Quantitative Predictive Modelling Approaches to Understanding Rheumatoid Arthritis: A Brief Review



Abstract
:1. Introduction
2. Key Biology in RA
2.1. Disease Risk and Initiation
2.2. Disease Progression
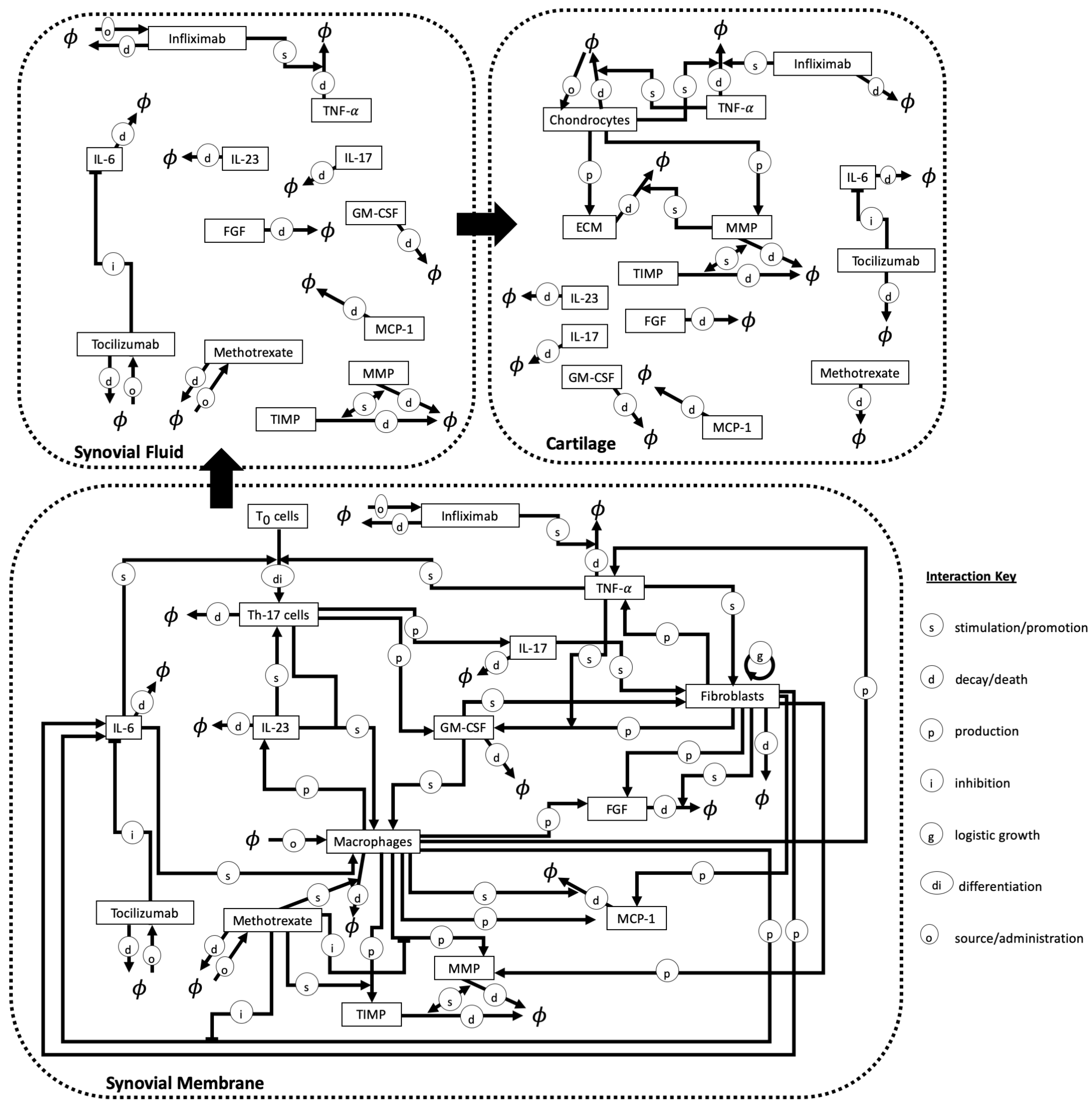
2.3. Treatment Approaches
- Methotrexate (MTX) can induce downregulation of MMPs and can be used to modify the patient’s cytokine profile. It is administered weekly at a low dose, and requires regular monitoring, through blood tests, to assess the immunosuppressive and hepatotoxic effects of the drug [6]. MTX is generally the first drug prescribed to treat RA in the UK and has been shown to reduce pain, joint damage and other symptoms within a short time period. Common side effects can include sickness, loss of appetite and diarrhoea. However, more severe effects, such as kidney, lung and liver problems, can be experienced. Furthermore, there can be a high discontinuation (e.g., 16% of patients) of the drug due to adverse effects [28].
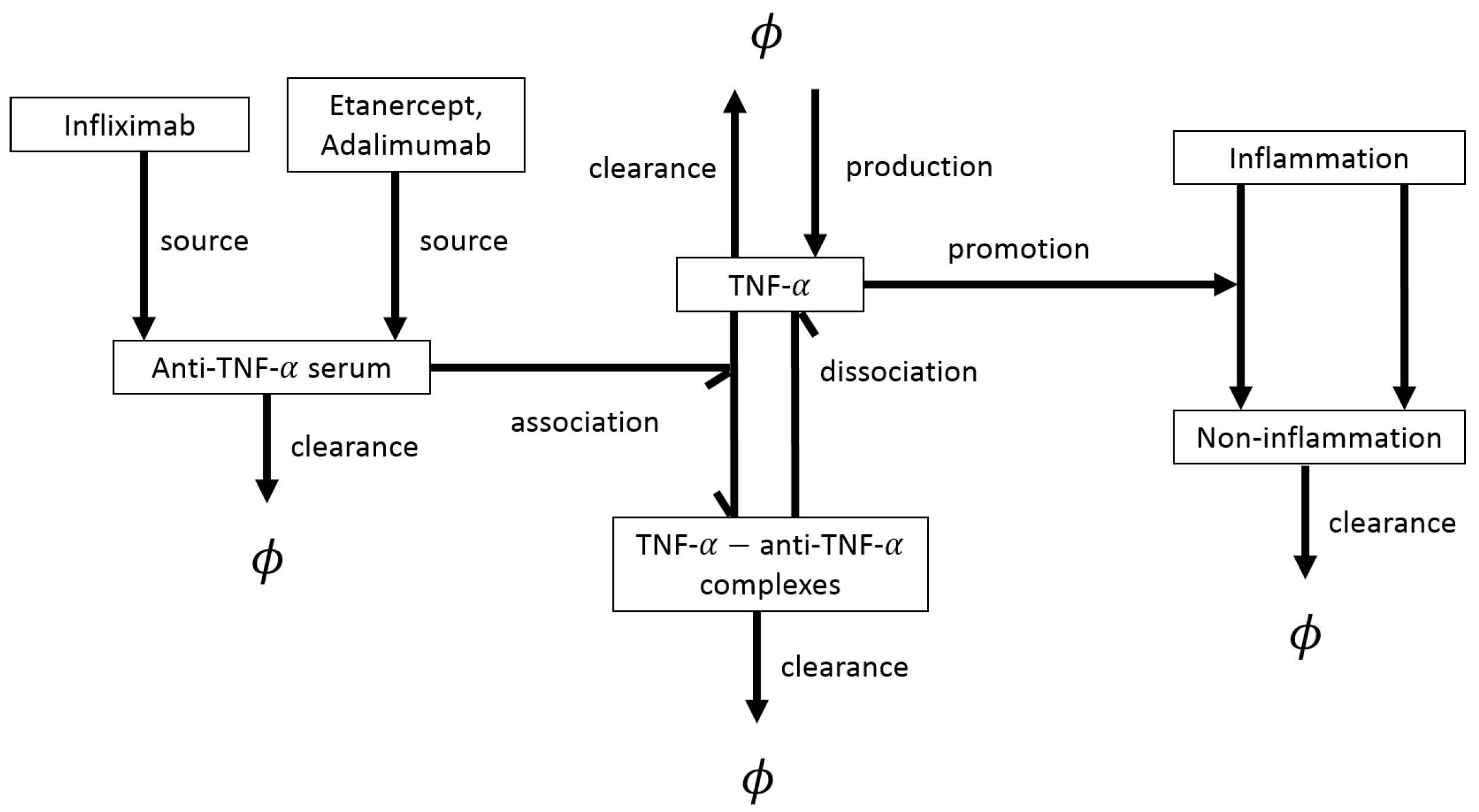
- Infliximab (IFX) is a TNF- inhibitor (TNFi) which reduces the thickness of the synovial layer and has also been shown to lead to a decrease in the levels of IL-6 [6].
