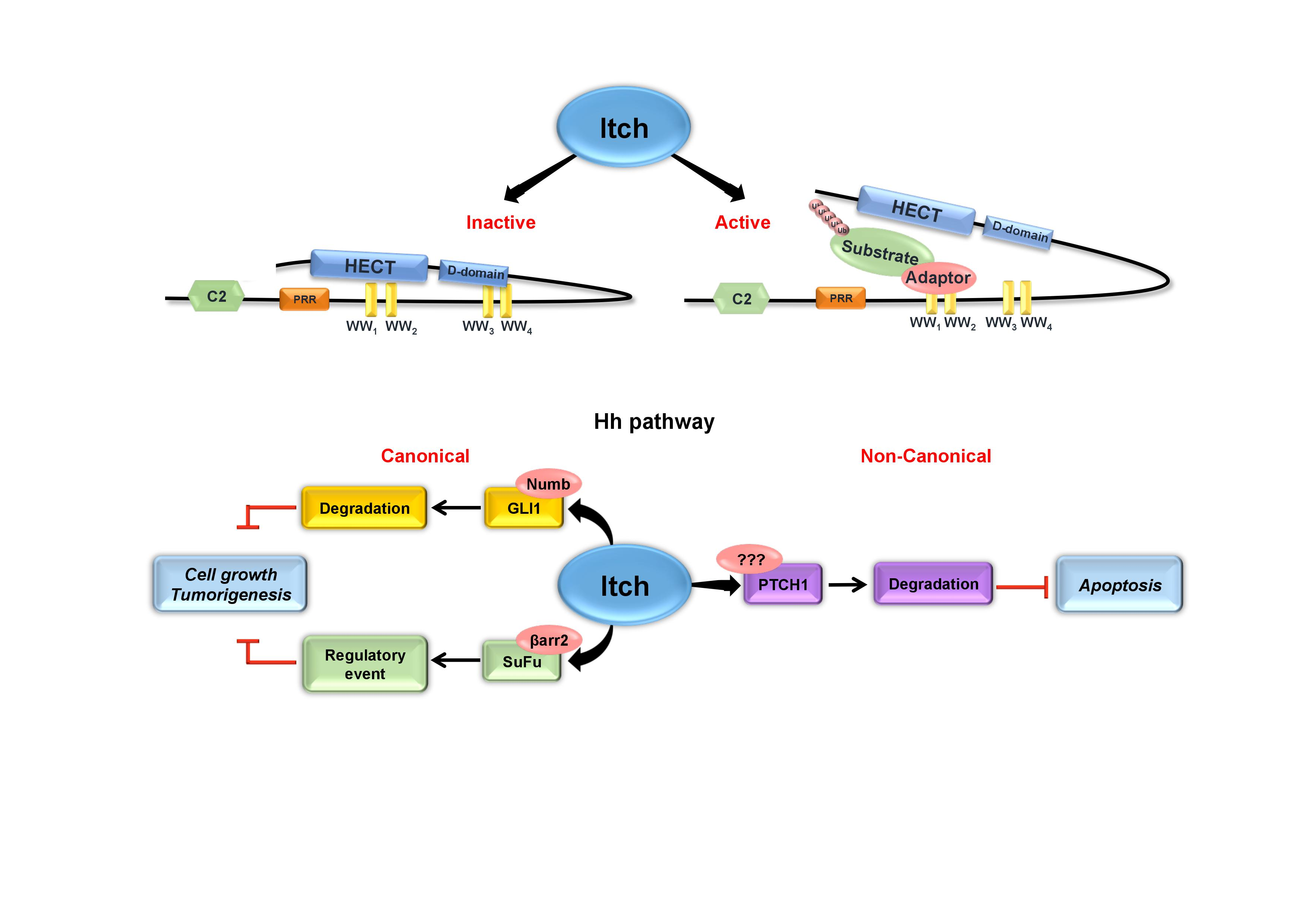
3.1. Structure of Itch
3.2. Regulation of Itch
4. Itch and Its Role in Canonical Hh Pathway
4.1. Itch/Numb/GLI1 Axis
4.2. Itch/β-Arrestin2/SuFu Axis
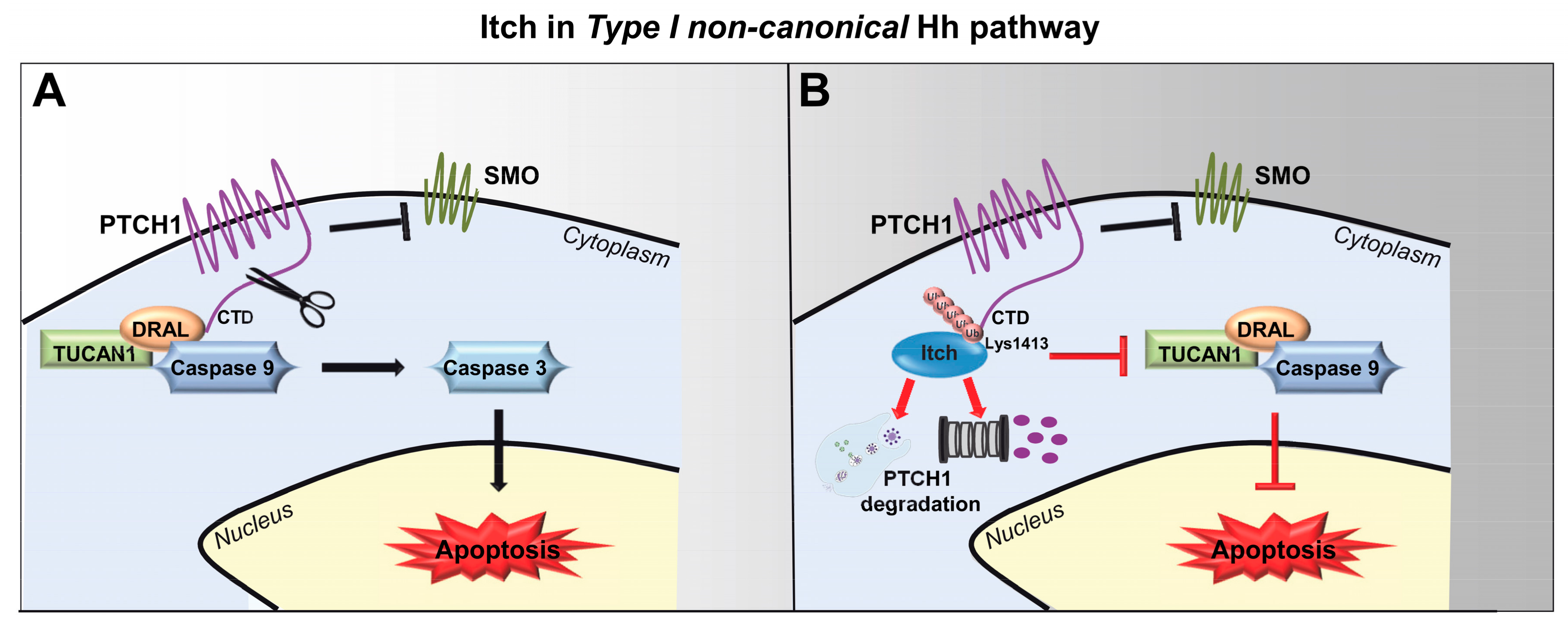
5. Itch and Its Role in Type I Non-Canonical Hh Pathway
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Hh | Hedgehog |
| CNS | Central Nervous System |
| GCPs | Granule Cells Progenitors |
| MB | Medulloblastoma |
| PTCH1 | Patched1 |
| SMO | Smoothened |
| SuFu | Suppressor of Fused |
| Ub | Ubiquitin |
| E3s | E3 ubiquitin ligases |
| RING | Really Interesting New Gene |
| HECT | Homologous to the E6-associated protein Carboxyl-Terminus |
| AIP4 or Itch | Atrophin-1 interacting protein 4 |
| NEDD4 | Neural precursor cell-expressed developmentally downregulated protein 4 |
| NSCLCs | Non Small Cell Lung Carcinomas |
| EGL | External Granule Layer |
| IGL | Internal Granule Layer |
| PTB | Phospho-tyrosine-binding domain |
| PRR | Proline Rich Region |
| pSP | Proline residues preceded by phosphoserine |
| GPCRs | G protein-coupled receptors |
| GRKs | GPCR kinases |
| Kif3A | Kinesin-2 motor complex |
| GLI3R | GLI3 repressor form |
| Hh-MBs | Hedgehog-group medulloblastoma |
| CTD | C-terminal domain |
| TCR | T-cell receptor |
| NDFIPs | NEDD4 Family Interacting Proteins |
References
- Ruiz i Altaba, A.; Mas, C.; Stecca, B. The Gli code: An information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007, 17, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Ruiz i Altaba, A.; Sánchez, P.; Dahmane, N. Gli and hedgehog in cancer: Tumours, embryos and stem cells. Nat. Rev. Cancer 2002, 2, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Wechsler-Reya, R.J.; Scott, M.P. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999, 22, 103–114. [Google Scholar] [CrossRef]
- Schüller, U.; Heine, V.M.; Mao, J.; Kho, A.T.; Dillon, A.K.; Han, Y.G.; Huillard, E.; Sun, T.; Ligon, A.H.; Qian, Y.; et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 2008, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Ellis, T.; Markant, S.L.; Read, T.A.; Kessler, J.D.; Bourboulas, M.; Schüller, U.; Machold, R.; Fishell, G.; Rowitch, D.H.; et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 2008, 14, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.S.; Scott, M.P. Sonic hedgehog in the nervous system: Functions, modifications and mechanisms. Curr. Opin. Neurobiol. 2002, 12, 57–63. [Google Scholar] [CrossRef]
- Kieran, M.W. Targeted treatment for sonic hedgehog-dependent medulloblastoma. Neuro Oncol. 2014, 16, 1037–1047. [Google Scholar] [CrossRef] [Green Version]
