Intranasal Administration of Mesenchymal Stem Cells Ameliorates the Abnormal Dopamine Transmission System and Inflammatory Reaction in the R6/2 Mouse Model of Huntington Disease

 ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Isolation, Cultivation and Characterization of MSC in Vitro
2.2. HD Animals
2.3. Intranasal Cell Transplantation
2.4. Rotarod Test
2.5. Locomotor Activities and Food Intake
2.6. Quantitative PCR
2.7. Immunohistochemical Staining and Immunofluorescence Staining
2.8. Quantification of Striatal Area
2.9. Western Blotting Analysis
2.10. Statistical Analysis
3. Results
3.1. Cell Characterization in Vitro
3.2. Cell Tracking in the Brain after Intranasal Administration
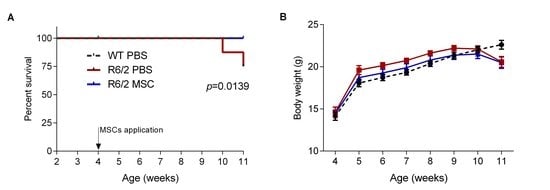
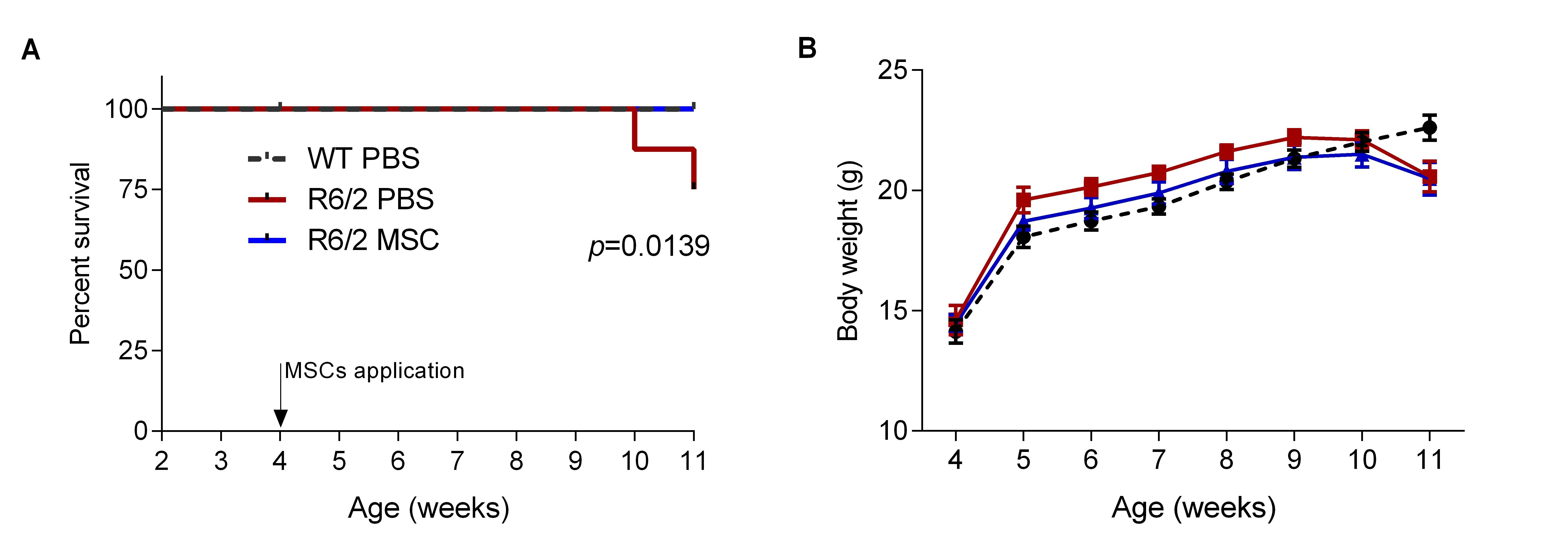
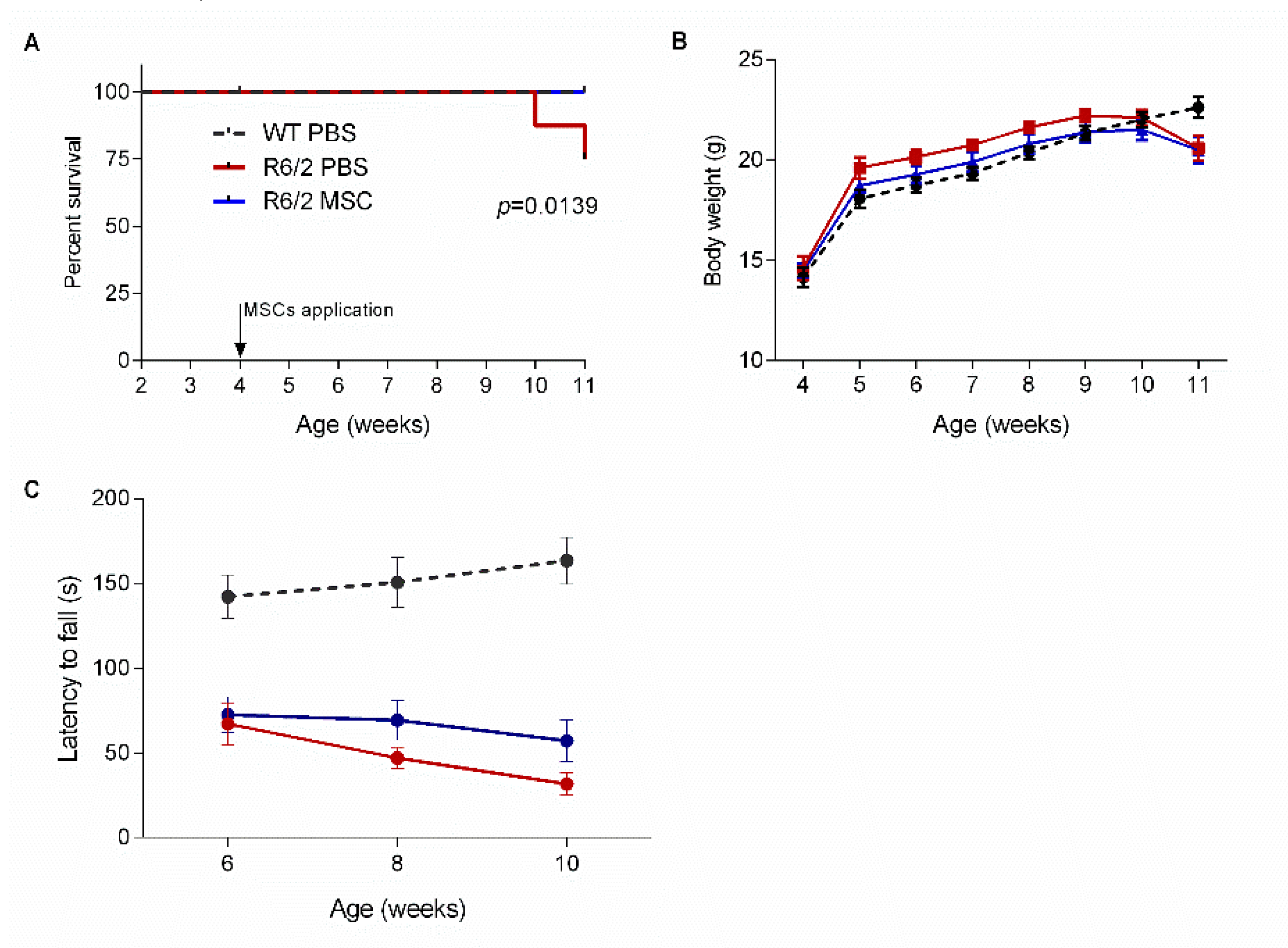
3.3. Intranasal Administration of MSCs Prolonged Survival of R6/2 Mice with Potentially Improved Motor Function
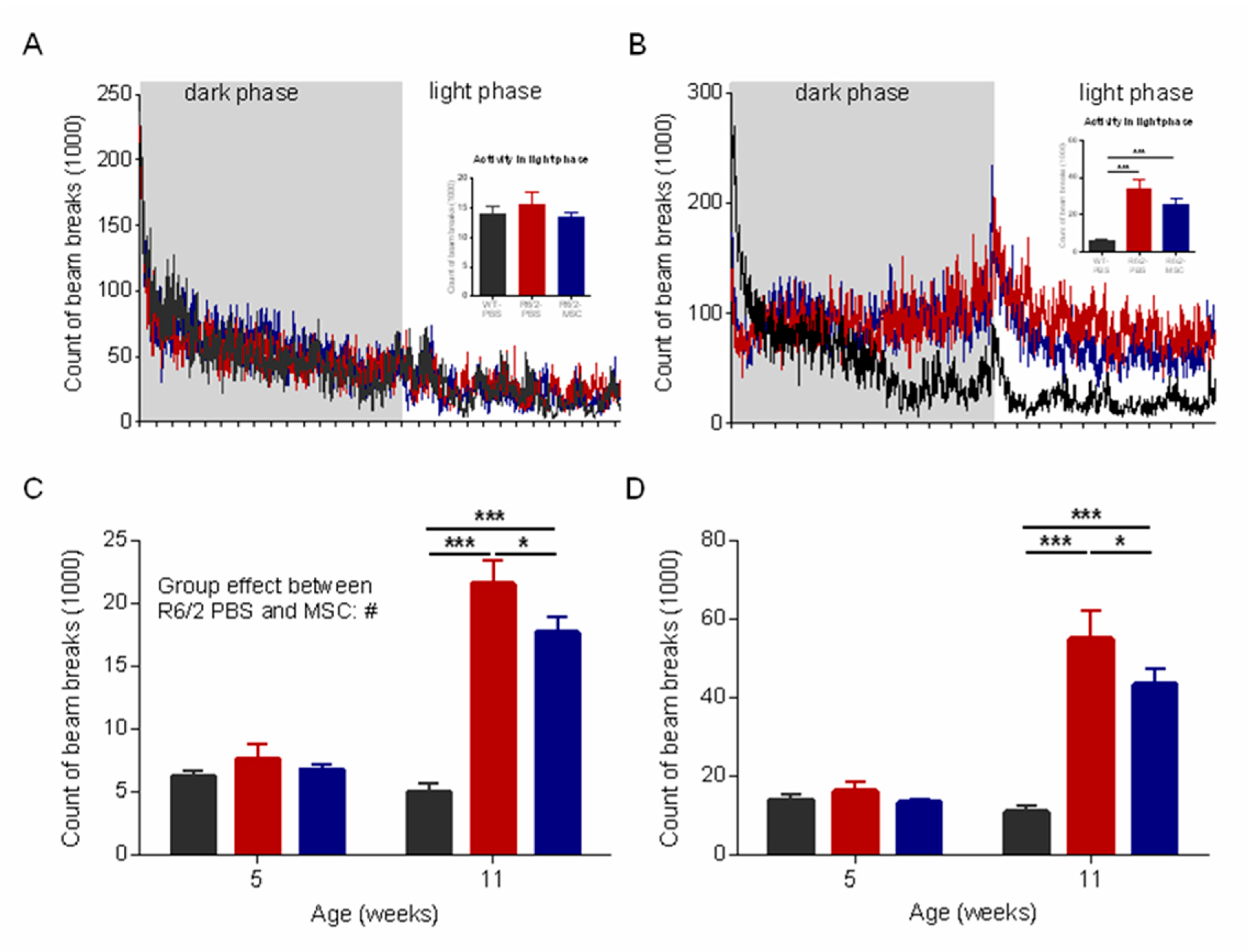
3.4. Ameliorated Circadian Rhythm in the MSC-Treated R6/2 Mice
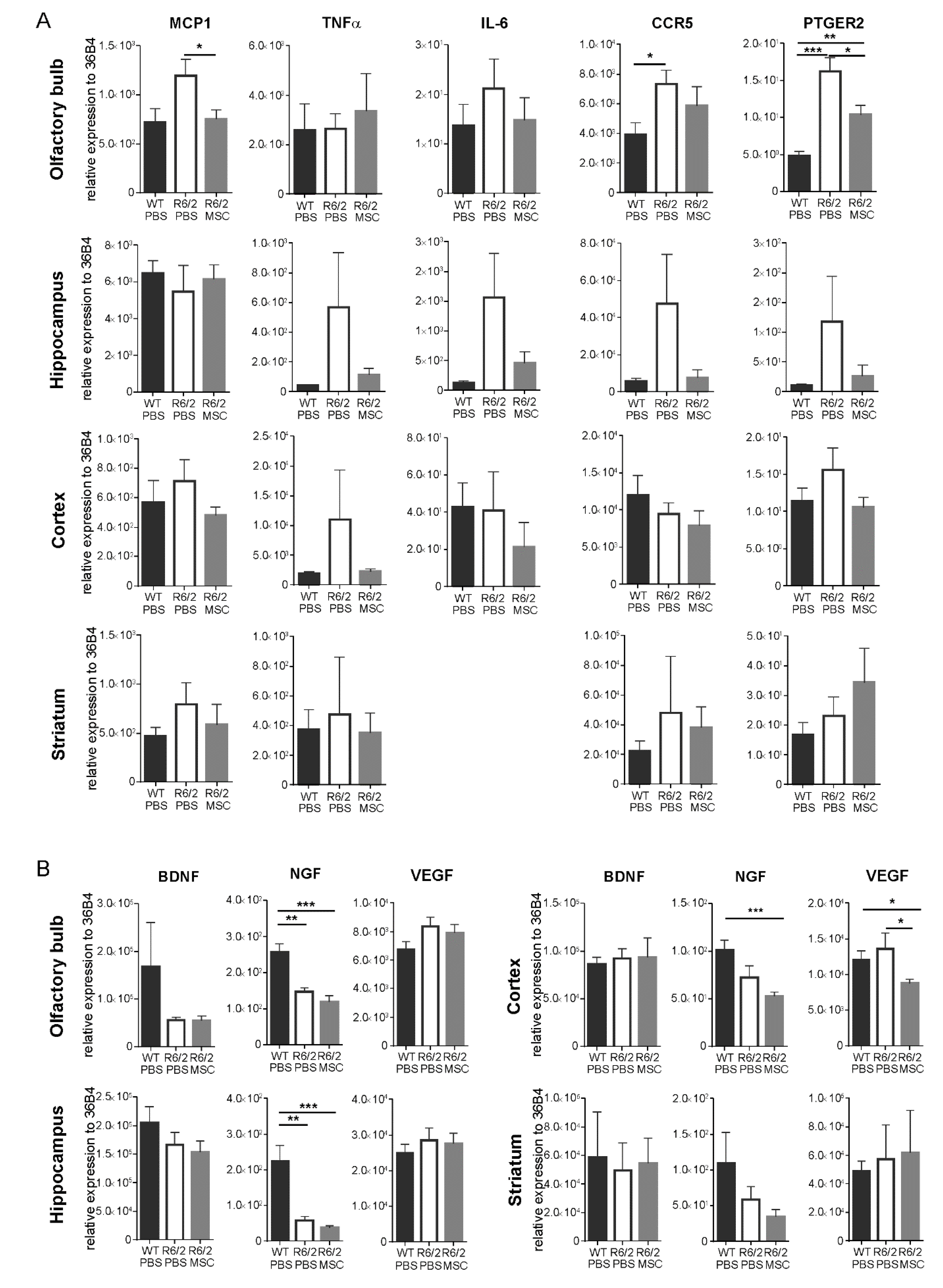
3.5. Gene Expression Profiles of Inflammatory Regulators and Neurotrophic Factors
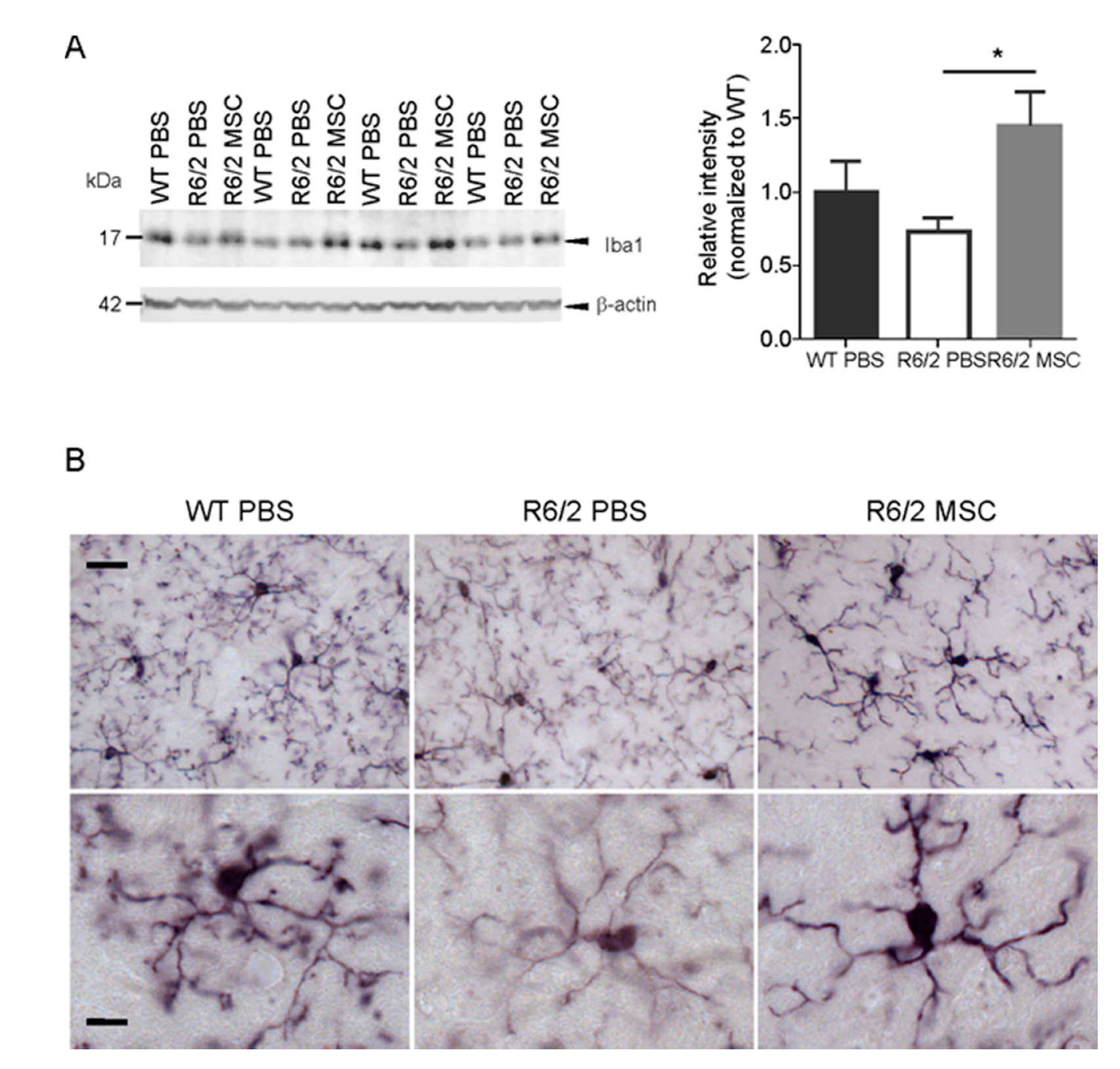
3.6. Microglial Changes in MSC-Treated R6/2 Mice
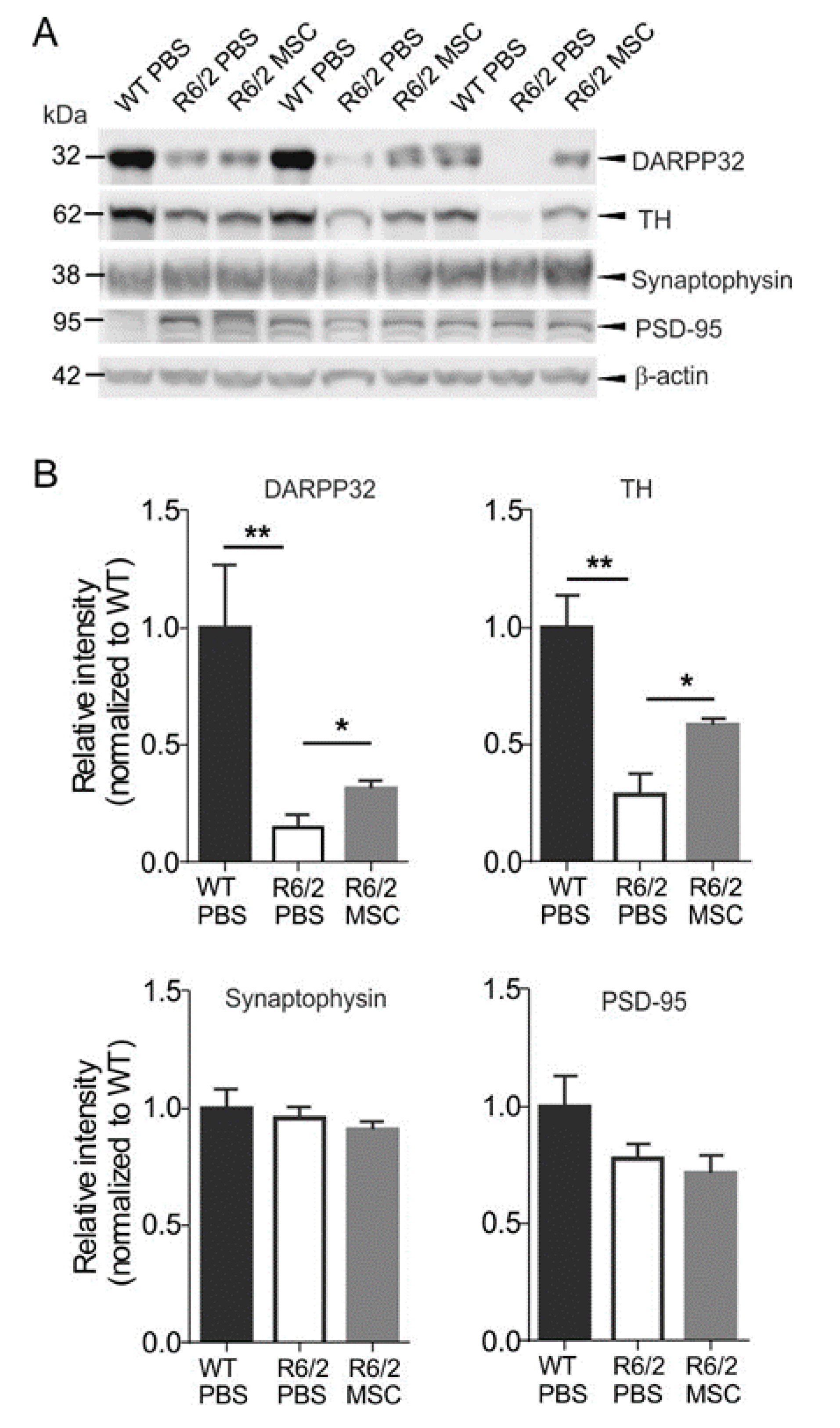
3.7. Neuropathological Changes in MSC-Treated R6/2 Mice
4. Discussion
4.1. Migration Pattern and Survival of Intranasally Delivered MSCs in the Brain
4.2. Increased TH and DARPP-32 Expressions and Attenuated Circadian Rhythm Disturbances Indicate An Amelioration of Dopamine Signaling in MSC-Treated Mice
4.3. Intranasal Administration of MSCs Reduced Neuroinflammation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Schapira, A.H.; Olanow, C.W.; Greenamyre, J.T.; Bezard, E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: Future therapeutic perspectives. Lancet 2014, 384, 545–555. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Albin, R.L.; Tagle, D.A. Genetics and molecular biology of Huntington’s disease. Trends Neurosci. 1995, 18, 11–14. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Kordower, J.H. Gene therapy for Huntington’s disease. Neurobiol. Dis. 2012, 48, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.L.; Barker, R.A. Advancing pharmacotherapy for treating Huntington’s disease: A review of the existing literature. Expert Opin Pharm. 2016, 17, 41–52. [Google Scholar] [CrossRef]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s disease collaborative research group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Rodda, R.A. Cerebellar atrophy in Huntington’s disease. J. Neurol. Sci. 1981, 50, 147–157. [Google Scholar] [CrossRef]
- Zielonka, D.; Mielcarek, M.; Landwehrmeyer, G.B. Update on Huntington’s disease: Advances in care and emerging therapeutic options. Parkinsonism Relat. Disord. 2015, 21, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Frank, S. Treatment of Huntington’s disease. Neurother. J. Am. Soc. Exp. Neurother. 2014, 11, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Handley, O.J.; Naji, J.J.; Dunnett, S.B.; Rosser, A.E. Pharmaceutical, cellular and genetic therapies for Huntington’s disease. Clin. Sci. 2006, 110, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lv, H.; Huang, S.; Tan, H.; Zhang, Y.; Li, H. Bone marrow mesenchymal stem cells can improve the motor function of a Huntington’s disease rat model. Neurol. Res. 2011, 33, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Aleynik, A.; Gernavage, K.M.; Mourad, Y.; Sherman, L.S.; Liu, K.; Gubenko, Y.A.; Rameshwar, P. Stem cell delivery of therapies for brain disorders. Clin. Transl. Med. 2014, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Herberts, C.A.; Kwa, M.S.; Hermsen, H.P. Risk factors in the development of stem cell therapy. J. Transl. Med. 2011, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Tanna, T.; Sachan, V. Mesenchymal stem cells: Potential in treatment of neurodegenerative diseases. Curr. Stem Cell Res. Ther. 2014, 9, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Chern, Y.; Shen, C.K.; Wen, H.L.; Chang, Y.C.; Li, H.; Cheng, T.H.; Hsieh-Li, H.M. Human mesenchymal stem cells prolong survival and ameliorate motor deficit through trophic support in Huntington’s disease mouse models. PLoS ONE 2011, 6, e22924. [Google Scholar] [CrossRef]
