Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity




Abstract
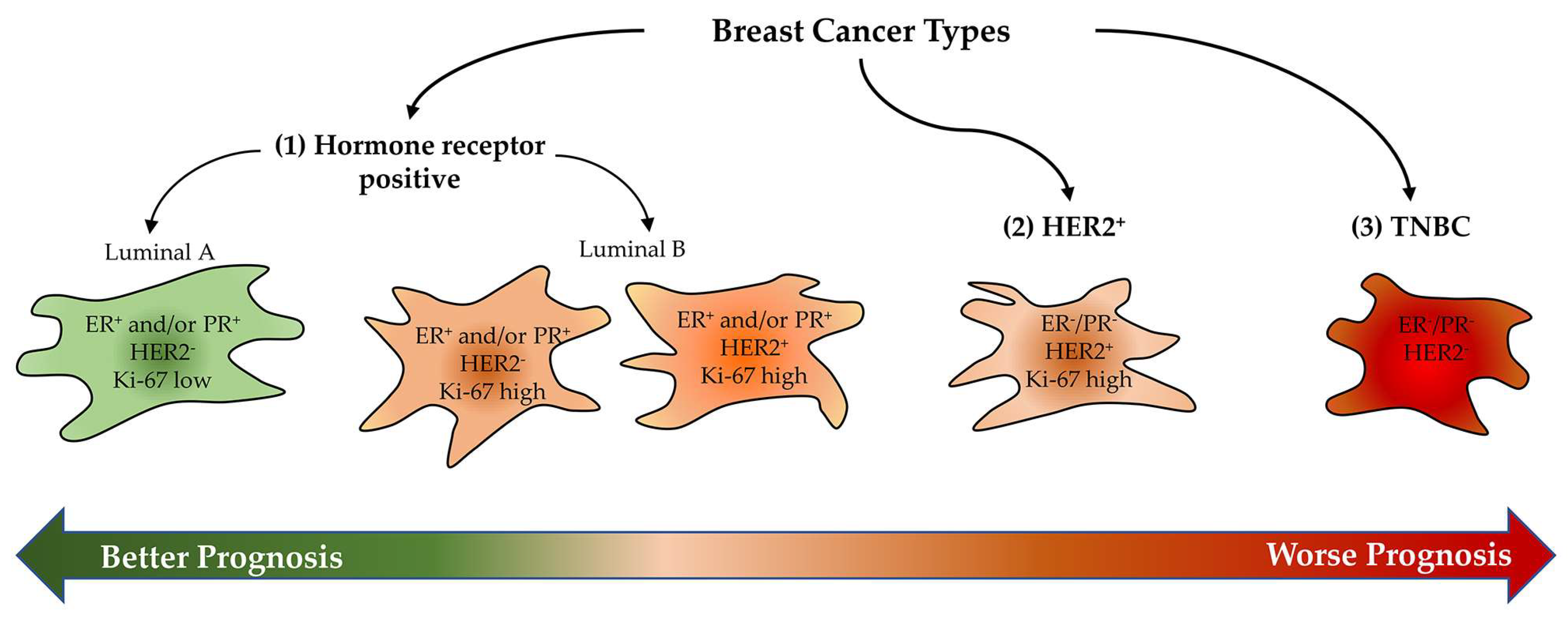
:1. Introduction
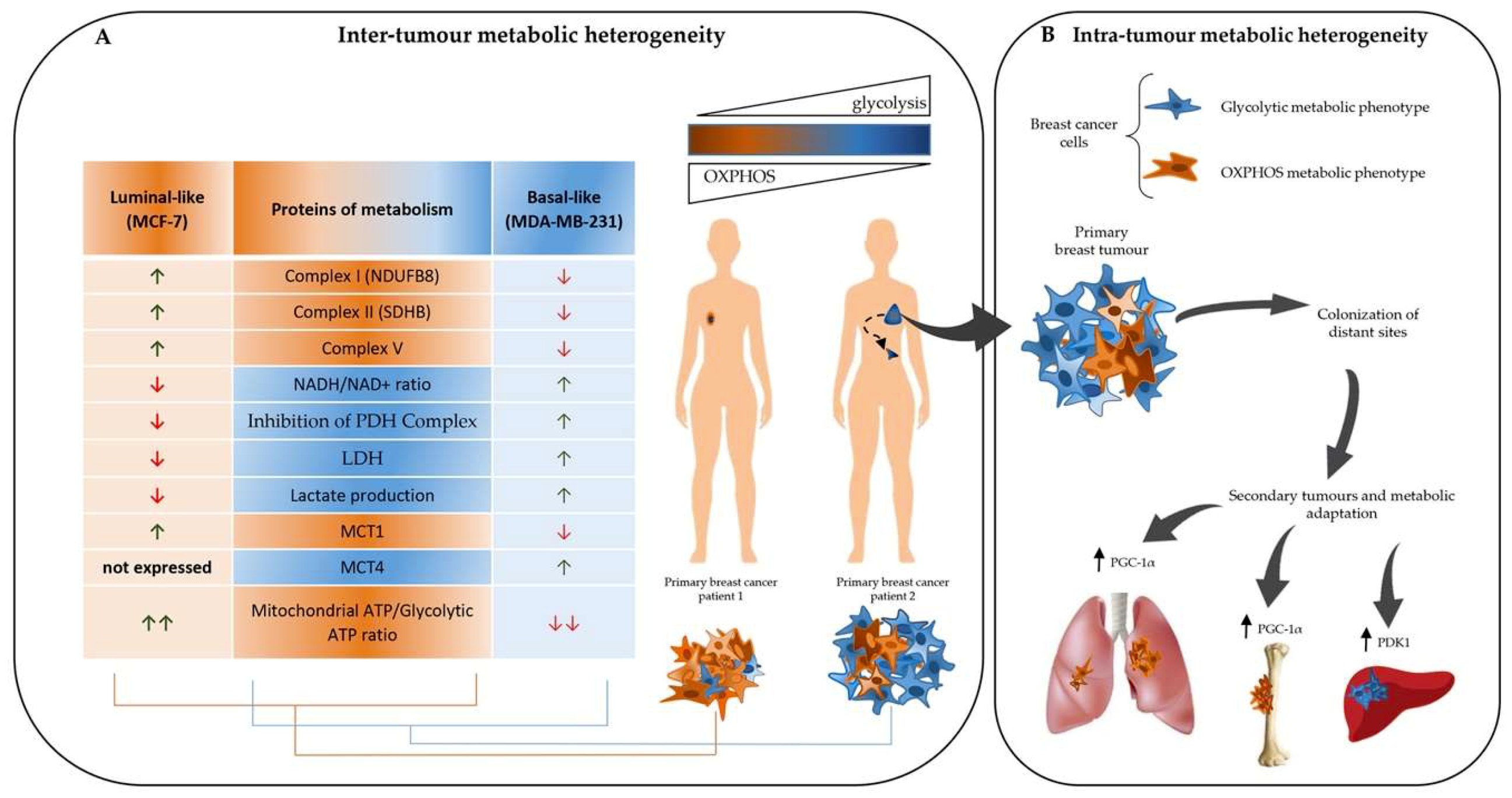
2. Metabolic Flexibility of Breast Cancers
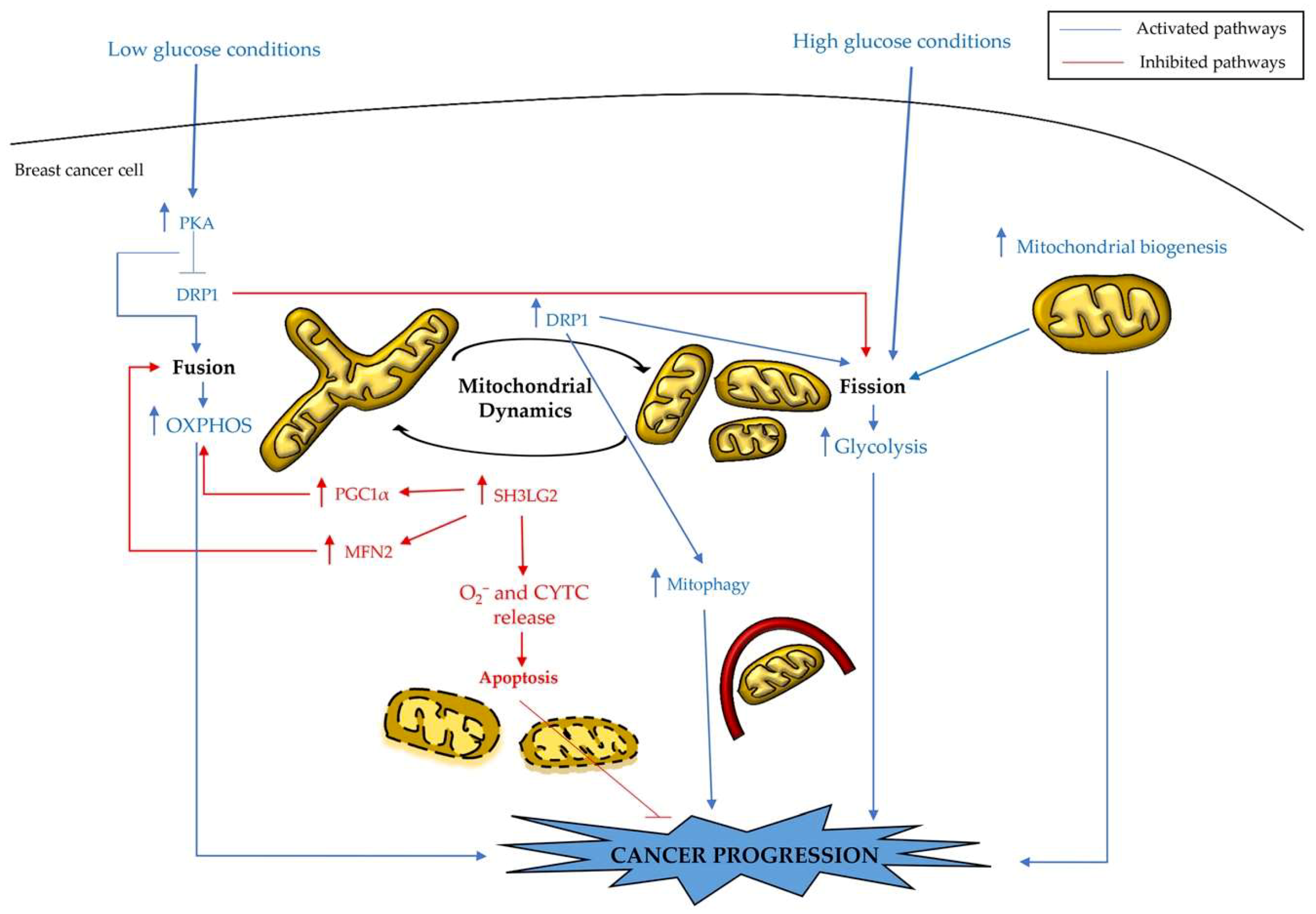
3. Mitochondrial Reprogramming in Breast Cancer Cells
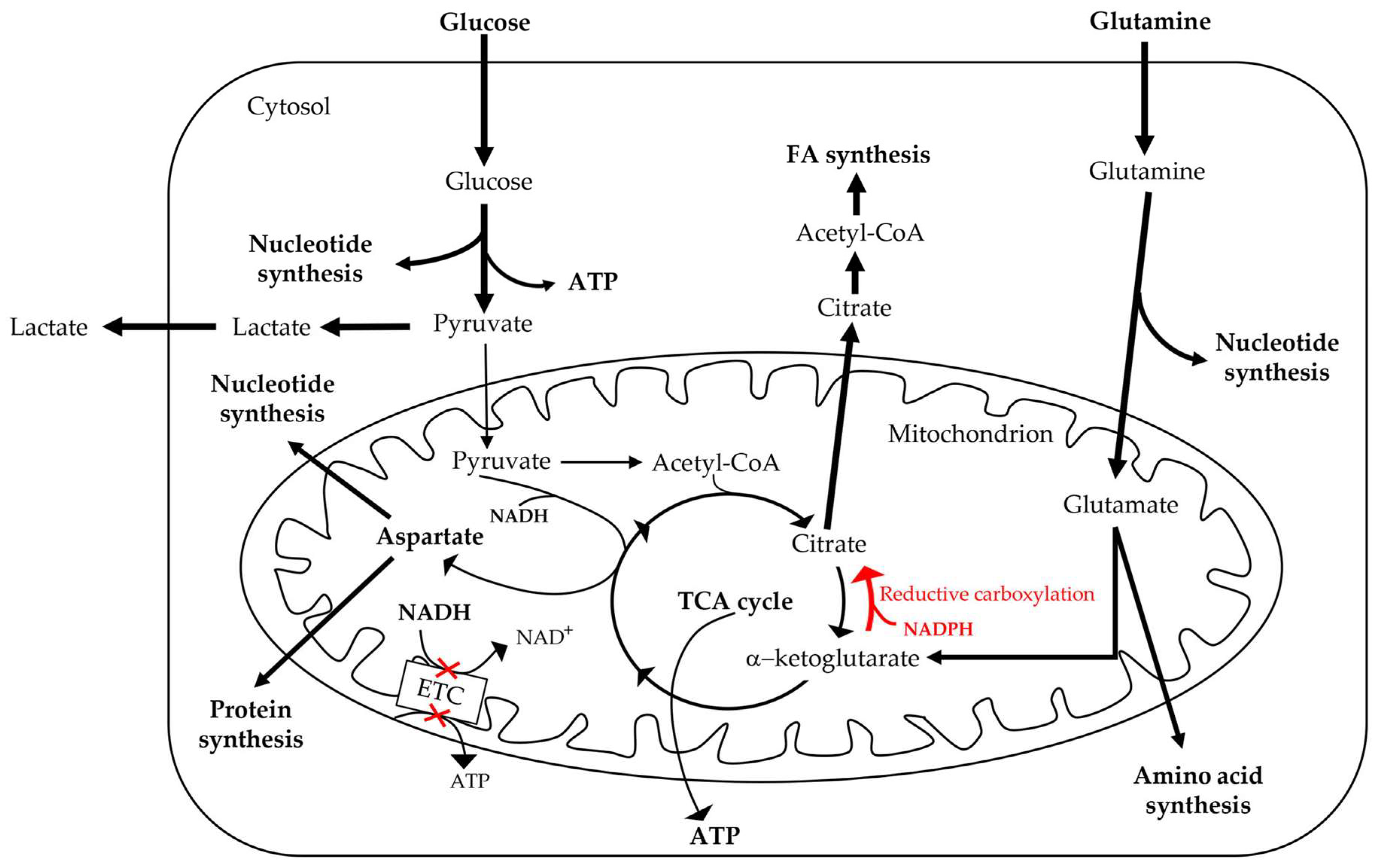
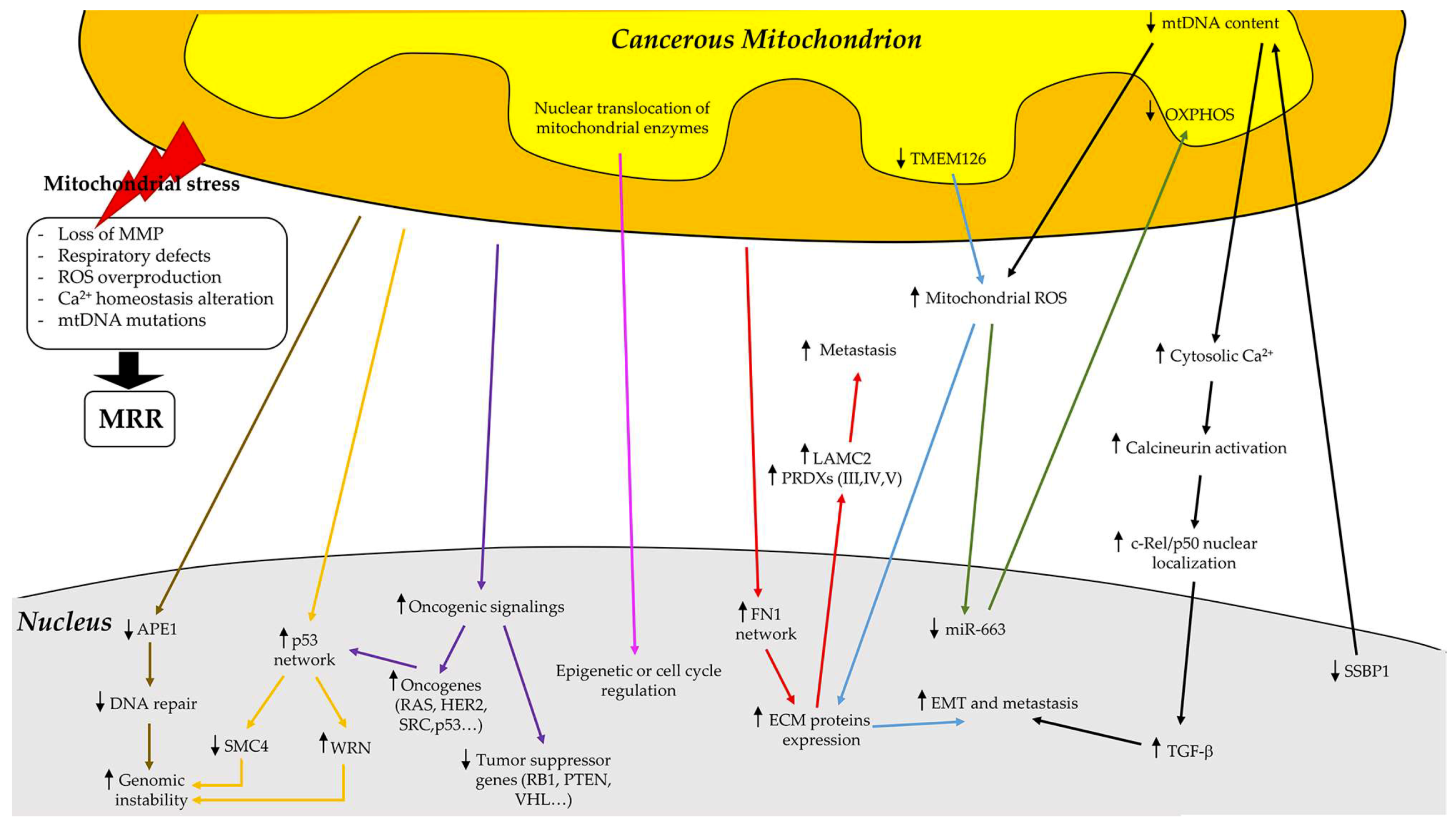
4. Mitochondrial Retrograde Regulation
5. Therapeutic Strategies Targeting Mitochondrial Alterations
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Imparato, V.; Masone, S.; Masullo, M.; Nasso, R.; Montagnani, S.; Arcucci, A. Involvement of Breast Cancer-Associated Fibroblasts in Tumor Development, Therapy Resistance and Evaluation of Potential Therapeutic Strategies. Curr. Med. Chem. 2018, 25, 3414–3434. [Google Scholar] [CrossRef] [PubMed]
- Arcucci, A.; Ruocco, M.R.; Granato, G.; Sacco, A.M.; Montagnani, S. Cancer: An Oxidative Crosstalk between Solid Tumor Cells and Cancer Associated Fibroblasts. Biomed. Res. Int. 2016, 2016, 4502846. [Google Scholar] [CrossRef]
- Sotgia, F.; Ozsvari, B.; Fiorillo, M.; De Francesco, E.M.; Bonuccelli, G.; Lisanti, M.P. A mitochondrial based oncology platform for targeting cancer stem cells (CSCs): MITO-ONC-RX. Cell Cycle 2018, 17, 2091–2100. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. Biomed. Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef]
- Morris, B.A.; Burkel, B.; Ponik, S.M.; Fan, J.; John, S.; Condeelis, J.S.; Aguire-Ghiso, J.A.; Castracane, J.; Denu, J.M. and Keely, P.J. Collagen Matrix Density Drives the Metabolic Shift in Breast Cancer Cells. EBioMedicine 2016, 13, 146–156. [Google Scholar] [CrossRef]
- Ferreira, L.M.R. Cancer metabolism: The Warburg effect today. Exp. Mol. Pathol. 2010, 89, 372–380. [Google Scholar] [CrossRef]
- Guaragnella, N.; Giannattasio, S.; Moro, L. Mitochondrial dysfunction in cancer chemoresistance. Biochem. Pharmacol. 2014, 92, 62–72. [Google Scholar] [CrossRef]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and Mitochondrial Respiration: A Neglected Connection. Cancer Res. 2015, 75, 3687–3691. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Cazzaniga, M.; Bonanni, B. Breast Cancer Metabolism and Mitochondrial Activity: The Possibility of Chemoprevention with Metformin. BioMed. Res. Int. 2015, 2015, 972193. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Enríquez, S.; Carreño-Fuentes, L.; Gallardo-Pérez, J.C.; Saavedra, E.; Quezada, H.; Vega, A.; Marín-Hernández, A.; Olín-Sandoval, V.; Torres-Márquez, M.E.; Moreno-Sánchez, R. Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int. J. Biochem. Cell Biol. 2010, 42, 1744–1751. [Google Scholar] [CrossRef] [PubMed]
- Cox, E.; Bonner, J. The advantages of togetherness. Science 2001, 292, 448–449. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Serguienko, A.; Grad, I.; Wennerstrøm, A.B.; Meza-Zepeda, L.A.; Thiede, B.; Stratford, E.W.; Myklebost, O.; Munthe, E. Metabolic reprogramming of metastatic breast cancer and melanoma by let-7a microRNA. Oncotarget 2014, 6, 2451–2465. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, A.; Serizawa, S.; Tachibana, K.; Sakurada, K.; Samejima, H.; Kuchino, Y.; Kitanaka, C. Critical Role for Mitochondrial Oxidative Phosphorylation in the Activation of Tumor Suppressors Bax and Bak. JNCI 2006, 98, 1462–1473. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Aragó, M.; Chamorro, M.; Cuezva, J.M. Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis 2010, 31, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef]
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Bajzikova, M.; Kovarova, J.; Coelho, A.R.; Boukalova, S.; Oh, S.; Rohlenova, K.; Svec, D.; Hubackova, S.; Endaya, B.; Judasova, K.; et al. Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab. 2019, 29, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [PubMed]
- Lunetti, P.; Di Giacomo, M.; Vergara, D.; De Domenico, S.; Maffia, M.; Zara, V.; Capobianco, L.; Ferramosca, A. Metabolic reprogramming in breast cancer results in distinct mitochondrial bioenergetics between luminal and basal subtypes. FEBS J. 2019, 286, 688–709. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Reue, K.; Frohnen, P.; Chan, M.; Alhiyar, Y.; Dratver, M.B.; Pajonk, F. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res. Treat. 2014, 146, 525–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Bennet, B.; Mu, E.; Rabinowitz, J.; Kang, Y. Metabolomic changes accompanying transformation and acquisition of metastatic potential in a syngeneic mouse mammary tumor model. J. Biol. Chem. 2010, 285, 9317–9321. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 10, 992–1003. [Google Scholar] [CrossRef]