- Etanercept is another TNFi, with a similar toxicity profile to IFX [6].
- Adalimumab is a TNFi which is considered safe to use, with minor side effects [31].
- Tocilizumab (TCZ) targets and inhibits the IL-6 receptor which is produced by B cells, FLSs and monocytes. Patients, generally, exhibit a positive response to the drug, however, increased cholesterol and increased (minor) adverse effects are common side effects [32].
3. Open Questions in Rheumatoid Arthritis and the Role of Mathematical Modelling
4. Mathematical Modelling Approaches
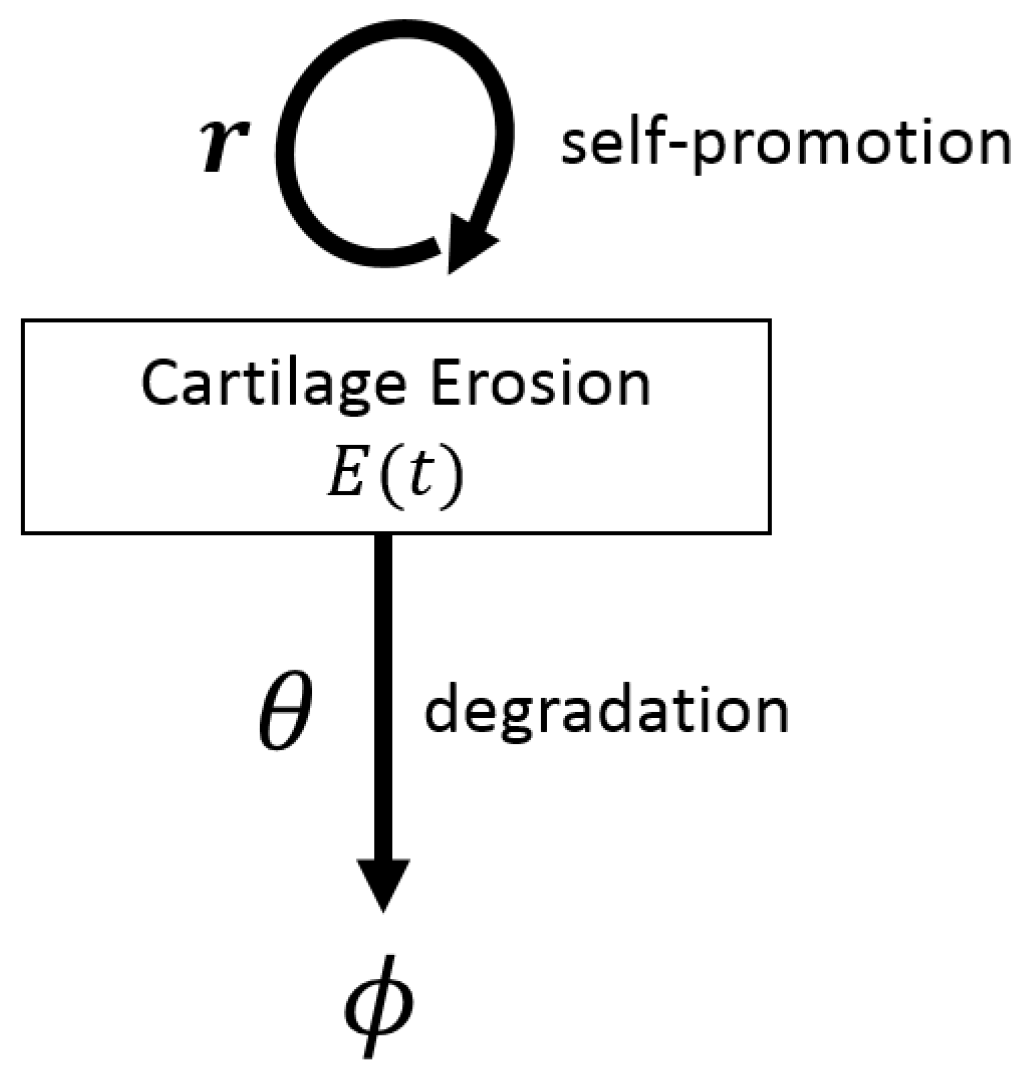
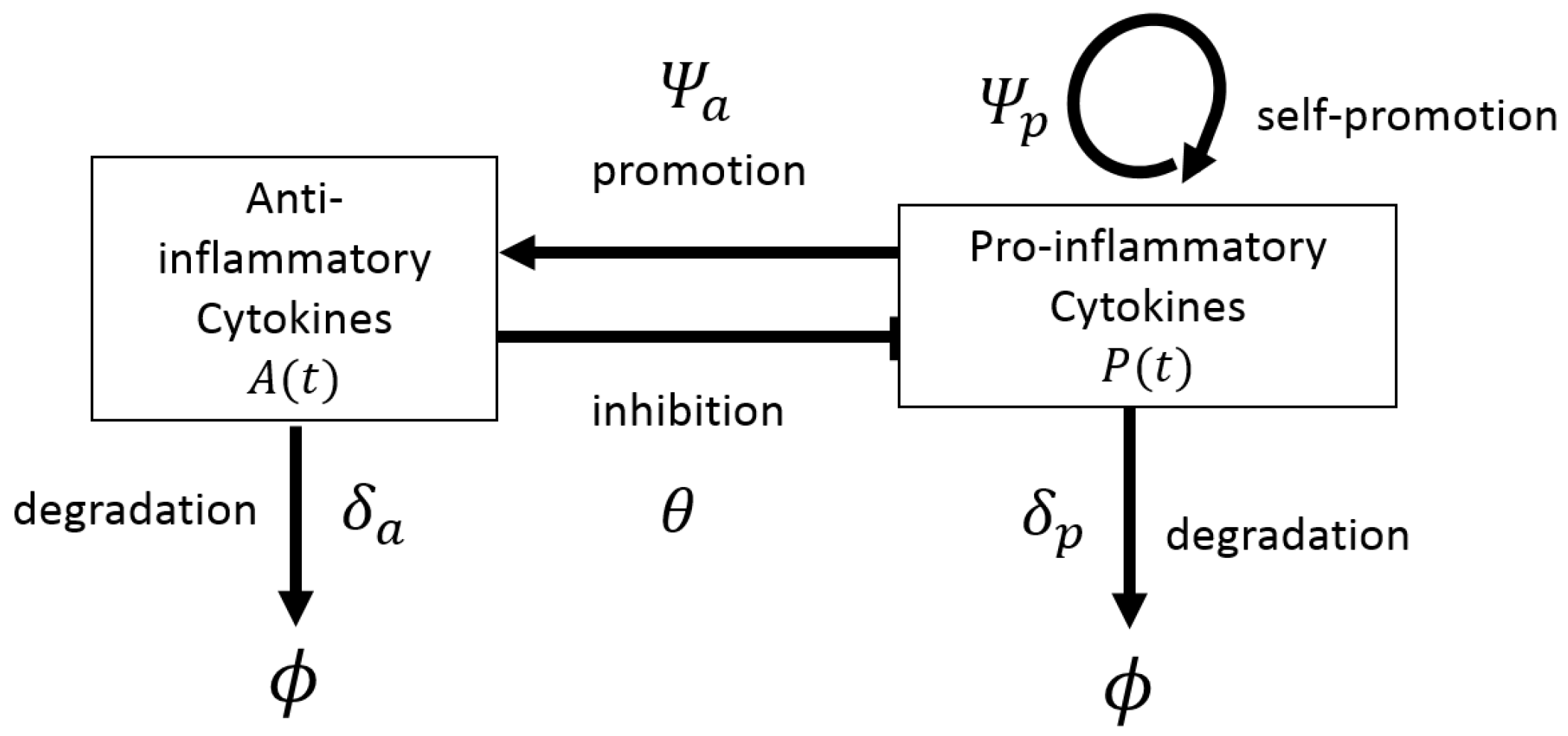
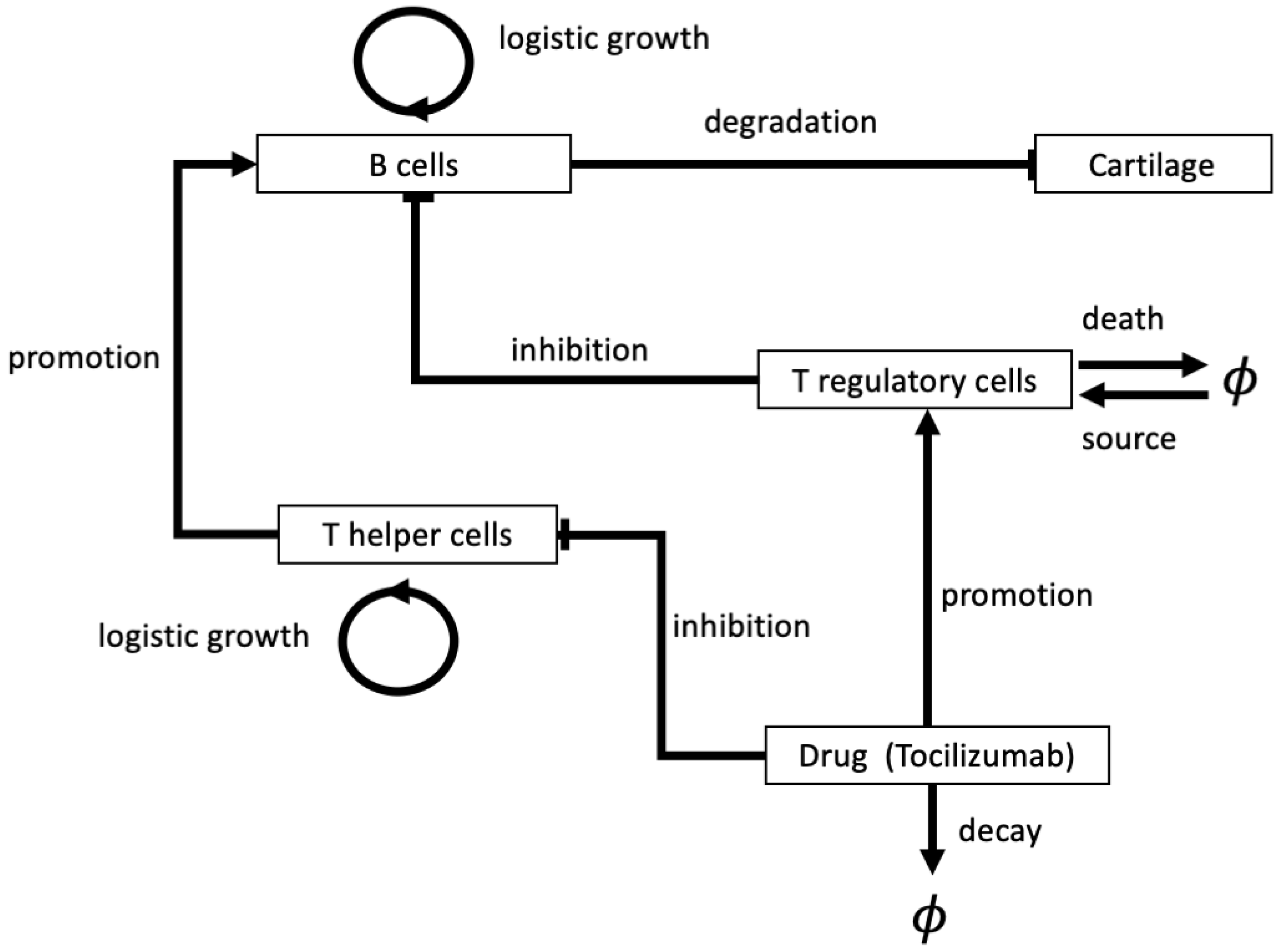
4.1. Deterministic Models for Disease Progression and Treatment: ODEs
4.2. Deterministic Models for Disease Progression and Treatment: PDEs
4.3. Stochastic Models
4.4. Probabilistic Cost-Effectiveness Models for RA Treatment Strategies
4.5. Parameter Estimation and Availability of Data
- The mathematical models used to describe the pharmacokinetics and pharmacodynamics of RA drugs have all been parametrised using patients data from clinical trials, as well as using different laboratory analyses; see [48,83,84,86]. Similarly, the cost-effectiveness models for various RA therapies have all been parametrised using patients data from clinical trials; see [49,97,98]. Given the large number of clinical trials on RA (e.g., there are currently more that 2,300 studies on RA listed on the website “ClinicalTrials.gov”), this might explain the very fast development of these two classes of mathematical models (i.e., pharmacokinetic/pharmacodynamics models, and cost-effectiveness models) over the last two decades.