- Rossignol, J.; Boyer, C.; Leveque, X.; Fink, K.D.; Thinard, R.; Blanchard, F.; Dunbar, G.L.; Lescaudron, L. Mesenchymal stem cell transplantation and DMEM administration in a 3np rat model of Huntington’s disease: Morphological and behavioral outcomes. Behav. Brain Res. 2011, 217, 369–378. [Google Scholar] [CrossRef]
- Pollock, K.; Dahlenburg, H.; Nelson, H.; Fink, K.D.; Cary, W.; Hendrix, K.; Annett, G.; Torrest, A.; Deng, P.; Gutierrez, J.; et al. Human mesenchymal stem cells genetically engineered to overexpress brain-derived neurotrophic factor improve outcomes in Huntington’s disease mouse models. Mol. Ther. 2016, 24, 965–977. [Google Scholar] [CrossRef]
- Fink, K.D.; Rossignol, J.; Crane, A.T.; Davis, K.K.; Bombard, M.C.; Bavar, A.M.; Clerc, S.; Lowrance, S.A.; Song, C.; Lescaudron, L.; et al. Transplantation of umbilical cord-derived mesenchymal stem cells into the striata of r6/2 mice: Behavioral and neuropathological analysis. Stem Cell Res. Ther. 2013, 4, 130. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, F.G.; Carvalho, M.M.; Panchalingam, K.M.; Rodrigues, A.J.; Mendes-Pinheiro, B.; Anjo, S.; Manadas, B.; Behie, L.A.; Sousa, N.; Salgado, A.J. Impact of the secretome of human mesenchymal stem cells on brain structure and animal behavior in a rat model of Parkinson’s disease. Stem Cells Transl. Med. 2017, 6, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Sadan, O.; Bahat-Stromza, M.; Barhum, Y.; Levy, Y.S.; Pisnevsky, A.; Peretz, H.; Ilan, A.B.; Bulvik, S.; Shemesh, N.; Krepel, D.; et al. Protective effects of neurotrophic factor-secreting cells in a 6-OHDA rat model of Parkinson disease. Stem Cells Dev. 2009, 18, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.S.; Bahat-Stroomza, M.; Barzilay, R.; Burshtein, A.; Bulvik, S.; Barhum, Y.; Panet, H.; Melamed, E.; Offen, D. Regenerative effect of neural-induced human mesenchymal stromal cells in rat models of Parkinson’s disease. Cytotherapy 2008, 10, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Cova, L.; Armentero, M.T.; Zennaro, E.; Calzarossa, C.; Bossolasco, P.; Busca, G.; Lambertenghi Deliliers, G.; Polli, E.; Nappi, G.; Silani, V.; et al. Multiple neurogenic and neurorescue effects of human mesenchymal stem cell after transplantation in an experimental model of Parkinson’s disease. Brain Res. 2010, 1311, 12–27. [Google Scholar] [CrossRef]
- Blandini, F.; Cova, L.; Armentero, M.T.; Zennaro, E.; Levandis, G.; Bossolasco, P.; Calzarossa, C.; Mellone, M.; Giuseppe, B.; Deliliers, G.L.; et al. Transplantation of undifferentiated human mesenchymal stem cells protects against 6-hydroxydopamine neurotoxicity in the rat. Cell Transplant. 2010, 19, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Ma, J.; Yu, L.; Yuan, C. Grafted neural stem cells migrate to substantia nigra and improve behavior in Parkinsonian rats. Neurosci. Lett. 2009, 462, 213–218. [Google Scholar] [CrossRef]
- Bae, J.S.; Jin, H.K.; Lee, J.K.; Richardson, J.C.; Carter, J.E. Bone marrow-derived mesenchymal stem cells contribute to the reduction of amyloid-beta deposits and the improvement of synaptic transmission in a mouse model of pre-dementia Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 524–531. [Google Scholar] [CrossRef]
- Yun, H.M.; Kim, H.S.; Park, K.R.; Shin, J.M.; Kang, A.R.; Lee, K.i.; Song, S.; Kim, Y.B.; Han, S.B.; Chung, H.M.; et al. Placenta-derived mesenchymal stem cells improve memory dysfunction in an abeta1-42-infused mouse model of Alzheimer’s disease. Cell Death Dis. 2013, 4, e958. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.A.; Kim, H.J.; Joo, Y.; Ha, S.; Suh, Y.H. The therapeutic effects of human adipose-derived stem cells in Alzheimer’s disease mouse models. Neuro-Degener. Dis. 2014, 13, 99–102. [Google Scholar] [CrossRef]