- Dupuy, F.; Bastien Tabariè, S.; Andrzejewski, S.; Pierre, J.S.; Jones, R.G.; Siegel, P.M.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.H.; Jung, W.H.; Koo, J.S. Metabolic phenotypes in triple-negative breast cancer. Tumor Biol. 2013, 34, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Urra, F.A.; Muñoz, F.; Córdova-Delgado, M.; Ramírez, M.P.; Peña-Ahumada, B.; Rios, M.; Cruz, P.; Ahumada-Castro, U.; Bustos, G.; Silva-Pavez, E.; et al. FR58P1a; a new uncoupler of OXPHOS that inhibits migration in triple-negative breast cancer cells via Sirt1/AMPK/β1-integrin pathway. Sci. Rep. 2018, 8, 13190. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M. Importance of critical metabolites and cellular interactions in the biology of microregions of tumors. Cancer 1986, 58, 1668–1680. [Google Scholar] [CrossRef] [Green Version]
- Kallinowski, F.; Vaupel, P.; Runkel, S.; Berg, G.; Fortmeyer, H.P.; Baessler, K.H.; Wagner, K.; Mueller-Klieser, W.; Walenta, S. Glucose uptake, lactate release, ketone body turnover, metabolic micromilieu, and pH distributions in human breast cancer xenografts in nude rats. Cancer Res. 1988, 48, 7264–7272. [Google Scholar] [PubMed]
- Witney, T.H.; Brindle, K.M. Imaging tumour cell metabolism using hyperpolarized 13 C magnetic resonance spectroscopy. Biochem. Soc. Trans. 2010, 38, 1220–1224. [Google Scholar] [CrossRef]
- Vaupel, P. Pathophysiology of Solid Tumors. In The Impact of Tumor Biology on Cancer Treatment and Multidisciplinary Strategies; Molls, M., Vaupel, P., Nieder, C., Anscher, M.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 52–85. [Google Scholar]
- Simões, R.V.; Serganova, I.S.; Kruchevsky, N.; Leftin, A.; Shestov, A.A.; Thaler, H.T.; Sukenick, G.; Locasale, J.W.; Blasberg, R.G.; Koutcher, J.A.; et al. Metabolic plasticity of metastatic breast cancer cells: Adaptation to changes in the microenvironment. Neoplasia 2015, 17, 671–684. [Google Scholar] [CrossRef]
- Otto, A.M.; Hintermair, J.; Janzon, C. NADH-linked metabolic plasticity of MCF-7 breast cancer cells surviving in a nutrient-deprived microenvironment. J. Cell Biochem. 2015, 116, 822–835. [Google Scholar] [CrossRef]
- Lyons, A.; Coleman, M.; Riis, S.; Favre, C.; O’Flanagan, C.H.; Zhdanov, A.V.; Papkovsky, D.B.; Hursting, S.D.; O’Connor, R. Insulin-like growth factor 1 signaling is essential for mitochondrial biogenesis and mitophagy in cancer cells. J. Biol. Chem. 2017, 292, 16983–16998. [Google Scholar] [CrossRef]
- Moulder, D.E.; Hatoum, D.; Tay, E.; Lin, Y.; McGowan, E.M. The Roles of p53 in Mitochondrial Dynamics and Cancer Metabolism: The Pendulum between Survival and Death in Breast Cancer? Cancers 2018, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. p53 regulates mitochondrial respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Harrelson, J.P.; Lee, M.W. Expanding the view of breast cancer metabolism: Promising molecular targets and therapeutic opportunities. Pharmacol. Ther. 2016, 167, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Ogrodzinski, M.P.; Bernard, J.J.; Lunt, S.Y. Deciphering metabolic rewiring in breast cancer subtypes. Transl. Res. 2017, 189, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Konduri, S.D.; Bansal, S.; Nayak, B.K.; Rajasekaran, S.A.; Karuppayil, S.M.; Rajasekaran, A.K.; Das, G.M. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 2006, 281, 9837–9840. [Google Scholar] [CrossRef]
- Li, J.; Huang, Q.; Long, X.; Guo, X.; Sun, X.; Jin, X.; Li, Z.; Ren, T.; Yuan, P.; Huang, X.; et al. Mitochondrial elongation-mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene 2017, 36, 4901–4912. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Indo, H.P.; Yen, H.C.; Nakanishi, I.; Matsumoto, K.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell. Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef]
- Zou, P.; Liu, L.; Zheng, L.D.; Payne, K.K.; Manjili, M.H.; Idowu, M.O.; Zhang, J.; Schmelz, E.M.; Cheng, Z. Coordinated Upregulation of Mitochondrial Biogenesis and Autophagy in Breast Cancer Cells: The Role of Dynamin Related Protein-1 and Implication for Breast Cancer Treatment. Oxid. M. Cell Longev. 2016, 2016, 4085727. [Google Scholar] [CrossRef] [PubMed]