- De Smaele, E.; Di Marcotullio, L.; Ferretti, E.; Screpanti, I.; Alesse, E.; Gulino, A. Chromosome 17p deletion in human medulloblastoma: A missing checkpoint in the Hedgehog pathway. Cell Cycle 2004, 3, 1263–1266. [Google Scholar] [CrossRef]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Di Marcotullio, L.; Ferretti, E.; Greco, A.; De Smaele, E.; Screpanti, I.; Gulino, A. Multiple ubiquitin-dependent processing pathways regulate hedgehog/gli signaling: Implications for cell development and tumorigenesis. Cell Cycle 2007, 6, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Di Marcotullio, L.; Ferretti, E.; De Smaele, E.; Screpanti, I.; Gulino, A. Suppressors of hedgehog signaling: Linking aberrant development of neural progenitors and tumorigenesis. Mol. Neurobiol. 2006, 34, 193–204. [Google Scholar] [CrossRef]
- Bhatia, N.; Thiyagarajan, S.; Elcheva, I.; Saleem, M.; Dlugosz, A.; Mukhtar, H.; Spiegelman, V.S. Gli2 is targeted for ubiquitination and degradation by beta-TrCP ubiquitin ligase. J. Biol. Chem. 2006, 281, 19320–19326. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Huntzicker, E.G.; Estay, I.S.; Zhen, H.; Lokteva, L.A.; Jackson, P.K.; Oro, A.E. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006, 20, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef]
- Welchman, R.L.; Gordon, C.; Mayer, R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 2005, 6, 599–609. [Google Scholar] [CrossRef]
- Gulino, A.; Di Marcotullio, L.; Canettieri, G.; De Smaele, E.; Screpanti, I. Hedgehog/Gli control by ubiquitination/acetylation interplay. Vitam. Horm. 2012, 88, 211–227. [Google Scholar] [CrossRef]
- Po, A.; Silvano, M.; Miele, E.; Capalbo, C.; Eramo, A.; Salvati, V.; Todaro, M.; Besharat, Z.M.; Catanzaro, G.; Cucchi, D.; et al. Noncanonical GLI1 signaling promotes stemness features and in vivo growth in lung adenocarcinoma. Oncogene 2017, 36, 4641–4652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Pan, Y.; Wang, B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 2010, 137, 2001–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.H.; Wilson, C.W.; Li, Y.J.; Law, K.K.; Lu, C.S.; Gacayan, R.; Zhang, X.; Hui, C.C.; Chuang, P.T. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef]
- Di Marcotullio, L.; Ferretti, E.; Greco, A.; De Smaele, E.; Po, A.; Sico, M.A.; Alimandi, M.; Giannini, G.; Maroder, M.; Screpanti, I.; et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat. Cell Biol. 2006, 8, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Di Marcotullio, L.; Greco, A.; Mazzà, D.; Canettieri, G.; Pietrosanti, L.; Infante, P.; Coni, S.; Moretti, M.; De Smaele, E.; Ferretti, E.; et al. Numb activates the E3 ligase Itch to control Gli1 function through a novel degradation signal. Oncogene 2011, 30, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Mazzà, D.; Infante, P.; Colicchia, V.; Greco, A.; Alfonsi, R.; Siler, M.; Antonucci, L.; Po, A.; De Smaele, E.; Ferretti, E.; et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013, 20, 1688–1697. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A.; Orian, A.; Schwartz, A.L. Ubiquitin-mediated proteolysis: Biological regulation via destruction. Bioessays 2000, 22, 442–451. [Google Scholar] [CrossRef]
- Hsia, E.Y.; Gui, Y.; Zheng, X. Regulation of Hedgehog signaling by ubiquitination. Front. Biol. 2015, 10, 203–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Lee, T.L.; Shyu, Y.C.; Hsu, T.Y.; Shen, C.K. Itch regulates p45/NF-E2 in vivo by Lys63-linked ubiquitination. Biochem. Biophys. Res. Commun. 2008, 375, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Scialpi, F.; Malatesta, M.; Peschiaroli, A.; Rossi, M.; Melino, G.; Bernassola, F. Itch self-polyubiquitylation occurs through lysine-63 linkages. Biochem. Pharmacol. 2008, 76, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Faedda, R.; Bernardi, F.; Bufalieri, F.; Lospinoso Severini, L.; Alfonsi, R.; Mazzà, D.; Siler, M.; Coni, S.; Po, A.; et al. Itch/β-arrestin2-dependent non-proteolytic ubiquitylation of SuFu controls Hedgehog signalling and medulloblastoma tumorigenesis. Nat. Commun. 2018, 9, 976. [Google Scholar] [CrossRef] [PubMed]
- Perry, W.L.; Hustad, C.M.; Swing, D.A.; O’Sullivan, T.N.; Jenkins, N.A.; Copeland, N.G. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat. Genet. 1998, 18, 143–146. [Google Scholar] [CrossRef]
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. The HECT family of E3 ubiquitin ligases: Multiple players in cancer development. Cancer Cell 2008, 14, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Dunn, R.; Klos, D.A.; Adler, A.S.; Hicke, L. The C2 domain of the Rsp5 ubiquitin ligase binds membrane phosphoinositides and directs ubiquitination of endosomal cargo. J. Cell Biol. 2004, 165, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, S.; Cantor, A.J.; Rape, M.; Kuriyan, J. Macromolecular juggling by ubiquitylation enzymes. BMC Biol. 2013, 11, 65. [Google Scholar] [CrossRef]
- Raimondo, D.; Giorgetti, A.; Bernassola, F.; Melino, G.; Tramontano, A. Modelling and molecular dynamics of the interaction between the E3 ubiquitin ligase Itch and the E2 UbcH7. Biochem. Pharmacol. 2008, 76, 1620–1627. [Google Scholar] [CrossRef]
- Zhu, K.; Shan, Z.; Chen, X.; Cai, Y.; Cui, L.; Yao, W.; Wang, Z.; Shi, P.; Tian, C.; Lou, J.; et al. Allosteric auto-inhibition and activation of the Nedd4 family E3 ligase Itch. EMBO Rep. 2017, 18, 1618–1630. [Google Scholar] [CrossRef]