- Lee, J.K.; Jin, H.K.; Bae, J.S. Bone marrow-derived mesenchymal stem cells reduce brain amyloid-beta deposition and accelerate the activation of microglia in an acutely induced Alzheimer’s disease mouse model. Neurosci. Lett. 2009, 450, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Babaei, P.; Soltani Tehrani, B.; Alizadeh, A. Transplanted bone marrow mesenchymal stem cells improve memory in rat models of Alzheimer’s disease. Stem Cells Int. 2012, 2012, 369417. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Park, H.J.; Kim, H.N.; Oh, S.H.; Bae, J.S.; Ha, H.J.; Lee, P.H. Mesenchymal stem cells enhance autophagy and increase beta-amyloid clearance in Alzheimer disease models. Autophagy 2014, 10, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jin, H.K.; Endo, S.; Schuchman, E.H.; Carter, J.E.; Bae, J.S. Intracerebral transplantation of bone marrow-derived mesenchymal stem cells reduces amyloid-beta deposition and rescues memory deficits in Alzheimer’s disease mice by modulation of immune responses. Stem Cells 2010, 28, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, Z.; Wei, L.; Yang, H.; Yang, S.; Zhu, Z.; Wang, P.; Zhao, C.; Bi, J. Human umbilical cord mesenchymal stem cell-derived neuron-like cells rescue memory deficits and reduce amyloid-beta deposition in an AbetaPP/PS1 transgenic mouse model. Stem Cell Res. Ther. 2013, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Gong, K.; Ao, Q.; Yan, Y.; Song, B.; Huang, H.; Zhang, X.; Gong, Y. Intracerebral transplantation of adipose-derived mesenchymal stem cells alternatively activates microglia and ameliorates neuropathological deficits in Alzheimer’s disease mice. Cell Transplant. 2013, 22 (Suppl. 1), S113–S126. [Google Scholar] [CrossRef]
- Favre, J.; Taha, J.M.; Burchiel, K.J. An analysis of the respective risks of hematoma formation in 361 consecutive morphological and functional stereotactic procedures. Neurosurgery 2002, 50, 48–57. [Google Scholar] [PubMed]
- Sansur, C.A.; Frysinger, R.C.; Pouratian, N.; Fu, K.M.; Bittl, M.; Oskouian, R.J.; Laws, E.R.; Elias, W.J. Incidence of symptomatic hemorrhage after stereotactic electrode placement. J. Neurosurg. 2007, 107, 998–1003. [Google Scholar] [CrossRef]
- Frey, W.H., II. Method of Administering Neurologic Agents to the Brain. U.S. Patent 5,624,898, 29 April 1997. [Google Scholar]
- Donega, V.; Nijboer, C.H.; van Velthoven, C.T.; Youssef, S.A.; de Bruin, A.; van Bel, F.; Kavelaars, A.; Heijnen, C.J. Assessment of long-term safety and efficacy of intranasal mesenchymal stem cell treatment for neonatal brain injury in the mouse. Pediatr. Res. 2015, 78, 520–526. [Google Scholar] [CrossRef]
- Danielyan, L.; Schafer, R.; von Ameln-Mayerhofer, A.; Buadze, M.; Geisler, J.; Klopfer, T.; Burkhardt, U.; Proksch, B.; Verleysdonk, S.; Ayturan, M.; et al. Intranasal delivery of cells to the brain. Eur. J. Cell Biol. 2009, 88, 315–324. [Google Scholar] [CrossRef]
- Galeano, C.; Qiu, Z.; Mishra, A.; Farnsworth, S.L.; Hemmi, J.J.; Moreira, A.; Edenhoffer, P.; Hornsby, P.J. The route by which intranasally delivered stem cells enter the central nervous system. Cell Transplant. 2018, 27, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Danielyan, L.; Schafer, R.; von Ameln-Mayerhofer, A.; Bernhard, F.; Verleysdonk, S.; Buadze, M.; Lourhmati, A.; Klopfer, T.; Schaumann, F.; Schmid, B.; et al. Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson disease. Rejuvenation Res. 2011, 14, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Salama, M.; Sobh, M.; Emam, M.; Abdalla, A.; Sabry, D.; El-Gamal, M.; Lotfy, A.; El-Husseiny, M.; Sobh, M.; Shalash, A.; et al. Effect of intranasal stem cell administration on the nigrostriatal system in a mouse model of Parkinson’s disease. Exp. Ther. Med. 2017, 13, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, K.; Iwatsuki, K.; Ohnishi, Y.; Ohkawa, T.; Yoshimine, T. Intranasal delivery of bone marrow stromal cells to spinal cord lesions. J. Neurosurg. Spine 2015, 23, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Mahal, A.; Mott, R.; Seller, M.; Bates, G.P. Instability of highly expanded cag repeats in mice transgenic for the Huntington’s disease mutation. Nat. Genet. 1997, 15, 197–200. [Google Scholar] [CrossRef]
- Sathasivam, K.; Hobbs, C.; Mangiarini, L.; Mahal, A.; Turmaine, M.; Doherty, P.; Davies, S.W.; Bates, G.P. Transgenic models of Huntington’s disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 963–969. [Google Scholar] [CrossRef]
- Stricker-Shaver, J.; Novati, A.; Yu-Taeger, L.; Nguyen, H.P. Genetic rodent models of Huntington disease. In Polyglutamine Disorders; Nóbrega, C., Pereira de Almeida, L., Eds.; Springer: Cham, Switzerland, 2018; Volume 1049, pp. 29–57. [Google Scholar]