- Kjaerulff, O.; Brodin, L.; Jung, A. The structure and function of endophilin proteins. Cell. Biochem. Biophys. 2011, 60, 137–154. [Google Scholar] [CrossRef]
- Kannan, A.; Wells, R.B.; Sivakumar, S.; Komatsu1, S.; Singh, K.P.; Samten, B.; Philley, J.V.; Sauter, E.R.; Ikebe, M.; Idell, S.; et al. Mitochondrial Reprogramming Regulates Breast Cancer Progression. Clin. Cancer. Res. 2016, 22, 3348–3360. [Google Scholar] [CrossRef] [Green Version]
- Guppy, M.; Leedman, P.; Zu, X.; Russell, V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem. J. 2002, 364, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.; Luiken, J.J. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie 2017, 136, 21–26. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [Green Version]
- Farese, R.V., Jr.; Walther, T.C. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 2009, 139, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Louie, S.M.; Roberts, L.S.; Mulvihill, M.M.; Luo, K.; Nomura, D.K. Cancer cells incorporate and remodel exogenous palmitate into structural and oncogenic signaling lipids. Biochim. Biophys. Acta 2013, 1831, 1566–1572. [Google Scholar] [CrossRef]
- Bruce, C.R.; Hoy, A.J.; Turner, N.; Watt, M.J.; Allen, T.L.; Carpenter, K.; Cooney, G.J.; Febbraio, M.A.; Kraegen, E.W. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat dietinduced insulin resistance. Diabetes 2009, 58, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Feola, A.; Ricci, S.; Kouidhi, S.; Rizzo, A.; Penon, A.; Formisano, P.; Giordano, A.; Di Carlo, A.; Di Domenico, M. Multifaceted breast cancer: The molecular connection with obesity. J. Cell. Physiol. 2017, 232, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer. Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Blanquer-Rosselló, M.D.M.; Oliver, J.; Sastre-Serra, J.; Valle, A.; Roca, P. Leptin regulates energy metabolism in MCF-7 breast cancer cells. Int. J. Biochem. Cell Biol. 2016, 72, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Vayalil, P.K.; Zhou, F.; Benavides, G.A.; Beggs, R.R.; Golzarian, H.; Nijampatnam, B.; Oliver, P.G.; Smith, R.A.J.; Murphy, M.P.; et al. Mitochondrial thiol modification by a targeted electrophile inhibits metabolism in breast adenocarcinoma cells by inhibiting enzyme activity and protein levels. Redox Biol. 2016, 8, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Cruz, R.S.; Carney, E.J.; Clarke, J.; Cao, H.; Cruz, M.I.; Benitez, C.; Jin, L.; Fu, Y.; Cheng, Z.; Wang, Y.; et al. Paternal malnutrition programs breast cancer risk and tumor metabolism in offspring. Breast Cancer Res. 2018, 20, 99. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef]
- Dumitrescu, R.; Cotarla, I. Understanding breast cancer risk-where do we stand in 2005? J. Cell. Mol. Med. 2005, 9, 208–221. [Google Scholar] [CrossRef]
- Boyd, N.F.; Dite, G.S.; Stone, J.; Gunasekara, A.; English, D.R.; Mccredie, M.R.; Giles, G.G.; Tritchler, D.; Chiarelli, A.; Yaffe, M.J. Heritability of mammographic density, a risk factor for breast cancer. N. Engl. J. Med. 2002, 347, 886–894. [Google Scholar] [CrossRef]
- Boyd, N.F.; Guo, H.; Martin, L.J.; Sun, L.; Stone, J.; Fishell, E.; Jong, R.A.; Hislop, G.; Chiarelli, A.; Minkin, S. Mammographic density and the risk and detection of breast cancer. N. Engl. J. Med. 2007, 356, 227–236. [Google Scholar] [CrossRef]
- Guo, Y.P.; Martin, L.J.; Hanna, W.; Banerjee, D.; Miller, N.; Fishell, E.; Khokha, R.; Boyd, N.F. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer Epidemiol. Biomark. Prev. 2001, 10, 243–248. [Google Scholar]
- Daye, D.; Wellen, K.E. Metabolic reprogramming in cancer: Unraveling the role of glutamine in tumorigenesis. Semin. Cell. Dev. Biol. 2012, 23, 362–369. [Google Scholar] [CrossRef]
- Wise, D.R.; Deberardinis, R.J.; Mancuso, A.; Sayeda, N.; Zhangc, X.Y.; Pfeifferc, H.K.; Nissimd, I.; Daikhind, E.; Yudkoffd, M.; McMahonc, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. PNAS 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bai, C.; Ruan, Y.; Liu, M.; Chu, Q.; Qiu, L.; Yang, C.; Li, B. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 2019, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.B.; Dias, S.M.G.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.R.; Lane, A.N.; Robertson, B.; Kemp, S.; Liu, Y.; Hill, B.G.; Douglas, C.; Dean, D.C.; Clem, B.F. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. 2014, 33, 556–566. [Google Scholar] [CrossRef] [PubMed]