- Fang, D.; Elly, C.; Gao, B.; Fang, N.; Altman, Y.; Joazeiro, C.; Hunter, T.; Copeland, N.; Jenkins, N.; Liu, Y.C. Dysregulation of T lymphocyte function in itchy mice: A role for Itch in TH2 differentiation. Nat. Immunol. 2002, 3, 281–287. [Google Scholar] [CrossRef]
- Venuprasad, K.; Zeng, M.; Baughan, S.L.; Massoumi, R. Multifaceted role of the ubiquitin ligase Itch in immune regulation. Immunol. Cell Biol. 2015, 93, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Labuda, T.; Xia, Y.; Gallagher, E.; Fang, D.; Liu, Y.C.; Karin, M. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 2004, 306, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Wegierski, T.; Hill, K.; Schaefer, M.; Walz, G. The HECT ubiquitin ligase AIP4 regulates the cell surface expression of select TRP channels. EMBO J. 2006, 25, 5659–5669. [Google Scholar] [CrossRef] [Green Version]
- Courbard, J.R.; Fiore, F.; Adélaïde, J.; Borg, J.P.; Birnbaum, D.; Ollendorff, V. Interaction between two ubiquitin-protein isopeptide ligases of different classes, CBLC and AIP4/ITCH. J. Biol. Chem. 2002, 277, 45267–45275. [Google Scholar] [CrossRef] [PubMed]
- Angers, A.; Ramjaun, A.R.; McPherson, P.S. The HECT domain ligase itch ubiquitinates endophilin and localizes to the trans-Golgi network and endosomal system. J. Biol. Chem. 2004, 279, 11471–11479. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Yang, C.; Hu, K.; Elly, C.; Liu, Y.C. Itch E3 ligase-mediated regulation of TGF-beta signaling by modulating smad2 phosphorylation. Mol. Cell 2004, 15, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Stagni, V.; Giambruno, R.; Fianco, G.; Di Benedetto, A.; Mottolese, M.; Pellegrini, M.; Barilà, D. ATM kinase activity modulates ITCH E3-ubiquitin ligase activity. Oncogene 2014, 33, 1113–1123. [Google Scholar] [CrossRef]
- Gallagher, E.; Gao, M.; Liu, Y.C.; Karin, M. Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc. Natl. Acad. Sci. USA 2006, 103, 1717–1722. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Zhou, W.; Jeon, M.S.; Demydenko, D.; Harada, Y.; Zhou, H.; Liu, Y.C. Negative regulation of the E3 ubiquitin ligase itch via Fyn-mediated tyrosine phosphorylation. Mol. Cell 2006, 21, 135–141. [Google Scholar] [CrossRef]
- Mouchantaf, R.; Azakir, B.A.; McPherson, P.S.; Millard, S.M.; Wood, S.A.; Angers, A. The ubiquitin ligase itch is auto-ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X. J. Biol. Chem. 2006, 281, 38738–38747. [Google Scholar] [CrossRef] [PubMed]