- Carter, R.J.; Lione, L.A.; Humby, T.; Mangiarini, L.; Mahal, A.; Bates, G.P.; Dunnett, S.B.; Morton, A.J. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J. Neurosci. J. Soc. Neurosci. 1999, 19, 3248–3257. [Google Scholar] [CrossRef]
- Stack, E.C.; Kubilus, J.K.; Smith, K.; Cormier, K.; Del Signore, S.J.; Guelin, E.; Ryu, H.; Hersch, S.M.; Ferrante, R.J. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington’s disease transgenic mice. J. Comp. Neurol. 2005, 490, 354–370. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Lione, L.A.; Carter, R.J.; Hunt, M.J.; Bates, G.P.; Morton, A.J.; Dunnett, S.B. Selective discrimination learning impairments in mice expressing the human Huntington’s disease mutation. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 10428–10437. [Google Scholar] [CrossRef]
- Bolivar, V.J.; Manley, K.; Messer, A. Exploratory activity and fear conditioning abnormalities develop early in r6/2 Huntington’s disease transgenic mice. Behav. Neurosci. 2003, 117, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Pla, P.; Orvoen, S.; Saudou, F.; David, D.J.; Humbert, S. Mood disorders in Huntington’s disease: From behavior to cellular and molecular mechanisms. Front. Behav. Neurosci. 2014, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.A.; Gallant, K.; Gross, G.G.; Levine, M.S.; Chesselet, M.F. Early behavioral deficits in r6/2 mice suitable for use in preclinical drug testing. Neurobiol. Dis. 2005, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maywood, E.S.; Fraenkel, E.; McAllister, C.J.; Wood, N.; Reddy, A.B.; Hastings, M.H.; Morton, A.J. Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J. Neurosci. J. Soc. Neurosci. 2010, 30, 10199–10204. [Google Scholar] [CrossRef] [PubMed]
- Luthi-Carter, R.; Hanson, S.A.; Strand, A.D.; Bergstrom, D.A.; Chun, W.; Peters, N.L.; Woods, A.M.; Chan, E.Y.; Kooperberg, C.; Krainc, D.; et al. Dysregulation of gene expression in the r6/2 model of polyglutamine disease: Parallel changes in muscle and brain. Hum. Mol. Genet. 2002, 11, 1911–1926. [Google Scholar] [CrossRef]
- Dalrymple, A.; Wild, E.J.; Joubert, R.; Sathasivam, K.; Bjorkqvist, M.; Petersen, A.; Jackson, G.S.; Isaacs, J.D.; Kristiansen, M.; Bates, G.P.; et al. Proteomic profiling of plasma in Huntington’s disease reveals neuroinflammatory activation and biomarker candidates. J. Proteome Res. 2007, 6, 2833–2840. [Google Scholar] [CrossRef]
- Silvestroni, A.; Faull, R.L.; Strand, A.D.; Moller, T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport 2009, 20, 1098–1103. [Google Scholar] [CrossRef]
- Moller, T. Neuroinflammation in Huntington’s disease. J. Neural Transm. 2010, 117, 1001–1008. [Google Scholar] [CrossRef]
- Hsiao, H.Y.; Chiu, F.L.; Chen, C.M.; Wu, Y.R.; Chen, H.M.; Chen, Y.C.; Kuo, H.C.; Chern, Y. Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 2014, 23, 4328–4344. [Google Scholar] [CrossRef]
- Yang, H.M.; Yang, S.; Huang, S.S.; Tang, B.S.; Guo, J.F. Microglial activation in the pathogenesis of Huntington’s disease. Front. Aging Neurosci. 2017, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Singhrao, S.K.; Neal, J.W.; Morgan, B.P.; Gasque, P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp. Neurol. 1999, 159, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Sapp, E.; Kegel, K.B.; Aronin, N.; Hashikawa, T.; Uchiyama, Y.; Tohyama, K.; Bhide, P.G.; Vonsattel, J.P.; DiFiglia, M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 2001, 60, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Glennie, S.; Soeiro, I.; Dyson, P.J.; Lam, E.W.; Dazzi, F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood 2005, 105, 2821–2827. [Google Scholar] [CrossRef] [PubMed]