- De Berardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Deblois, G.; St-Pierre, J.; Giguère, V. The PGC-1/ERR signaling axis in cancer. Oncogene 2013, 32, 3483–3490. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. Punctum on two different transcription factors regulated by PGC-1α: Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim. Biophys. Acta 2013, 1830, 4137–4146. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.Y.; Kazmin, D.; Jasper, J.S.; Kunder, R.; Zuercher, W.J.; McDonnell, D.P. The metabolic regulator ERRα, a downstream target of HER2/IGF-1R, as a therapeutic target in breast cancer. Cancer Cell 2011, 20, 500–510. [Google Scholar] [CrossRef]
- Hayashi, J.; Takemitsu, M.; Nonaka, I. Recovery of the missing tumorigenicity in mitochondrial DNA-less HeLa cells by introduction of mitochondrial DNA from normal human cells. Somat. Cell Mol. Genet. 1992, 18, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, L.R.; Varella-Garcia, M.; Liang, B.C. Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 1997, 8, 1189–1198. [Google Scholar]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Regulation of Oncogenic Properties inTriple Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef]
- Dong, Y.; Yoshitomi, T.; Hu, J.F.; Cui, J. Long noncoding RNAs coordinate functions between mitochondria and the nucleus. Epigenetics Chromatin 2017, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Chandel, N.S. Evolution of mitochondria as signaling organelles. Cell Metab. 2015, 22, 204–206. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Craig, B.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saki, M.; Prakash, A. DNA damage related crosstalk between the nucleus and mitochondria. Radic. Biol. Med. 2016, 107, 216–227. [Google Scholar] [CrossRef]
- Parikh, V.S.; Morgan, M.M.; Scott, R.; Clements, L.S.; Butow, R.A. The mitochondrial genotype can influence nuclear gene expression in yeast. Science 1987, 235, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Butow, R.A.; Avadhani, N.G. Mitochondrial signaling: The retrograde response. Mol Cell. 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Liu, Z.; Butow, R.A. Mitochondrial retrograde signaling. Annu. Rev. Genet. 2006, 40, 159–185. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Kim, J. Mitochondrial Retrograde Signalling and Metabolic Alterations in the Tumour Microenvironment. Cells 2019, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jeon, J.H.; Min, B.K.; Ha, C.M.; Thoudam, T.; Park, B.Y.; Lee, I.K. Role of the Pyruvate Dehydrogenase Complex in Metabolic Remodeling: Differential Pyruvate Dehydrogenase Complex Functions in Metabolism. Diabetes Metab. J. 2018, 42, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A Nuclear Pyruvate Dehydrogenase Complex Is Important for the Generation of Acetyl-CoA and Histone Acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukouris, A.E.; Zervopoulos, S.D.; Michelakis, E.D. Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends Biochem. Sci. 2016, 41, 712–730. [Google Scholar] [CrossRef]
- Ma, Y.; Bai, R.K.; Trieu, R.; Wong, L.J. Mitochondrial dysfunction in human breast cancer cells and their transmitochondrial cybrids. Biochim. Biophys. Acta 2010, 1797, 29–37. [Google Scholar] [CrossRef]
- Singh, K.K.; Kulawiec, M.; Still, I.; Desouki, M.M.; Geradts, J.; Matsui, S. Inter-genomic cross talk between mitochondria and the nucleus plays an important role in tumorigenesis. Gene 2005, 354, 140–146. [Google Scholar] [CrossRef]
- Liu, C.; Srihari, S.; Lal, S.; Gautier, B.; Simpson, P.T.; Khanna, K.K.; Ragan, M.A.; Lê Cao, K.A. Personalised pathway analysis reveals association between DNA repair pathway dysregulation and chromosomal instability in sporadic breast cancer. Mol. Oncol. 2016, 10, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Aboalela, N.; Lyon, D.; Elswick, R.K.; Kelly, D.L.; Brumelle, J.; Bear, H.D.; Jackson-Cook, C. Perceived Stress Levels, Chemotherapy, Radiation Treatment and Tumor Characteristics Are Associated with a Persistent Increased Frequency of Somatic Chromosomal Instability in Women Diagnosed with Breast Cancer: A One Year Longitudinal Study. PLoS ONE 2015, 10, e0133380. [Google Scholar] [CrossRef]