- Shearwin-Whyatt, L.; Dalton, H.E.; Foot, N.; Kumar, S. Regulation of functional diversity within the Nedd4 family by accessory and adaptor proteins. Bioessays 2006, 28, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Gallagher, E.; Aqeilan, R.I.; Knight, R.; Peschiaroli, A.; Rossi, M.; Scialpi, F.; Malatesta, M.; Zocchi, L.; Browne, G.; et al. Itch: A HECT-type E3 ligase regulating immunity, skin and cancer. Cell Death Differ. 2008, 15, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Mund, T.; Pelham, H.R. Control of the activity of WW-HECT domain E3 ubiquitin ligases by NDFIP proteins. EMBO Rep. 2009, 10, 501–507. [Google Scholar] [CrossRef]
- Shukla, A.K.; Kim, J.; Ahn, S.; Xiao, K.; Shenoy, S.K.; Liedtke, W.; Lefkowitz, R.J. Arresting a transient receptor potential (TRP) channel: Beta-arrestin 1 mediates ubiquitination and functional down-regulation of TRPV4. J. Biol. Chem. 2010, 285, 30115–30125. [Google Scholar] [CrossRef] [PubMed]
- Dho, S.E.; French, M.B.; Woods, S.A.; McGlade, C.J. Characterization of four mammalian numb protein isoforms. Identification of cytoplasmic and membrane-associated variants of the phosphotyrosine binding domain. J. Biol. Chem. 1999, 274, 33097–33104. [Google Scholar] [CrossRef] [PubMed]
- Verdi, J.M.; Bashirullah, A.; Goldhawk, D.E.; Kubu, C.J.; Jamali, M.; Meakin, S.O.; Lipshitz, H.D. Distinct human NUMB isoforms regulate differentiation vs. proliferation in the neuronal lineage. Proc. Natl. Acad. Sci. USA 1999, 96, 10472–10476. [Google Scholar] [CrossRef] [Green Version]
- Gulino, A.; Di Marcotullio, L.; Screpanti, I. The multiple functions of Numb. Exp. Cell Res. 2010, 316, 900–906. [Google Scholar] [CrossRef]
- Pece, S.; Confalonieri, S.; Romano, P.R.; Di Fiore, P.P. NUMB-ing down cancer by more than just a NOTCH. Biochim. Biophys. Acta 2011, 1815, 26–43. [Google Scholar] [CrossRef]
- Colaluca, I.N.; Basile, A.; Freiburger, L.; D’Uva, V.; Disalvatore, D.; Vecchi, M.; Confalonieri, S.; Tosoni, D.; Cecatiello, V.; Malabarba, M.G.; et al. A Numb-Mdm2 fuzzy complex reveals an isoform-specific involvement of Numb in breast cancer. J. Cell Biol. 2018, 217, 745–762. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, K.; Cheng, C.; Ji, Z.; Wang, X.; Wang, M.; Chu, M.; Tang, D.G.; Zhu, H.H.; Gao, W.Q. Numb−/low Enriches a Castration-Resistant Prostate Cancer Cell Subpopulation Associated with Enhanced Notch and Hedgehog Signaling. Clin. Cancer Res. 2017, 23, 6744–6756. [Google Scholar] [CrossRef] [PubMed]