- Jouhou, H.; Yamamoto, K.; Homma, A.; Hara, M.; Kaneko, A.; Yamada, M. Depolarization of isolated horizontal cells of fish acidifies their immediate surrounding by activating V-ATPase. J. Physiol. 2007, 585, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Corcione, A.; Benvenuto, F.; Ferretti, E.; Giunti, D.; Cappiello, V.; Cazzanti, F.; Risso, M.; Gualandi, F.; Mancardi, G.L.; Pistoia, V.; et al. Human mesenchymal stem cells modulate B-cell functions. Blood 2006, 107, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Burchell, J.T.; Strickland, D.H.; Stumbles, P.A. The role of dendritic cells and regulatory t cells in the regulation of allergic asthma. Pharmacol. Ther. 2010, 125, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Capobianco, A.; Becchetti, S.; Mingari, M.C.; Moretta, L. Mesenchymal stem cell-natural killer cell interactions: Evidence that activated NK cells are capable of killing mscs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood 2006, 107, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Meyerrose, T.; Olson, S.; Pontow, S.; Kalomoiris, S.; Jung, Y.; Annett, G.; Bauer, G.; Nolta, J.A. Mesenchymal stem cells for the sustained in vivo delivery of bioactive factors. Adv. Drug Deliv. Rev. 2010, 62, 1167–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, A.; Kemp, K.; Ginty, M.; Hares, K.; Mallam, E.; Scolding, N. Human bone marrow-derived mesenchymal stem cells secrete brain-derived neurotrophic factor which promotes neuronal survival in vitro. Stem Cell Res. 2009, 3, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, W.; Trager, U.; Davalos, D.; Chou, A.; Bouchard, J.; Andre, R.; Miller, A.; Weiss, A.; Giorgini, F.; Cheah, C.; et al. Mutant huntingtin impairs immune cell migration in Huntington disease. J. Clin. Investig. 2012, 122, 4737–4747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpaugh, M.; Galleguillos, D.; Forero, J.; Morales, L.C.; Lackey, S.W.; Kar, P.; Di Pardo, A.; Holt, A.; Kerr, B.J.; Todd, K.G.; et al. Disease-modifying effects of ganglioside GM1 in Huntington’s disease models. EMBO Mol. Med. 2017, 9, 1537–1557. [Google Scholar] [CrossRef] [PubMed]
- Bibb, J.A.; Yan, Z.; Svenningsson, P.; Snyder, G.L.; Pieribone, V.A.; Horiuchi, A.; Nairn, A.C.; Messer, A.; Greengard, P. Severe deficiencies in dopamine signaling in presymptomatic Huntington’s disease mice. Proc. Natl. Acad. Sci. USA 2000, 97, 6809–6814. [Google Scholar] [CrossRef] [PubMed]
- Callahan, J.W.; Abercrombie, E.D. In vivo dopamine efflux is decreased in striatum of both fragment (R6/2) and full-length (YAC128) transgenic mouse models of Huntington’s disease. Front. Syst. Neurosci. 2011, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Kosinski, C.M.; Kerner, J.A.; Alsdorf, S.A.; Mangiarini, L.; Davies, S.W.; Penney, J.B.; Bates, G.P.; Young, A.B. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc. Natl. Acad. Sci. USA 1998, 95, 6480–6485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, M.; Schimmelpfennig, C.; Stolzing, A. Influence of murine mesenchymal stem cells on proliferation, phenotype, vitality, and cytotoxicity of murine cytokine-induced killer cells in coculture. PLoS ONE 2014, 9, e88115. [Google Scholar] [CrossRef] [PubMed]
- Hockly, E.; Woodman, B.; Mahal, A.; Lewis, C.M.; Bates, G. Standardization and statistical approaches to therapeutic trials in the r6/2 mouse. Brain Res. Bull. 2003, 61, 469–479. [Google Scholar] [CrossRef]
- Naaldijk, Y.; Jager, C.; Fabian, C.; Leovsky, C.; Bluher, A.; Rudolph, L.; Hinze, A.; Stolzing, A. Effect of systemic transplantation of bone marrow-derived mesenchymal stem cells on neuropathology markers in APP/PS1 Alzheimer mice. Neuropathol. Appl. Neurobiol. 2017, 43, 299–314. [Google Scholar] [CrossRef]
- Menalled, L.; El-Khodor, B.F.; Patry, M.; Suarez-Farinas, M.; Orenstein, S.J.; Zahasky, B.; Leahy, C.; Wheeler, V.; Yang, X.W.; MacDonald, M.; et al. Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol. Dis. 2009, 35, 319–336. [Google Scholar] [CrossRef]
- Goodman, A.O.; Morton, A.J.; Barker, R.A. Identifying sleep disturbances in Huntington’s disease using a simple disease-focused questionnaire. PLoS Curr. 2010, 2, RRN1189. [Google Scholar] [CrossRef]
- Morton, A.J.; Wood, N.I.; Hastings, M.H.; Hurelbrink, C.; Barker, R.A.; Maywood, E.S. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci. J. Soc. Neurosci. 2005, 25, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, M.; Aungier, J.; Morton, A.J. The methamphetamine-sensitive circadian oscillator is dysfunctional in a transgenic mouse model of Huntington’s disease. Neurobiol. Dis. 2012, 45, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Morton, A.J.; Nicholson, L.F. Microglia density decreases with age in a mouse model of Huntington’s disease. Glia 2003, 43, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Spires, T.L.; Grote, H.E.; Varshney, N.K.; Cordery, P.M.; van Dellen, A.; Blakemore, C.; Hannan, A.J. Environmental enrichment rescues protein deficits in a mouse model of Huntington’s disease, indicating a possible disease mechanism. J. Neurosci. J. Soc. Neurosci. 