- Kulawiec, M.; Safina, A.; Desouki, M.M.; Still, I.; Matsui, S.I.; Bakin, A.; Singh, K.K. Tumorigenic transformation of human breast epithelial cells induced by mitochondrial DNA depletion. Cancer Biol. Ther. 2008, 7, 1732–1743. [Google Scholar] [CrossRef] [Green Version]
- Tseng, L.M.; Yin, P.H.; Chi, C.W.; Hsu, C.Y.; Wu, C.W.; Lee, L.M.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosom. Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef]
- Yu, M.; Zhou, Y.; Shi, Y.; Ning, L.; Yang, Y.; Wei, X.; Zhang, N.; Hao, X.; Niu, R. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life 2007, 59, 450–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delsite, R.; Kachhap, S.; Anbazhagan, R.; Gabrielson, E.; Singh, K.K. Nuclear genes involved in mitochondria-to-nucleus communication in breast cancer cells. Mol. Cancer 2002, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Kaipparettu, B.A.; Ma, Y.; Park, J.H.; Lee, T.L.; Zhang, Y.; Yotnda, P. Crosstalk from Non-Cancerous Mitochondria Can Inhibit Tumor Properties of Metastatic Cells by Suppressing Oncogenic Pathways. PLoS ONE 2013, 8, e61747. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L. The distribution of fibronectin, laminin and tetranectin in human breast cancer with special attention to the extracellular matrix. APMIS Suppl. 1992, 26, 1–39. [Google Scholar]
- Koukoulis, G.K.; Howeedy, A.A.; Korhonen, M.; Virtanen, I.; Gould, V.E. Distribution of tenascin, cellular fibronectins and integrins in the normal, hyperplastic and neoplastic breast. J. Submicrosc. Cytol. Pathol. 1993, 25, 285–295. [Google Scholar] [PubMed]
- Karihtala, P.; Mäntyniemi, A.; Kang, S.W.; Kinnula, V.L.; Soini, Y. Peroxiredoxins in breast carcinoma. Clin. Cancer Res. 2003, 9, 3418–3424. [Google Scholar] [PubMed]
- Jiang, H.L.; Sun, H.F.; Gao, S.P.; Li, L.D.; Huang, S.; Hu, X.; Liu, S.; Wu, J.; Shao, Z.M.; Jin, W. SSBP1 Suppresses TGFβ-Driven Epithelial-to-Mesenchymal Transition and Metastasis in Triple-Negative Breast Cancer by Regulating Mitochondrial Retrograde Signaling. Cancer Res. 2016, 76, 952–964. [Google Scholar] [CrossRef]
- Wong, C.C.; Gilkes, D.M.; Zhang, H.; Chen, J.; Wei, H.; Chaturvedi, P.; Fraley, S.I.; Wong, C.M.; Khoo, U.S.; Ng, I.O.; et al. Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. PNAS 2011, 108, 16369–16374. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.Y.; Cheng, C.T.; Hou, P.; Lin, Y.P.; Ma, H.; Chung, Y.; Chi, K.; Chen, Y.; Li, W.; Kung, H.J.; Ann, D.K. HIF-1-alpha links mitochondrial perturbation to the dynamic acquisition of breast cancer tumorigenicity. Oncotarget 2016, 7, 34052–34069. [Google Scholar] [CrossRef] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef]
- Hanein, S.; Garcia, M.; Fares-Taie, L.; Serre, V.; De Keyzer, Y.; Delaveau, T.; Perrault, I.; Delphin, N.; Gerber, S.; Schmitt, A.; et al. TMEM126A is a mitochondrial located mRNA (MLR) protein of the mitochondrial inner membrane. Biochim. Biophys. Acta 2013, 1830, 3719–3733. [Google Scholar] [CrossRef]
- Sun, H.F.; Yang, X.L.; Zhao, Y.; Tian, Q.; Chen, M.T.; Zhao, Y.Y.; Jin, W. Loss of TMEM126A promotes extracellular matrix remodeling, epithelial-to-mesenchymal transition, and breast cancer metastasis by regulating mitochondrial retrograde signaling. Cancer Lett. 2019, 440-441, 189–201. [Google Scholar] [CrossRef]
- Singh, R.K.; Srivastava, A.; Kalaiarasan, P.; Manvati, S.; Chopra, R.; Bamezai, R.N. mtDNA germ line variation mediated ROS generates retrograde signaling and induces pro-cancerous metabolic features. Sci. Rep. 2014, 4, 6571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indo, H.P.; Hawkins, C.L.; Nakanishi, I.; Matsumoto, K.; Matsui, H.; Suenaga, S.; Davies, M.J.; St Clair, D.K.; Ozawa, T.; Majima, H.J. Role of Mitochondrial Reactive Oxygen Species in the Activation of Cellular Signals, Molecules and Function. In Pharmacology of Mitochondria; Handb Exp Pharmacol (HEP); Singh, H., Sheu, S.-S., Eds.; Springer Nature Switzerland AG: Basel, Switzerland, 2017; Volume 240, pp. 439–456. [Google Scholar] [CrossRef]
- Carden, T.; Singh, B.; Mooga, V.; Bajpai, P.; Singh, K.K. Epigenetic modification of miR-663 controls mitochondria-to-nucleus retrograde signaling and tumor progression. J. Biol. Chem. 2017, 292, 20694–20706. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.T.; Kuo, C.Y.; Ouyang, C.; Li, C.F.; Chung, Y.; Chan, D.C.; Kung, H.J.; Ann, D.K. Metabolic stress-induced phosphorylation of KAP1ser473 blocks mitochondrial fusion in breast cancer cells. Cancer Res. 2016, 76, 5006–5018. [Google Scholar] [CrossRef]