- The majority of mathematical models used to describe disease progression used a mixture of in vitro, in vivo and human data, with additional unknown parameters being estimated, as in some of the models we have described earlier in this work [70,74]. In these models, data such as cytokine decay rates comes from non-RA specific in vitro studies, while cytokine concentrations are taken from RA specific studies [74]. Combining different types of data to parametrise the models can lead to uncertainty in the parameter values. This uncertainty is increased by variability in the data from different patients (and cohorts of patients), or by variability in the experimental set-ups. Therefore, using a (reasonable) range of values for each parameter and undertaking sensitivity analysis may prove beneficial.
5. Conclusions
Future Predictive Modelling Approaches
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACPA | Anti-citrullinated protein antibody |
| CRP | C-reactive protein |
| DMARD | Disease modifying anti-rheumatic drug |
| ECM | Extracellular matrix |
| ESR | Erthrocyte sedimentation rate |
| FLS | Fibroblast-like synoviocyte |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| IFN- | Interferon- |
| IFX | Infliximab |
| IL-‘x’ | Interleukin-‘x’ |
| MMP | Matrix metalloproteinase |
| MTX | Methotrexate |
| NF-B | Nuclear factor- B |
| NSAID | Non-steroidal anti-inflammatory drug |
| ODE | Ordinary differential equation |
| PDE | Partial differential equation |
| RA | Rheumatoid arthritis |
| RANK(L) | Receptor activator of NF-B (ligand) |
| TCZ | Tocilzumab |
| TIMP | Tissue inhibitor of metalloproteinase |
| TNF- | Tumour necrosis factor- |
| TNFi | TNF- inhibitor |
References
- Merola, J.F.; Espinoza, L.R.; Fleischmann, R. Distinguishing rheumatoid arthritis from psoriatic arthritis. RMD Open 2018, 4, e000656. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, E.; Petrelli, F.; Bonifacio, A.F.; Puxeddu, I.; Alunno, A. One year in review 2018: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2018, 36, 175–184. [Google Scholar] [PubMed]
- National Rheumatoid Arthritis Society. What is RA? Available online: https://www.nras.org.uk/what-is-ra-article (accessed on 1 November 2019).
- Chimenti, M.S.; Triggianese, P.; Conigliaro, P.; Candi, E.; Melino, G.; Perricone, R. The interplay between inflammation and metabolism in rheumatoid arthritis. Cell Death Dis. 2015, 6, e1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NHS Choices. Rheumatoid Arthritis. Available online: https://www.nhs.uk/conditions/rheumatoid-arthritis/ (accessed on 1 October 2019).
- Guo, Q.; Wang, Y.; Xu, D.; Nossent, J.; Pavlos, N.J.; Xu, J. Rheumatoid arthritis: Pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018, 6, 15. [Google Scholar] [CrossRef]
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterising the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000, 43, 30–37. [Google Scholar] [CrossRef]
- Okada, Y.; Eyre, S.; Suzuki, A.; Kochi, Y.; Yamamoto, K. Genetics of rheumatoid arthritis: 2018 status. Ann. Rheum. Dis. 2019, 78, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Weyand, C.M.; Klimiuk, P.A.; Goronzy, J.J. Heterogeneity of rheumatoid arthritis: From phenotypes to genotypes. Springer Semin. Immunopathol. 1998, 20, 5–22. [Google Scholar] [CrossRef]
- Chang, K.; Yang, S.M.; Kim, S.H.; Han, K.H.; Park, S.J.; Shin, J.I. Smoking and rheumatoid arthritis. Int. J. Mol. Sci. 2014, 15, 22279–22295. [Google Scholar] [CrossRef] [Green Version]
- Castro-Sánchez, P.; Roda-Navarro, P. Role of CD4+ T cells in Rheumatoid Arthritis, Physiology and Pathology of Immunology. In Physiology and Pathology of Autoimmune Diseases; INTECH Open: London, UK, 2017; pp. 149–171. [Google Scholar]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of rheumatoid arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scrivo, R.; Massaro, L.; Barbati, C.; Vomero, M.; Ceccarelli, F.; Spinelli, F.R.; Riccieri, V.; Spagnoli, A.; Alessandri, C.; Desideri, G.; et al. The role of dietary sodium intake on the modulation of T helper 17 cells and regulatory T cells in patients with rheumatoid arthritis and systemic lupus erythematosus. PLoS ONE 2017, 12, e0184449. [Google Scholar] [CrossRef] [PubMed]
- Azzi, L.; Rania, S.; Vinci, R.; Spadari, F.; Coveri, F.; Scognamiglio, C.; Farronato, D.; Tettamanti, L.; Tagliabue, A.; Silvestre-Rangil, J.; et al. Periodontal microbioma and rheumatoid arthritis: The role of Porphyromonas gingivalis. J. Biol. Regul. Homeost. Agents 2017, 31, 97–103. [Google Scholar] [PubMed]
- Balandraud, N.; Roudier, J. Epstein-Barr virus and rheumatoid arthritis. Jt. Bone Spine 2018, 85, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Picchianti-Diamanti, A.; Rosado, M.M.; D’Amelio, R. Infectious agents and inflammation: The role of microbiota in autoimmune arthritis. Front. Microbiol. 2018, 8, 2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Steenbergen, H.W.; Da Silva, J.A.P.; Huizinga, T.W.J.; Van Der Helm-van, A.H.M. Preventing progression from arthralgia to arthritis: Targeting the right patients. Nat. Rev. Rheumatol. 2018, 14, 32. [Google Scholar] [CrossRef]
- Kokkonen, H.; Söderström, I.; Rocklöv, J.; Hallmans, G.; Lejon, K.; Dahlqvist, S.R. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010, 62, 383–391. [Google Scholar] [CrossRef]
- Chalan, P.; Bijzet, J.; van den Berg, A.; Kluiver, J.; Kroesen, B.J.; Boots, A.H.M.; Brouwer, E. Analysis of serum immune markers in seropositive and seronegative rheumatoid arthritis and in high-risk seropositive arthralgia patients. Sci. Rep. 2016, 6, 26021. [Google Scholar] [CrossRef] [Green Version]
- Burska, A.; Boissinot, M.; Ponchel, F. Cytokines as biomarkers in rheumatoid arthritis. Mediat. Inflamm. 2014, 2014, 545493. [Google Scholar] [CrossRef]
- Heidari, B. Rheumatoid arthritis: Early diagnosis and treatment outcomes. Caspian J. Intern. Med. 2011, 2, 161. [Google Scholar]
- Alarcón, G.S.; Koopman, W.J.; Acton, R.T.; Barger, B.O. Seronegative rheumatoid arthritis. Arthritis Rheum. 1982, 25, 502–507. [Google Scholar] [CrossRef]