2004, 24, 2270–2276. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Carreton, O.; Lao-Peregrin, C.; Martin, E.D.; Alberch, J. Conditional BDNF release under pathological conditions improves Huntington’s disease pathology by delaying neuronal dysfunction. Mol. Neurodegener. 2011, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Ellrichmann, G.; Blusch, A.; Fatoba, O.; Brunner, J.; Hayardeny, L.; Hayden, M.; Sehr, D.; Winklhofer, K.F.; Saft, C.; Gold, R. Laquinimod treatment in the R6/2 mouse model. Sci. Rep. 2017, 7, 4947. [Google Scholar] [CrossRef]
- Yohrling, G.J.t.; Jiang, G.C.; DeJohn, M.M.; Miller, D.W.; Young, A.B.; Vrana, K.E.; Cha, J.H. Analysis of cellular, transgenic and human models of Huntington’s disease reveals tyrosine hydroxylase alterations and substantia nigra neuropathology. Brain Res. Mol. Brain Res. 2003, 119, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Jung, K.H.; Im, W.S.; Park, J.E.; Lim, H.C.; Won, C.H.; Shin, S.H.; Lee, S.K.; Kim, M.; et al. Slowed progression in models of Huntington disease by adipose stem cell transplantation. Ann. Neurol. 2009, 66, 671–681. [Google Scholar] [CrossRef]
- Linares, G.R.; Chiu, C.T.; Scheuing, L.; Leng, Y.; Liao, H.M.; Maric, D.; Chuang, D.M. Preconditioning mesenchymal stem cells with the mood stabilizers lithium and valproic acid enhances therapeutic efficacy in a mouse model of Huntington’s disease. Exp. Neurol. 2016, 281, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Danielyan, L.; Beer-Hammer, S.; Stolzing, A.; Schafer, R.; Siegel, G.; Fabian, C.; Kahle, P.; Biedermann, T.; Lourhmati, A.; Buadze, M.; et al. Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer’s and Parkinson’s disease. Cell Transplant. 2014, 23 (Suppl. 1), S123–S139. [Google Scholar] [CrossRef]
- Fransson, M.; Piras, E.; Wang, H.; Burman, J.; Duprez, I.; Harris, R.A.; LeBlanc, K.; Magnusson, P.U.; Brittebo, E.; Loskog, A.S. Intranasal delivery of central nervous system-retargeted human mesenchymal stromal cells prolongs treatment efficacy of experimental autoimmune encephalomyelitis. Immunology 2014, 142, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Grassin-Delyle, S.; Buenestado, A.; Naline, E.; Faisy, C.; Blouquit-Laye, S.; Couderc, L.J.; Le Guen, M.; Fischler, M.; Devillier, P. Intranasal drug delivery: An efficient and non-invasive route for systemic administration: Focus on opioids. Pharmacol. Ther. 2012, 134, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Mishra, G.; Sharma, A.K.; Gothwal, A.; Kesharwani, P.; Gupta, U. Intranasal drug delivery: A non-invasive approach for the better delivery of neurotherapeutics. Pharm. Nanotechnol. 2017, 5, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef]
- Mahmood, A.; Lu, D.; Qu, C.; Goussev, A.; Chopp, M. Long-term recovery after bone marrow stromal cell treatment of traumatic brain injury in rats. J. Neurosurg. 2006, 104, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Donega, V.; van Velthoven, C.T.; Nijboer, C.H.; van Bel, F.; Kas, M.J.; Kavelaars, A.; Heijnen, C.J. Intranasal mesenchymal stem cell treatment for neonatal brain damage: Long-term cognitive and sensorimotor improvement. PLoS ONE 2013, 8, e51253. [Google Scholar] [CrossRef]
- Liang, X.; Ding, Y.; Zhang, Y.; Tse, H.F.; Lian, Q. Paracrine mechanisms of mesenchymal stem cell-based therapy: Current status and perspectives. Cell Transplant. 2014, 23, 1045–1059. [Google Scholar] [CrossRef]
- Tsai, M.J.; Tsai, S.K.; Hu, B.R.; Liou, D.Y.; Huang, S.L.; Huang, M.C.; Huang, W.C.; Cheng, H.; Huang, S.S. Recovery of neurological function of ischemic stroke by application of conditioned medium of bone marrow mesenchymal stem cells derived from normal and cerebral ischemia rats. J. Biomed. Sci. 2014, 21, 5. [Google Scholar] [CrossRef]
- Thomi, G.; Surbek, D.; Haesler, V.; Joerger-Messerli, M.; Schoeberlein, A. Exosomes derived from umbilical cord mesenchymal stem cells reduce microglia-mediated neuroinflammation in perinatal brain injury. Stem Cell Res. Ther. 2019, 10, 105. [Google Scholar] [CrossRef]