- Qian, W.; Wang, J.; Van Houten, B. The role of dynamin-related protein 1 in cancer growth: A promising therapeutic target? Expert Opin. Ther. Targets 2013, 17, 997–1001. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Delage, B.; Fennell, D.A.; Nicholson, L.; McNeish, I.; Lemoine, N.R.; Crook, T.; Szlosarek, P.W. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 2010, 126, 2762–2772. [Google Scholar] [CrossRef]
- Qiu, F.; Chen, Y.R.; Liu, X.; Chu, C.Y.; Shen, L.J.; Xu, J.; Gaur, S.; Forman, H.J.; Zhang, H.; Zheng, S.; et al. Arginine starvation impairs mitochondrial respiratory function in ASS1-deficient breast cancer cells. Sci. Signal. 2014, 7, ra31. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.T.; Qi, Y.; Wang, Y.C.; Chi, K.K.; Chung, Y.; Ouyang, C.; Chen, Y.R.; Oh, M.E.; Sheng, X.; Tang, Y.; et al. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun. Biol. 2018, 1, 178. [Google Scholar] [CrossRef] [PubMed]
- Ayyanathan, K.; Kesaraju, S.; Dawson-Scully, K.; Weissbach, H. Combination of sulindac and dichloroacetate kills cancer cells via oxidative damage. PLoS ONE 2012, 7, e39949. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. j. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Goliasova, T.; Denko, N.C. Anticancer drugs that target metabolism: Is dichloroacetate the new paradigm? Int. j. cancer 2011, 128, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Agostinis, P. ER stress, autophagy and immunogenic cell death in photodynamic therapyinduced anti-cancer immune responses. Photochem. Photobiol. Sci. 2014, 13, 474–487. [Google Scholar] [CrossRef]
- Lu, K.; He, C.; Guo, N.; Chan, C.; Ni, K.; Lan, G.; Tang, H.; Pelizzari, C.; Fu, Y.X.; Spiotto, M.T.; et al. Low-dose X-ray radiotherapy–radiodynamic therapy via nanoscale metal–organic frameworks enhances checkpoint blockade immunotherapy. Nat. Biomed. Eng. 2018, 2, 600–610. [Google Scholar] [CrossRef]
- Alkarakooly, Z.; Al-Anbaky, Q.A.; Kannan, K.; Ali, N. Metabolic reprogramming by Dichloroacetic acid potentiates photodynamic therapy of human breast adenocarcinoma MCF-7 cells. PLoS ONE 2018, 13, e0206182. [Google Scholar] [CrossRef] [PubMed]
- Schramm, L. Going Green: The Role of the Green Tea Component EGCG in Chemoprevention. J. Carcinog. Mutagen. 2013, 4, 1000142. [Google Scholar] [CrossRef]
- Cabrera, C.; Artacho, R.; Giménez, R. Beneficial effects of green tea--a review. J. Am. Coll. Nutr. 2006, 25, 79–99. [Google Scholar] [CrossRef]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Olenchock, B.A.; Mayers, J.R.; Wasylenko, T.M.; Vokes, N.I.; Guarente, L.; Vander Heiden, M.G.; Stephanopoulos, G. Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat. Commun. 2013, 4, 2236. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Invest. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.K.; Najyb, O.; Gravel, S.P.; McGuirk, S.; Ahn, R.; Avizonis, D.Z.; Chénard, V.; Sabourin, V.; Hudson, J.; Pawson, T.; et al. Interplay between ShcA signaling and PGC-1α triggers targetable metabolic vulnerabilities in breast cancer. Cancer Res. 2018, 78, 4826–4838. [Google Scholar] [CrossRef] [PubMed]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Davidson, S.M.; Wirth, G.J.; Fiske, B.; Mayers, J.R.; Schwab, M.; Bellinger, G.; Csibi, A.; et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013, 73, 4429–4438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrzejewski, S.; Gravel, S.P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Gravel, S.P.; Hulea, L.; Toban, N.; Birman, E.; Blouin, M.J.; Zakikhani, M.; Zhao, Y.; Topisirovic, I.; St-Pierre, J.; Pollak, M. Serine deprivation enhances antineoplastic activity of biguanides. Cancer Res. 2014, 74, 7521–7533. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Kroemer, G.; Petit, P.; Zamzami, N.; Vayssière, J.L.; Mignotte, B. The biochemistry of programmed cell death. FASEB J. 1995, 9, 1277–1287. [Google Scholar] [CrossRef]