- Tosoni, D.; Pambianco, S.; Ekalle Soppo, B.; Zecchini, S.; Bertalot, G.; Pruneri, G.; Viale, G.; Di Fiore, P.P.; Pece, S. Pre-clinical validation of a selective anti-cancer stem cell therapy for Numb-deficient human breast cancers. EMBO Mol. Med. 2017, 9, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Feder, J.N.; Jiang, M.M.; Jan, L.Y.; Jan, Y.N. Asymmetric localization of a mammalian numb homolog during mouse cortical neurogenesis. Neuron 1996, 17, 43–53. [Google Scholar] [CrossRef]
- Zhong, W.; Jiang, M.M.; Weinmaster, G.; Jan, L.Y.; Jan, Y.N. Differential expression of mammalian Numb, Numblike and Notch1 suggests distinct roles during mouse cortical neurogenesis. Development 1997, 124, 1887–1897. [Google Scholar] [PubMed]
- Zhong, W.; Jiang, M.M.; Schonemann, M.D.; Meneses, J.J.; Pedersen, R.A.; Jan, L.Y.; Jan, Y.N. Mouse numb is an essential gene involved in cortical neurogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 6844–6849. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Zhong, W.; Jan, Y.N.; Temple, S. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development 2002, 129, 4843–4853. [Google Scholar] [PubMed]
- Klein, A.L.; Zilian, O.; Suter, U.; Taylor, V. Murine numb regulates granule cell maturation in the cerebellum. Dev. Biol. 2004, 266, 161–177. [Google Scholar] [CrossRef] [Green Version]
- McGill, M.A.; McGlade, C.J. Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J. Biol. Chem. 2003, 278, 23196–23203. [Google Scholar] [CrossRef] [PubMed]
- Ingham, R.J.; Gish, G.; Pawson, T. The Nedd4 family of E3 ubiquitin ligases: Functional diversity within a common modular architecture. Oncogene 2004, 23, 1972–1984. [Google Scholar] [CrossRef]
- Sudol, M. Structure and function of the WW domain. Prog. Biophys. Mol. Biol. 1996, 65, 113–132. [Google Scholar] [CrossRef]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G protein-coupled receptor kinases. Annu. Rev. Biochem. 1998, 67, 653–692. [Google Scholar] [CrossRef] [PubMed]
- Kohout, T.A.; Lefkowitz, R.J. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol. Pharmacol. 2003, 63, 9–18. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Gurevich, V.V. Arrestins: Ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006, 7, 236. [Google Scholar] [CrossRef]
- Chen, W.; Ren, X.R.; Nelson, C.D.; Barak, L.S.; Chen, J.K.; Beachy, P.A.; de Sauvage, F.; Lefkowitz, R.J. Activity-dependent internalization of smoothened mediated by beta-arrestin 2 and GRK2. Science 2004, 306, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Whalen, E.J.; Liu, R.; Xiao, K.; Kim, J.; Chen, M.; Wang, J.; Chen, W.; Lefkowitz, R.J. Beta-arrestin-mediated localization of smoothened to the primary cilium. Science 2008, 320, 1777–1781. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Barak, L.S.; Xiao, K.; Ahn, S.; Berthouze, M.; Shukla, A.K.; Luttrell, L.M.; Lefkowitz, R.J. Ubiquitination of beta-arrestin links seven-transmembrane receptor endocytosis and ERK activation. J. Biol. Chem. 2007, 282, 29549–29562. [Google Scholar] [CrossRef] [PubMed]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Jones, D.T.; Jäger, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D. Hedgehog signalling: Emerging evidence for non-canonical pathways. Cell Signal 2009, 21, 1023–1034. [Google Scholar] [CrossRef]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002, 418, 892–897. [Google Scholar] [CrossRef]
- Barnes, E.A.; Kong, M.; Ollendorff, V.; Donoghue, D.J. Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J. 2001, 20, 2214–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adolphe, C.; Hetherington, R.; Ellis, T.; Wainwright, B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006, 66, 2081–2088. [Google Scholar] [CrossRef]
- Wakabayashi, Y.; Mao, J.H.; Brown, K.; Girardi, M.; Balmain, A. Promotion of Hras-induced squamous carcinomas by a polymorphic variant of the Patched gene in FVB mice. Nature 2007, 445, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Li, Q.; Moraes, R.C.; Lewis, M.T.; Hamel, P.A. Activation of Erk by sonic hedgehog independent of canonical hedgehog signalling. Int. J. Biochem. Cell Biol. 2010, 42, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, M.C.; Fleet, A.; Okolowsky, N.; Hamel, P.A. Distinct effects of the mesenchymal dysplasia gene variant of murine Patched-1 protein on canonical and non-canonical Hedgehog signaling pathways. J. Biol. Chem. 2014, 289, 10939–10949. [Google Scholar] [CrossRef]
- Chen, X.L.; Chinchilla, P.; Fombonne, J.; Ho, L.; Guix, C.; Keen, J.H.; Mehlen, P.; Riobo, N.A. Patched-1 proapoptotic activity is downregulated by modification of K1413 by the E3 ubiquitin-protein ligase Itchy homolog. Mol. Cell Biol. 2014, 34, 3855–3866. [Google Scholar] [CrossRef] [PubMed]
- Thibert, C.; Teillet, M.A.; Lapointe, F.; Mazelin, L.; Le Douarin, N.M.; Mehlen, P. Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science 2003, 301, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Mille, F.; Thibert, C.; Fombonne, J.; Rama, N.; Guix, C.; Hayashi, H.; Corset, V.; Reed, J.C.; Mehlen, P. The Patched dependence receptor triggers apoptosis through a DRAL-caspase-9 complex. Nat. Cell Biol. 2009, 11, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical Hedgehog signaling. Vitam. Horm. 2012, 88, 55–72. [Google Scholar] [CrossRef]