- Long, Q.; Upadhya, D.; Hattiangady, B.; Kim, D.K.; An, S.Y.; Shuai, B.; Prockop, D.J.; Shetty, A.K. Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus. Proc. Natl. Acad. Sci. USA 2017, 114, E3536–E3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Heo, J.; Chun, J.Y.; Bae, H.S.; Kang, J.W.; Kang, H.; Cho, Y.M.; Kim, S.W.; Shin, D.M.; Choo, M.S. The paracrine effects of mesenchymal stem cells stimulate the regeneration capacity of endogenous stem cells in the repair of a bladder-outlet-obstruction-induced overactive bladder. Stem Cells Dev. 2014, 23, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Petersen, A.; Leist, M.; Nicotera, P.; Castilho, R.F.; Brundin, P. Transgenic mice expressing a Huntington’s disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc. Natl. Acad. Sci. USA 1999, 96, 8727–8732. [Google Scholar] [CrossRef] [PubMed]
- Nishi, A.; Snyder, G.L.; Greengard, P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J. Neurosci. J. Soc. Neurosci. 1997, 17, 8147–8155. [Google Scholar] [CrossRef]
- Meijer, J.H.; Rietveld, W.J. Neurophysiology of the suprachiasmatic circadian pacemaker in rodents. Physiol. Rev. 1989, 69, 671–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.D. On the nature of the circadian clock in mammals. Am. J. Physiol. 1993, 264, R821–R832. [Google Scholar] [CrossRef] [PubMed]
- Rietveld, W.J. Neurotransmitters and the pharmacology of the suprachiasmatic nuclei. Pharmacol. Ther. 1992, 56, 119–130. [Google Scholar] [CrossRef]
- Yujnovsky, I.; Hirayama, J.; Doi, M.; Borrelli, E.; Sassone-Corsi, P. Signaling mediated by the dopamine d2 receptor potentiates circadian regulation by CLOCK:BMAL1. Proc. Natl. Acad. Sci. USA 2006, 103, 6386–6391. [Google Scholar] [CrossRef] [PubMed]
- Bussi, I.L.; Levin, G.; Golombek, D.A.; Agostino, P.V. Involvement of dopamine signaling in the circadian modulation of interval timing. Eur. J. Neurosci. 2014, 40, 2299–2310. [Google Scholar] [CrossRef]
- Esposito, E.; Cuzzocrea, S. Antiinflammatory activity of melatonin in central nervous system. Curr. Neuropharmacol. 2010, 8, 228–242. [Google Scholar] [CrossRef]
- Graybeal, J.J.; Bozzelli, P.L.; Graybeal, L.L.; Groeber, C.M.; McKnight, P.E.; Cox, D.N.; Flinn, J.M. Human ApoE epsilon4 alters circadian rhythm activity, IL-1beta, and GFAP in CRND8 mice. J. Alzheimer’s Dis. 2015, 43, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Motzkus, D.; Albrecht, U.; Maronde, E. The human per1 gene is inducible by interleukin-6. J. Mol. Neurosci. 2002, 18, 105–109. [Google Scholar] [CrossRef]
- Bjorkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Puissant, B.; Barreau, C.; Bourin, P.; Clavel, C.; Corre, J.; Bousquet, C.; Taureau, C.; Cousin, B.; Abbal, M.; Laharrague, P.; et al. Immunomodulatory effect of human adipose tissue-derived adult stem cells: Comparison with bone marrow mesenchymal stem cells. Br. J. Haematol. 2005, 129, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Siegel, G.; Schafer, R.; Dazzi, F. The immunosuppressive properties of mesenchymal stem cells. Transplantation 2009, 87, S45–S49. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Moretta, L.; Pistoia, V. Immunoregulatory function of mesenchymal stem cells. Eur. J. Immunol. 2006, 36, 2566–2573. [Google Scholar] [CrossRef]
- Selmani, Z.; Naji, A.; Zidi, I.; Favier, B.; Gaiffe, E.; Obert, L.; Borg, C.; Saas, P.; Tiberghien, P.; Rouas-Freiss, N.; et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells 2008, 26, 212–222. [Google Scholar] [CrossRef]
- Crop, M.J.; Baan, C.C.; Korevaar, S.S.; Ijzermans, J.N.; Pescatori, M.; Stubbs, A.P.; van Ijcken, W.F.; Dahlke, M.H.; Eggenhofer, E.; Weimar, W.; et al. Inflammatory conditions affect gene expression and function of human adipose tissue-derived mesenchymal stem cells. Clin. Exp. Immunol. 2010, 162, 474–486. [Google Scholar] [CrossRef]
- Ren, G.; Zhao, X.; Zhang, L.; Zhang, J.; L’Huillier, A.; Ling, W.; Roberts, A.I.; Le, A.D.; Shi, S.; Shao, C.; et al. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef]
- Hardy, S.A.; Maltman, D.J.; Przyborski, S.A. Mesenchymal stem cells as mediators of neural differentiation. Curr. Stem Cell Res. Ther. 2008, 3, 43–52. [Google Scholar]