- Santidrian, A.F.; Matsuno-Yagi, A.; Ritland, M.; Seo, B.B.; LeBoeuf, S.E.; Gay, L.J.; Yagi, T.; Felding-Habermann, B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Invest. 2013, 123, 1068–1081. [Google Scholar] [CrossRef] [Green Version]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Birts, C.N.; Darley, M.; Parker, R.; Mirnezami, A.H.; West, J.; Cutress, R.I.; Beers, S.A.; Rose-Zerilli, M.J.; Blaydes, J.P. Stem cell-like breast cancer cells with acquired resistance to metformin are sensitive to inhibitors of NADH-dependent CtBP dimerization. Carcinogenesis 2019. [Google Scholar] [CrossRef]
- Rajh, M.; Dolinar, K.; Miš, K.; Pavlin, M.; Pirkmajer, S. Medium Renewal Blocks Anti-Proliferative Effects of Metformin in Cultured MDA-MB-231 Breast Cancer Cells. PLoS ONE 2016, 11, e0154747. [Google Scholar] [CrossRef]
- Lukey, M.J.; Greene, K.S.; Erickson, J.W.; Wilson, K.F.; Cerione, R.A. The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat. Commun. 2016, 7, 11321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.P.; Li, X.N.; Zhang, F. Targeting metabolism in breast cancer: How far we can go? World J. Clin. Oncol. 2016, 7, 122–130. [Google Scholar] [CrossRef]
- Karunakaran, S.; Ramachandran, S.; Coothankandaswamy, V.; Elangovan, S.; Babu, E.; Periyasamy-Thandavan, S.; Gurav, A.; Gnanaprakasam, J.P.; Singh, N.; Schoenlein, P.V.; et al. SLC6A14 (ATB0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J. Biol. Chem. 2011, 286, 31830–31838. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Simpson, N.E.; Tryndyak, V.P.; Pogribna, M.; Beland, F.A.; Pogribny, I.P. Modifying metabolically sensitive histone marks by inhibiting glutamine metabolism affects gene expression and alters cancer cell phenotype. Epigenetics 2012, 7, 1413–1420. [Google Scholar] [CrossRef] [Green Version]
- Pizer, E.S.; Thupari, J.; Han, W.F.; Pinn, M.L.; Chrest, F.J.; Frehywot, G.L.; Townsend, C.A.; Kuhajda, F.P. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000, 60, 213–218. [Google Scholar] [PubMed]
- Maddalena, F.; Sisinni, L.; Lettini, G.; Condelli, V.; Matassa, D.S.; Piscazzi, A.; La Torre, G.; Esposito, F.; Landriscina, M. Resistance to paclitxel in breast carcinoma cells requires a quality control of mitochondrial antiapoptotic proteins by TRAP1. Mol. Oncol. 2013, 7, 895–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matassa, D.S.; Agliarulo, I.; Avolio, R.; Landriscina, M.; Esposito, F. TRAP1 Regulation of Cancer Metabolism: Dual Role as Oncogene or Tumor Suppressor. Genes 2018, 9, 195. [Google Scholar] [CrossRef]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Huang, Z.; Wei, P.; Liu, Y.; Hao, J.; Zhao, L.; Zhang, F.; Tu, Y.; Wei, T. Aberrantly upregulated TRAP1 is required for tumorigenesis of breast cancer. Oncotarget 2015, 6, 44495–44508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amoroso, M.R.; Matassa, D.S.; Laudiero, G.; Egorova, A.V.; Polishchuk, R.S.; Maddalena, F.; Piscazzi, A.; Paladino, S.; Sarnataro, D.; Garbi, C.; et al. TRAP1 and the proteasome regulatory particle TBP7/Rpt3 interact in the endoplasmic reticulum and control cellular ubiquitination of specific mitochondrial proteins. Cell Death Differ. 2012, 19, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Sisinni, L.; Maddalena, F.; Lettini, G.; Condelli, V.; Matassa, D.S.; Esposito, F.; Landriscina, M. TRAP1 role in endoplasmic reticulum stress protection favors resistance to anthracyclins in breast carcinoma cells. Int. J. Oncol. 2014, 44, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept inantiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Østergaard, L.; Tietze, A.; Nielsen, T.; Drasbek, K.R.; Mouridsen, K.; Jespersen, S.N.; Horsman, M.R. The relationship between tumor blood flow, angiogenesis, tumor hypoxia, and aerobic glycolysis. Cancer Res. 2013, 73, 5618–5624. [Google Scholar] [CrossRef]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Antiangiogenic Therapy: A Universal Chemosensitization Strategy for Cancer? Science 2006, 312, 1171–1175. [Google Scholar] [CrossRef]
- Lu, H.P.; Chao, C.C. Cancer cells acquire resistance to anticancer drugs: An update. Biomed. J. 2012, 35, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.; Min, H.; Leal, J.; Yu, D.; Rao, S.; You, E.; Tang, X.; Kim, H.; Meyer, S.; Han, S.J.; et al. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell 2004, 6, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Navarro, P.; Bueno, M.J.; Zagorac, I.; Mondejar, T.; Sanchez, J.; Mourón, S.; Muñoz, J.; Gómez-López, G.; Jimenez-Renard, V.; Mulero, F.; et al. Targeting Tumor Mitochondrial Metabolism Overcomes Resistance to Antiangiogenics. Cell Rep. 2016, 15, 2705–2718. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [Green Version]