- Nordberg, L.B.; Lillegraven, S.; Lie, E.; Aga, A.B.; Olsen, I.C.; Hammer, H.; Uhlig, T.; Jonsson, M.K.; van der Heijde, D.; Kvien, T.K.; et al. Patients with seronegative RA have more inflammatory activity compared with patients with seropositive RA in an inception cohort of DMARD-naïve patients classified according to the 2010 ACR/EULAR criteria. Ann. Rheum. Dis. 2017, 76, 341–345. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; Van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.A.; Gizinski, A.; Morgan, R.; Lundy, S.K. Cell-cell interactions in rheumatoid arthritis synovium. Rheum. Dis. Clin. 2010, 36, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellado, M.; Martínez-Muñoz, L.; Cascio, G.; Lucas, P.; Pablos, J.L.; Rodríguez-Frade, J.M. T cell migration in rheumatoid arthritis. Front. Immunol. 2015, 6, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardar, S.; Andersson, Å. Old and new therapeutics for rheumatoid arthritis: In vivo models and drug development. Immunopharmacol. Immunotoxicol. 2016, 38, 2–13. [Google Scholar] [CrossRef]
- Lopez-Olivo, M.A.; Siddhanamatha, H.R.; Shea, B.; Tugwell, P.; Wells, G.A.; Suarez-Almazor, M.E. Methotrexate for treating rheumatoid arthritis. Cochrane Database Syst. Rev. 2014. [Google Scholar] [CrossRef]
- Singh, J.A.; Cameron, C.; Noorbaloochi, S.; Cullis, T.; Tucker, M.; Christensen, R.; Ghogomu, E.T.; Coyle, D.; Clifford, T.; Tugwell, P.; et al. Risk of serious infection in biological treatment of patients with rheumatoid arthritis: A systematic review and meta-analysis. Lancet 2015, 386, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.L.; Takase-Minegishi, K.; Ramiro, S.; Chatzidionysiou, K.; Smolen, J.S.; Van Der Heijde, D.; Bijlsma, J.W.; Burmester, G.R.; Dougados, M.; Scholte-Voshaar, M.; et al. Efficacy of biological disease-modifying antirheumatic drugs: a systematic literature review informing the 2016 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1113–1136. [Google Scholar] [CrossRef]
- Navarro-Sarabia, F.; Ariza-Ariza, R.; Hernandez-Cruz, B.; Villanueva, I. Adalimumab for treating rheumatoid arthritis. Cochrane Database Syst. Rev. 2005. [Google Scholar] [CrossRef]
- Singh, J.A.; Beg, S.; Lopez-Olivo, M.A. Tocilizumab for rheumatoid arthritis: A Cochrane systematic review. J. Rheumatol. 2011, 38, 10–20. [Google Scholar] [CrossRef]
- Mallen, C.D.; Helliwell, T.; Scott, I.C. How can primary care physicians enhance the early diagnosis of rheumatic diseases? Expert Rev. Clin. Immunol. 2018, 14, 171–173. [Google Scholar] [CrossRef]
- Fleischmann, R.M. Early diagnosis and treatment of rheumatoid arthritis for improved outcomes: Focus on etanercept, a new biologic response modifier. Clin. Ther. 1999, 21, 1429–1442. [Google Scholar] [CrossRef]
- da Mota, L.M.H.; Brenol, C.V.; Palominos, P.; Pinheiro, G.R.C. Rheumatoid arthritis in Latin America: The importance of an early diagnosis. Clin. Rheumatol. 2015, 34, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.K.; Kim, D.; Won, S.; Lee, J.; Choi, C.B.; Choe, J.Y.; Hong, S.J.; Jun, J.B.; Kim, T.H.; Koh, E.; et al. Factors associated with time to diagnosis from symptom onset in patients with early rheumatoid arthritis. Korean J. Intern. Med. 2019, 34, 910–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NHS Choices. Osteoarthritis. Available online: https://www.nhs.uk/conditions/osteoarthritis/ (accessed on 1 November 2019).
- Flores-Borja, F.; Mauri, C.; Ehrenstein, M.R. Restoring the balance: Harnessing regulatory T cells for therapy in rheumatoid arthritis. Eur. J. Immunol. 2008, 38, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Smilek, D.E.; Ehlers, M.R.; Nepom, G.T. Restoring the balance: Immunotherapeutic combinations for autoimmune disease. Dis. Mod. Mech. 2014, 7, 503–513. [Google Scholar] [CrossRef] [Green Version]
- de Hair, M.J.H.; Jacobs, J.W.G.; Schoneveld, J.L.M.; van Laar, J.M. Difficult-to-treat rheumatoid arthritis: An area of unmet clinical need. Rheumatology 2017, 57, 1135–1144. [Google Scholar]
- Campbell, L.; Chen, C.; Bhagat, S.S.; Parker, R.A.; Östör, A.J.K. Risk of adverse events including serious infections in rheumatoid arthritis patients treated with tocilizumab: A systematic literature review and meta-analysis of randomized controlled trials. Rheumatology 2010, 50, 552–562. [Google Scholar] [CrossRef] [Green Version]
- Lang, V.R.; Englbrecht, M.; Rech, J.; Nüsslein, H.; Manger, K.; Schuch, F.; Tony, H.P.; Fleck, M.; Manger, B.; Schett, G.; et al. Risk of infections in rheumatoid arthritis patients treated with tocilizumab. Rheumatology 2011, 51, 852–857. [Google Scholar] [CrossRef] [Green Version]
- Alder, H.; Michel, B.A.; Marx, C.; Tamborrini, G.; Langenegger, T.; Bruehlmann, P.; Steurer, J.; Wildi, L.M. Computer-based diagnostic expert systems in rheumatology: Where do we stand in 2014? Int. J. Rheumatol. 2014, 2014, 672714. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.; Denman-Johnson, S.; Brook, B.S.; Gaywood, I.; Owen, M.R. Mathematical modelling of cytokine-mediated inflammation in rheumatoid arthritis. Math. Med. Biol. 2013, 30, 311–337. [Google Scholar] [CrossRef]
- Helliwell, P.S.; Hetthen, J.; Sokoll, K.; Green, M.; Marchesoni, A.; Lubrano, E.; Veale, D.; Emery, P. Joint symmetry in early and late rheumatoid and psoriatic arthritis: Comparison with a mathematical model. Arthritis Rheum. 2000, 43, 865–871. [Google Scholar] [CrossRef]
- Witten, T.M.; del Rincon, I.; Escalante, A. Modelling the progression of articular erosion in rheumatoid arthritis (RA): Initial mathematical models. Math. Comput. Model. 2000, 31, 31–38. [Google Scholar] [CrossRef]
- Rao, R.; DuBois, D.; Almon, R.; Jusko, W.J.; Androulakis, I.P. Mathematical modelling of the circadian dynamics of the neuroendocrine-immune network in experimentally induced arthritis. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E310–E324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, M.; Grange, S.; Frey, N. Exposure-response relationship of tocilizumab, and anti-IL-6 receptor monoclonal antibody, in a large population of patients with rheumatoid arthritis. J. Clin. Pharmacol. 2013, 53, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Kobelt, G.; Lindgren, P.; Singh, A.; Klareskog, L. Cost effectiveness of etanercept (Enbrel) in combination with methotrexate in the treatment of active rheumatoid arthritis based on the TEMPO trial. Ann. Rheum. Dis. 2005, 64, 1174–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Takayanagi, R.; Yokoyama, H.; Yamada, Y. Theory-based analysis of the anti-inflammatory effect of TNF inhibitors on rheumatoid arthritis. Drug. Metab. Pharmacokinet. 2014, 29, 272–277. [Google Scholar] [CrossRef]