- Coni, S.; Mancuso, A.B.; Di Magno, L.; Sdruscia, G.; Manni, S.; Serrao, S.M.; Rotili, D.; Spiombi, E.; Bufalieri, F.; Petroni, M.; et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci. Rep. 2017, 7, 44079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Magno, L.; Coni, S.; Di Marcotullio, L.; Canettieri, G. Digging a hole under Hedgehog: Downstream inhibition as an emerging anticancer strategy. Biochim. Biophys. Acta 2015, 1856, 62–72. [Google Scholar] [CrossRef]
- Infante, P.; Alfonsi, R.; Botta, B.; Mori, M.; Di Marcotullio, L. Targeting GLI factors to inhibit the Hedgehog pathway. Trends Pharmacol. Sci. 2015, 36, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, N.J.; Hager-Theodorides, A.L.; Cebecauer, M.; Shah, D.K.; Drakopoulou, E.; Dyson, J.; Outram, S.V.; Crompton, T. Activation of the Hedgehog signaling pathway in T-lineage cells inhibits TCR repertoire selection in the thymus and peripheral T-cell activation. Blood 2007, 109, 3757–3766. [Google Scholar] [CrossRef] [Green Version]
- Furmanski, A.L.; Saldana, J.I.; Ono, M.; Sahni, H.; Paschalidis, N.; D’Acquisto, F.; Crompton, T. Tissue-derived hedgehog proteins modulate Th differentiation and disease. J. Immunol. 2013, 190, 2641–2649. [Google Scholar] [CrossRef] [PubMed]
- Furmanski, A.L.; Barbarulo, A.; Solanki, A.; Lau, C.I.; Sahni, H.; Saldana, J.I.; D’Acquisto, F.; Crompton, T. The transcriptional activator Gli2 modulates T-cell receptor signalling through attenuation of AP-1 and NFκB activity. J. Cell Sci. 2015, 128, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Ji, Z.; Mei, F.; Lu, M.; Ou, Y.; Cheng, X. Lithium inhibits tumorigenic potential of PDA cells through targeting hedgehog-GLI signaling pathway. PLoS ONE 2013, 8, e61457. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Infante, P.; Lospinoso Severini, L.; Bernardi, F.; Bufalieri, F.; Di Marcotullio, L. Targeting Hedgehog Signalling through the Ubiquitylation Process: The Multiple Roles of the HECT-E3 Ligase Itch. Cells 2019, 8, 98. https://doi.org/10.3390/cells8020098
Infante P, Lospinoso Severini L, Bernardi F, Bufalieri F, Di Marcotullio L. Targeting Hedgehog Signalling through the Ubiquitylation Process: The Multiple Roles of the HECT-E3 Ligase Itch. Cells. 2019; 8(2):98. https://doi.org/10.3390/cells8020098
Chicago/Turabian StyleInfante, Paola, Ludovica Lospinoso Severini, Flavia Bernardi, Francesca Bufalieri, and Lucia Di Marcotullio. 2019. "Targeting Hedgehog Signalling through the Ubiquitylation Process: The Multiple Roles of the HECT-E3 Ligase Itch" Cells 8, no. 2: 98. https://doi.org/10.3390/cells8020098
APA StyleInfante, P., Lospinoso Severini, L., Bernardi, F., Bufalieri, F., & Di Marcotullio, L. (2019). Targeting Hedgehog Signalling through the Ubiquitylation Process: The Multiple Roles of the HECT-E3 Ligase Itch. Cells, 8(2), 98. https://doi.org/10.3390/cells8020098