- Arif, T.; Vasilkovsky, L.; Refaely, Y.; Konson, A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e159. [Google Scholar] [CrossRef]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers (Basel) 2018, 10, 499. [Google Scholar] [CrossRef]
- Daurio, N.A.; Tuttle, S.W.; Worth, A.J.; Song, E.Y.; Davis, J.M.; Snyder, N.W.; Blair, I.A.; Koumenis, C. AMPK activation and metabolic reprogramming by tamoxifen through estrogen receptor-independent mechanisms suggests new uses for this therapeutic modality in cancer treatment. Cancer Res. 2016, 76, 3295–3306. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Custódio, J.; Moreno, A.; Oliveira, C.R.; Santos, M.S. Tamoxifen and estradiol interact with the flavin mononucleotide site of complex I leading to mitochondrial failure. J. Biol. Chem. 2006, 281, 10143–10152. [Google Scholar] [CrossRef]
- Rohlenova, K.; Sachaphibulkij, K.; Stursa, J.; Bezawork-Geleta, A.; Blecha, J.; Endaya, B.; Werner, L.; Cerny, J.; Zobalova, R.; Goodwin, J.; et al. Selective Disruption of Respiratory Supercomplexes as a New Strategy to Suppress Her2high Breast Cancer. Antioxid. Redox Signal. 2017, 26, 84–103. [Google Scholar] [CrossRef]
- Albano, F.; Arcucci, A.; Granato, G.; Romano, S.; Montagnani, S.; De Vendittis, E.; Ruocco, M.R. Markers of mitochondrial dysfunction during the diclofenac-induced apoptosis in melanoma cell lines. Biochimie. 2013, 95, 934–945. [Google Scholar] [CrossRef]
- Gelzo, M.; Granato, G.; Albano, F.; Arcucci, A.; Dello Russo, A.; De Vendittis, E.; Ruocco, M.R.; Corso, G. Evaluation of cytotoxic effects of 7-dehydrocholesterol on melanoma cells. Free Radic. Biol. Med. 2014, 70, 129–140. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic targets | Agents or approaches | Effect on breast cancer cells | References |
|---|---|---|---|
| Arginine | ADI-PEG20 | Autophagy-dependent cell death | [132] |
| Pyruvate metabolism | DCA | Re-establishment of mitochondrial function and OXPHOS metabolism. mitochondrial dysfunction and apoptotic cell death | [134,135,136] |
| Pyruvate metabolism | DCA in combination with PDT | ICD | [139] |
| SLC6A14 transporter | α-MT | Inhibition of amino acid transporter. Apoptosis and autophagy | [157] |
| VDAC1 | VDAC1 silencing | Cell growth arrest and tumour growth inhibition | [176] |
| Glutaminase | CB-839 Compound 968 BPTES | Inhibition of glutamine metabolism. Apoptotic cell death | [155,158,159] |
| FASN | FASN inhibition | Apoptotic cell death | [160] |
| OXPHOS | TAM Biguanides (Metformin, Phenformin) | Inhibition of Complex I of ETC and cytotoxicity. Increased glycolysis and pharmacological resistance induction. | [11,145,146,147,148,152,153,173,177] |
| OXPHOS | MitoTam | Inhibition of Complex I of ETC. Cancer cell death. | [179] |
| OXPHOS | ME344 | Tumour growth inhibition | [173] |
| OXPHOS | FR58P1a | Metastatic capability reduction | [35] |
| OXPHOS and glycolysis | MGT | Anti-proliferative, anti-oxidant, and chemopreventive role | [140,141] |
| OXPHOS and glycolysis | TAM in combination with glycolysis inhibitor | Increased anti-tumour effect | [177] |
| OXPHOS and glycolysis | Metformin in combination with glycolysis inhibitor | Increased anti-tumour effect | [154] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avagliano, A.; Ruocco, M.R.; Aliotta, F.; Belviso, I.; Accurso, A.; Masone, S.; Montagnani, S.; Arcucci, A. Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells 2019, 8, 401. https://doi.org/10.3390/cells8050401
Avagliano A, Ruocco MR, Aliotta F, Belviso I, Accurso A, Masone S, Montagnani S, Arcucci A. Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells. 2019; 8(5):401. https://doi.org/10.3390/cells8050401
Chicago/Turabian StyleAvagliano, Angelica, Maria Rosaria Ruocco, Federica Aliotta, Immacolata Belviso, Antonello Accurso, Stefania Masone, Stefania Montagnani, and Alessandro Arcucci. 2019. "Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity" Cells 8, no. 5: 401. https://doi.org/10.3390/cells8050401
APA StyleAvagliano, A., Ruocco, M. R., Aliotta, F., Belviso, I., Accurso, A., Masone, S., Montagnani, S., & Arcucci, A. (2019). Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells, 8(5), 401. https://doi.org/10.3390/cells8050401