- Anderson, G.; Bombardier, C. Estimating disease activity in Rheumatoid Arthritis. Med. Decis. Mak. 1984, 4, 469–487. [Google Scholar] [CrossRef]
- Breedveld, F.C.; Han, C.; Bala, M.; van der Heijde, D.; Baker, D.; Kavanaugh, A.F.; Maini, R.N.; Lipsky, P.E. Association between baseline radiographic damage and improvement in physical function after treatment of patients with rheumatoid arthritis. Ann. Rheum. Dis. 2005, 64, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Capela, R.C.; Corrente, J.E.; Magalhães, C.S. Comparison of the disease activity score and juvenile arthritis disease activity score in the juvenile idiopathic arthritis. Rev. Bras. Rheumatol. 2015, 55, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terao, C.; Brynedal, B.; Chen, Z.; Jiang, X.; Westerlind, H.; Hansson, M.; Jakobsson, P.J.; Lundberg, K.; Skriner, K.; Serre, G.; et al. Distinct HLA associations with rheumatoid arthritis subsets defined by serological subphenotype. Am. J. Hum. Genet. 2019, 105, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Lezcano-Valverde, J.M.; Salazar, F.; León, L.; Toledano, E.; Jover, J.A.; Fernandez-Gutierrez, B.; Soudah, E.; González-Älvaro, I.; Abasolo, L.; Rodriguez-Rodriguez, L. Development and validation of a multivariate predictive model for rheumatoid arthritis mortality using a machine learning approach. Sci. Rep. 2017, 7, 10189. [Google Scholar] [CrossRef] [PubMed]
- Norgeot, B.; Glicksberg, B.S.; Turpin, L.; Lituiev, D.; Gianfrancesco, M.; Oskotsky, B.; Schmajuk, G.; Yazdany, J.; Butte, A.J. Assessment of a deep learning model based on electronic health record data to forecast clinical outcomes in patients with rheumatoid arthritis. JAMA Netw. Open 2019, 2, e190606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.J.; Tagkopoulos, I. Application of machine learning in rheumatic disease research. Korean J. Intern. Med. 2019, 34, 708–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, H.K.; Wahl, L.M. Self-tolerance and autoimmunity in a regulatory T cell model. Bull. Math. Biol. 2011, 73, 33–71. [Google Scholar] [CrossRef]
- Arazi, A.; Neumann, A. Modelling immune complex-mediated autoimmune inflammation. J. Theor. Biol. 2010, 267, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Blyuss, K.B.; Nicholson, L.B. The role of tunable activation thresholds in the dynamics of autoimmunity. J. Theor. Biol. 2012, 308, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Delitala, M.; Dianzani, U.; Lorenzi, T.; Melensi, M. A mathematicl model for immune and autoimmune response mediated by T-cells. Comput. Math. Appl. 2013, 66, 1010–1023. [Google Scholar] [CrossRef]
- Fatehi, F.; Kyrychko, S.N.; Ross, A.; Kyrychko, Y.N.; Blyuss, K.B. Stochastic effects in autoimmune dynamics. Front. Physiol. 2018, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Fatehi, F.; Kyrychko, Y.N.; Blyuss, K.B. Effects of viral and cytokine delays on dynamics of autoimmunity. Mathematics 2018, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Iwami, S.; Takeuchi, Y.; Miura, Y.; Sasaki, T.; Kajiwara, T. Dynamical properties of autoimmune disease models: Tolerance, flare-up, dormancy. J. Theor. Biol. 2007, 246, 646–659. [Google Scholar] [CrossRef]
- Ramos, M.P.M.; Ribeiro, C.; Soares, A.J. A kinetic model of T cell autoreactivity in autoimmune diseases. J. Math. Biol. 2019, 79, 2005–2031. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Mosekilde, E.; Lund, O. Bistability in autoimmune diseases. Autoimmunity 2011, 44, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.A.; Stepanova, N.V. Mathematical model of autoimmunity [Article in Russian]. Biofizika 1975, 20, 1095–1098. [Google Scholar] [PubMed]
- Ridgley, L.A.; Anderson, A.E.; Pratt, A.G. What are the dominant cytokines in early rheumatoid arthritis? Curr. Opin. Rheumatol. 2018, 30, 207–214. [Google Scholar] [CrossRef] [Green Version]
- McInnes, I.B.; Buckley, C.D.; Isaacs, J.D. Cytokines in rheumatoid arthritis—Shaping the immunological landscape. Nat. Rev. Rheumatol. 2016, 12, 63–68. [Google Scholar] [CrossRef]
- Jit, M.; Henderson, B.; Stevens, M.; Seymour, R.M. TNF-α neutralisation in cytokine-driven diseases: A mathematical model to account for therapeutic success in rheumatoid arthritis but therapeutic failure in systemic inflammatory response syndrome. Rheumatology 2005, 44, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Matteucci, L.; Nucci, M.C. Solution of a mathematical model for the treatment of rheumatoid arthritis. Commun. Appl. Ind. Math 2019, 10, 12–24. [Google Scholar] [CrossRef] [Green Version]
- Odisharia, K.; Odisharia, V.; Tsereteli, P.; Janikashvili, N. On the Mathematical Model of Drug Treatment of Rheumatoid Arthritis. In Mathematics, Informatics, and Their Applications in Natural Sciences and Engineering; Jaiani, G., Natroshvili, D., Eds.; AMINSE 2017, Springer Proceedings in Mathematics & Statistics; Springer: Cham, Switzerland, 2017; Volume 276. [Google Scholar]
- Rullmann, J.A.C.; Struemper, H.; Defranoux, N.A.; Ramanujan, S.; Meeuwisse, C.M.L.; Van Elsas, A. Systems biology for battling rheumatoid arthritis: Application of the Entelos PhysioLab platform. IEE Proc. Syst. Biol. 2005, 152, 256–262. [Google Scholar] [CrossRef]
- Moise, N.; Friedman, A. Rheumatoid arthritis—A mathematical model. J. Theor. Biol. 2019, 461, 17–33. [Google Scholar] [CrossRef]
- Scholz, S.; Mittendorf, T. Modelling rheumatoid arthritis using different techniques—A review of model construction and results. Health Econ. Rev. 2014, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, H.A.; Pincus, T. Radiographic damage in rheumatoid arthritis: Description by nonlinear models. J. Rheumatol. 1992, 19, 1655–1658. [Google Scholar] [PubMed]
- Salaffi, F.; Ferraccioli, G.; Peroni, M.; Carotti, M.; Bartoli, E.; Cervini, C. Progression of erosion and joint space narrowing scores in rheumatoid arthritis assesed by nonlinear models. J. Rheumatol. 1994, 21, 1626–1630. [Google Scholar] [PubMed]
- Sharp, J.T. Attempts to apply curve fitting models to the analysis of progression of radiographic damage in rheumatoid arthritis are laudable, but are the results believable? J. Rheumatol. 1994, 21, 1589–1590. [Google Scholar] [PubMed]
- Graudal, N.A.; Jurik, A.G.; de Carvalho, A.; Graudal, H.K. Radiographic progression in rheumatoid arthritis. Arthritis Rheum. 1998, 41, 1470–1480. [Google Scholar] [CrossRef]