- Rossignol, J.; Fink, K.D.; Crane, A.T.; Davis, K.K.; Bombard, M.C.; Clerc, S.; Bavar, A.M.; Lowrance, S.A.; Song, C.; Witte, S.; et al. Reductions in behavioral deficits and neuropathology in the R6/2 mouse model of Huntington’s disease following transplantation of bone-marrow-derived mesenchymal stem cells is dependent on passage number. Stem Cell Res. Ther. 2015, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; Kaltenbach, L.S.; Lo, D.C.; Harry, G.J. Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiol. Aging 2012, 33, 621.e17–621.e33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benraiss, A.; Wang, S.; Herrlinger, S.; Li, X.; Chandler-Militello, D.; Mauceri, J.; Burm, H.B.; Toner, M.; Osipovitch, M.; Jim Xu, Q.; et al. Human glia can both induce and rescue aspects of disease phenotype in Huntington disease. Nat. Commun. 2016, 7, 11758. [Google Scholar] [CrossRef] [PubMed]
- Noh, M.Y.; Lim, S.M.; Oh, K.W.; Cho, K.A.; Park, J.; Kim, K.S.; Lee, S.J.; Kwon, M.S.; Kim, S.H. Mesenchymal stem cells modulate the functional properties of microglia via tgf-beta secretion. Stem Cells Transl. Med. 2016, 5, 1538–1549. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, R.; Yan, K.; Chen, F.; Huang, W.; Lv, B.; Sun, C.; Xu, L.; Li, F.; Jiang, X. Mesenchymal stem cells inhibit lipopolysaccharide-induced inflammatory responses of BV2 microglial cells through TSG-6. J. Neuroinflamm. 2014, 11, 135. [Google Scholar] [CrossRef] [PubMed]
- Giunti, D.; Parodi, B.; Usai, C.; Vergani, L.; Casazza, S.; Bruzzone, S.; Mancardi, G.; Uccelli, A. Mesenchymal stem cells shape microglia effector functions through the release of CX3CL1. Stem Cells 2012, 30, 2044–2053. [Google Scholar] [CrossRef]
- Jaimes, Y.; Naaldijk, Y.; Wenk, K.; Leovsky, C.; Emmrich, F. Mesenchymal stem cell-derived microvesicles modulate lipopolysaccharides-induced inflammatory responses to microglia cells. Stem Cells 2017, 35, 812–823. [Google Scholar] [CrossRef]
- Saijo, K.; Glass, C.K. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef]
- Welberg, L. Synaptic plasticity: A synaptic role for microglia. Nat. Rev. Neurosci. 2014, 15, 69. [Google Scholar] [CrossRef]
- Baron, R.; Babcock, A.A.; Nemirovsky, A.; Finsen, B.; Monsonego, A. Accelerated microglial pathology is associated with abeta plaques in mouse models of Alzheimer’s disease. Aging Cell 2014, 13, 584–595. [Google Scholar] [CrossRef]
- Dey, N.D.; Bombard, M.C.; Roland, B.P.; Davidson, S.; Lu, M.; Rossignol, J.; Sandstrom, M.I.; Skeel, R.L.; Lescaudron, L.; Dunbar, G.L. Genetically engineered mesenchymal stem cells reduce behavioral deficits in the YAC 128 mouse model of Huntington’s disease. Behav. Brain Res. 2010, 214, 193–200. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu-Taeger, L.; Stricker-Shaver, J.; Arnold, K.; Bambynek-Dziuk, P.; Novati, A.; Singer, E.; Lourhmati, A.; Fabian, C.; Magg, J.; Riess, O.; et al. Intranasal Administration of Mesenchymal Stem Cells Ameliorates the Abnormal Dopamine Transmission System and Inflammatory Reaction in the R6/2 Mouse Model of Huntington Disease. Cells 2019, 8, 595. https://doi.org/10.3390/cells8060595
Yu-Taeger L, Stricker-Shaver J, Arnold K, Bambynek-Dziuk P, Novati A, Singer E, Lourhmati A, Fabian C, Magg J, Riess O, et al. Intranasal Administration of Mesenchymal Stem Cells Ameliorates the Abnormal Dopamine Transmission System and Inflammatory Reaction in the R6/2 Mouse Model of Huntington Disease. Cells. 2019; 8(6):595. https://doi.org/10.3390/cells8060595
Chicago/Turabian StyleYu-Taeger, Libo, Janice Stricker-Shaver, Katrin Arnold, Patrycja Bambynek-Dziuk, Arianna Novati, Elisabeth Singer, Ali Lourhmati, Claire Fabian, Janine Magg, Olaf Riess, and et al. 2019. "Intranasal Administration of Mesenchymal Stem Cells Ameliorates the Abnormal Dopamine Transmission System and Inflammatory Reaction in the R6/2 Mouse Model of Huntington Disease" Cells 8, no. 6: 595. https://doi.org/10.3390/cells8060595
APA StyleYu-Taeger, L., Stricker-Shaver, J., Arnold, K., Bambynek-Dziuk, P., Novati, A., Singer, E., Lourhmati, A., Fabian, C., Magg, J., Riess, O., Schwab, M., Stolzing, A., Danielyan, L., & Nguyen, H. H. P. (2019). Intranasal Administration of Mesenchymal Stem Cells Ameliorates the Abnormal Dopamine Transmission System and Inflammatory Reaction in the R6/2 Mouse Model of Huntington Disease. Cells, 8(6), 595. https://doi.org/10.3390/cells8060595