- Defranoux, N.A.; Stokes, C.L.; Young, D.L.; Kahn, A.J. In silico modeling and simulation of bone biology: A proposal. J. Bone Miner. Res. 2005, 20, 1079–1084. [Google Scholar] [CrossRef]
- Mould, D.R.; Davis, C.B.; Minthorn, E.A.; Kwok, D.C.; Elliott, M.J.; Luggen, M.E.; Totoritis, M.C. A population pharmacokinetic-pharmacodynamic analysis of single doses of clenoliximab in patients with rheumatoid arthritis. Pharmacokinet. Drug Dispos. 1999, 66, 246–257. [Google Scholar] [CrossRef]
- Ng, C.M.; Bruno, R.; Combs, D.; Davies, B. Population pharmacokinetics of rituximab (anti-CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J. Clin. Pharmacol. 2005, 45, 792–801. [Google Scholar] [CrossRef]
- Kimura, K.; Takayanagi, R.; Yokoyama, H.; Yamada, Y. Theory-based analysis of anti-inflammatory effect of infliximab on Crohn’s disease and rheumatoid arthritis. Rheumatol. Int. 2012, 32, 145–150. [Google Scholar] [CrossRef]
- Ternant, D.; Ducourau, E.; Perdriger, A.; Corondan, A.; Goff, B.L.; Devauchelle-Pensec, V.; Solau-Gervais, E.; Watier, H.; Goupille, P.; Paintaud, G.; et al. Realtionship between inflammation and infliximab pharmacokinetics in rheumatoid arthritis. Br. J. Clin Pharmacol 2013, 78, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.Y.; Lon, H.K.; Wang, Y.L.; DuBois, D.C.; Almon, R.A.; Jusko, W.J. Pharmacokinetics, pharmacodynamics, and toxicities of methotrexate in healthy and collagen-induced arthritic rats. Biopharm. Drug Dispos. 2013, 34, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Namour, F.; Diderichsen, P.M.; Cox, E.; Vayssière, B.; der Aa, A.V.; Tasset, C.; Klooster, G.V. Pharmacokinetics and pharmacokinetic/pharmacodynamic modelling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIB dose selection. Clin. Pharmacokinet. 2015, 54, 859–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workie, D.W.; Dardzinski, B.J.; Laor, T.B.G.T.; Bommer, W.A.; O’Brien, K.J. Quantification of dynamic contrast-enhanced MR imaging of the knee in children with juvenile rheumatoid arthritis based on pharmacokinetic modelling. Magn. Reson. Imaging 2004, 22, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Eseonu, O.I.; Bari, C.D. Homing of mesenchymal stem cells: Mechanistic or stochastic? Implications for targeted delivery in arthritis. Rheumatology 2015, 54, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts-Thompson, P.J.; Jones, M.E.; Walker, J.G.; Macfarlane, J.G.; Smith, M.D.; Ahem, M.J. Stochastic processes in the causation of rheumatic disease. J. Rheumatol. 2002, 29, 2628–2634. [Google Scholar]
- Wick, M.C.; Lindblad, S.; Klareskog, L.; Van Vollenhoven, R.F. Relationship between inflammation and joint destruction in early rheumatoid arthritis: A mathematical description. Ann. Rheum. Dis. 2004, 63, 848–852. [Google Scholar] [CrossRef]
- Kalden, J.R. Emerging therapies for rheumatoid arthritis. Rheumatol. Ther. 2016, 3, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Nurmohamed, M.T.; Dijkmans, B.A. Efficacy, tolerability and cost effectiveness of disease-modifying antirheumatic drugs and biologic agents in rheumatoid arthritis. Drugs 2005, 65, 661–694. [Google Scholar] [CrossRef]
- Geyer, M.; Müller-Ladner, U. Rationale of using different biological therapies in rheumatoid arthritis. Arthritis Res.Ther. 2010, 12, 214. [Google Scholar] [CrossRef] [Green Version]
- Schipper, L.G.; Kievit, W.; den Broeder, A.A.; van der Laar, M.A.; Adang, E.M.M.; Fransen, J.; van Riel, P.L.C.M. Treatment strategies aiming at remission in early rheumatoid arthritis patients: Starting with methotrexate monotherapy is cost-effective. Rheumatology 2011, 50, 1320–1330. [Google Scholar] [CrossRef] [Green Version]
- Spalding, J.R.; Hay, J. Cost effectiveness of tumour necrosis factor-α inhibitors as first-line agents in rheumatoid arthritis. Pharmacoeconomics 2006, 24, 1221–1232. [Google Scholar] [CrossRef]
- Lekander, I.; Kobelt, G.; Svarvar, P.; Ljung, T.; van Vollenhoven, R.; Borgström, F. The comparison of trial data-based and registry data-based cost-effectiveness of Infliximab treatment for rheumatoid arthritis in Sweden using a modeling approach. Value Health 2013, 16, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobelt, G.; Lekander, I.; Lang, A.; Raffeiner, B.; Botsios, C.; Geborek, P. Cost-effectiveness of etanercept treatment in early active rheumatoid arthritis followed by dose adjustment. Int. J. Technol. Assess. Health Care 2011, 27, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Maetzel, A.; Krahn, M.; Naglie, G. The cost effectiveness of rofecoxib and celecoxib in patients with osteoarthritis or rheumatoid arthritis. Arthritis Rheum. 2003, 49, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Freeman, I.; Yang, Y. Cost-effectiveness of sarilumab versus adalimumab for treating patients with active rheumatoid arthritis: A Markov model assessment. Value Health 2018, 21, S197. [Google Scholar] [CrossRef]
- Kostic, M. Cost-effectiveness analysis of etanercept in combination with methotrexate for rheumatoid arthritis—Markov model based on data from Serbia. Serb. J. Exp. Clin. Res. 2017, 18, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Brennan, A.; Bansback, N.; Reynolds, A.; Conway, P. Modelling the cost-effectiveness of etanercept in adults with rheumatoid arthritis in the UK. Rheumatology 2003, 43, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Jalal, H.; O’Dell, J.R.; Bridges, S.L., Jr.; Cofield, S.; Curtis, J.R.; Mikuls, T.R.; Moreland, L.W.; Michaud, K. Cost-effectiveness of triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis. Arthritis Care Res. 2016, 68, 1751–1757. [Google Scholar] [CrossRef]
- Alemao, E.; Al, M.J.; Boonen, A.A.; Stevenson, M.D.; Verstappen, S.M.M.; Michaud, K.; Weinblatt, M.E.; Rutten-van Mölken, M.P.M.H. Conceptual model for the health technology assessment of current and novel interventions in rheumatoid arthritis. PLoS ONE 2018, 13, e0205013. [Google Scholar] [CrossRef]
- ∅ksendal, B. Stochastic Differential Equations. An Introduction with Applications; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Dalang, R.; Khoshnevisan, D.; Mueller, C.; Nualart, D.; Xiao, Y. A Minicourse on Stochastic Partial Differential Equations; Springer: Berlin/Heidelberg, Germany, 2006. [Google Scholar]
- Mina-Osorio, P. Basics of drug development in rheumatology. Arthritis Rheum. 2015, 67, 2581–2590. [Google Scholar] [CrossRef] [Green Version]
- Peck, Y.; Leom, L.T.; Low, P.F.P.; Wang, D. Establishment of an in vitro three-dimensional model for cartilage damage in rheumatoid arthritis. J. Tissue Eng. Regen. Med. 2018, 12, e237–e249. [Google Scholar] [CrossRef]
- Kirkham, J.J.; Boers, M.; Tugwell, P.; Clarke, M.; Williamson, P.R. Outcome measures in rheumatoid arthritis randomised trials over the last 50 years. Trials 2013, 14, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawar, A.; Desai, R.J.; Solomon, D.H.; Ortiz, A.J.S.; Gale, S.; Bao, M.; Sarsour, K.; Schneeweiss, S.; Kim, S.C. Risk of serious infections in tocilizumab versus other biologic drugs in patients with rheumatoid arthritis: A multidatabase cohort study. Ann. Rheum. Dis. 2019, 78, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Lethaby, A.; Lopez-Olivo, M.A.; Maxwell, L.J.; Burls, A.; Tugwell, P.; Wells, G.A. Etanercept for the treatment of rheumatoid arthritis. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef] [PubMed]
- Blumenauer, B.B.T.B.; Judd, M.; Wells, G.A.; Burls, A.; Cranney, A.; Hochberg, M.C.; Tugwell, P.; Lopez-Olivo, M.A. Infliximab for the treatment of rheumatoid arthritis. Cochrane Database Syst. Rev. 2002. [Google Scholar] [CrossRef] [PubMed]
- Grimm, V.; Railsback, S.F. Individual-Based Models and Ecology; Princeton Series in Theoretical and Computational Biology; Princeton University Press: Princeton, NJ, USA, 2005. [Google Scholar]
- Van Liedekerke, P.; Palm, M.M.; Jagiella, N.; Drasdo, D. Simulating tissue mechanics with agent-based models: Concepts, perspectives and some novel results. Comput. Part. Mech. 2015, 2, 401–444. [Google Scholar] [CrossRef] [Green Version]
- An, G.; Mi, Q.; Dutta-Moscato, J.; Vodovotz, Y. Agent-Based Models in Translational Systems Biology. WIRES Syst. Biol. Med. 2009, 1, 159–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzcar, J.; Wang, Y.; Heiland, R.; Macklin, P. A review of cell-based computational modeling in cancer biology. Clin. Cancer Inform. 2019, 2, 1–13. [Google Scholar] [CrossRef]
- Martins, M.L.; Ferreira, S.C., Jr.; Vilela, M.J. Multiscale models for biological systems. Curr. Opin. Colloid Interface Sci. 2010, 15, 18–23. [Google Scholar] [CrossRef]
- Macklin, P.; Edgerton, M.E. Discrete Cell Modeling. In Multiscale Modelling of Cancer: An Integrated Experimental and Mathematical Modeling Approach; Cristini, V., Lowengrub, J.S., Eds.; Cambridge University Press: Cambridge, UK, 2010; pp. 88–122. [Google Scholar]
- Rejniak, K.A.; Anderson, A.R.A. Hybrid models of tumor growth. WIRES Syst. Biol. Med. 2011, 3, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Lowengrub, J.S.; Frieboes, H.B.; Jin, F.; Chuang, Y.L.; Li, X.; Macklin, P.; Wise, S.M.; Cristini, V. Nonlinear modelling of cancer: Bridging the gap between cells and tumours. Nonlinearity 2009, 23, R1. [Google Scholar] [CrossRef] [Green Version]
- Powathil, G.G.; Swat, M.; Chaplain, M.A.J. Systems oncology: Towards patient-specific treatment regimes informed by multiscale mathematical modelling. Semin. Cancer Biol. 2015, 30, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | Role |
|---|---|
| GM-CSF | Promotes inflammation [68] |
| Activates macrophages, neutrophils [69] | |
| IFN- | Increases antigen presentation [11] |
| Activates macrophages [11] | |
| Increases chemokine secretion [11] | |
| IL-1 | Induces osteoclastogenesis [11,21] |
| IL-6 | Activates leukocytes and osteoclasts [11,12,21] |
| Stimulates antibody production [11] | |
| Promotes pannus formation [6] | |
| IL-12 | Involved in the plasticity of Th17 (subset of T helper) cells [68] |
| IL-15 | Promotes T cell migration [25] |
| IL-17 | Induces production of inflammatory cytokines [11] |
| Activates innate immune cells [11] | |
| Induces osteoclastogenesis [11,21] | |
| Stimulates neutrophil recruitment [11] | |
| RANKL | Promotes bone erosion [11,12,21] |
| TNF- | Activates leukocytes, FLSs, endothelial cells and osteoclasts [11] |
| Induces production of inflammatory cytokines [11] | |
| Enhances MMP production [11,21] | |
| Suppresses T regulatory cells [11] | |
| Promotes T cell migration [25] | |
| Activates the RANKL pathway [21] | |
| Promotes osteoclastogenesis [11,12,21] | |
| Promotes angiogenesis [25] |
| Mathematical Models | Description/Advantages/Disadvantages |
|---|---|
| Ordinary Differential Equations (ODEs) | ODEs are deterministic mathematical equations that describe the time evolution of a variable of interest (e.g., the density of immune cells involved in RA, the density of chondrocytes in the cartilage, the concentration of some cytokines, or the concentration of a therapeutic drug). These are the most common models used to describe the evolution of RA [44,46,47,50,70,71,72,73]. |
| Advantages: These types of models require shorter simulation time, and are easily parametrised using experimental lab data or clinical patients data (because the large majority of collected data describes the temporal changes in some variable of interest; e.g., levels of pro-inflammatory cytokines). These models can also be investigated analytically; e.g., their long-term dynamics can be studied via the identification of possible steady states and their stability [44]. | |
| Disadvantages: These types of models cannot really capture the mechanisms behind the spatial degradation of the articular cartilage. Also, being deterministic, these models cannot capture the variability in the cytokine levels between different patients [20]. This variability in the cytokine level can impact also the variability in the evolution of the disease. | |
| Partial Differential Equations (PDEs) | PDEs are deterministic mathematical equations that describe the space and time evolution of a variable of interest (e.g., the density of immune cells involved in RA, the density of chondrocytes in the cartilage, the concentration of some cytokines, or the concentration of a therapeutic drug) [74]. They can also describe age-related aspects of the immune responses; however, this approach is not very common in the context of RA dynamics. |
| Advantages: These models can be used to test various hypotheses on the spatial dynamics of the components of immune system involved in RA evolution (e.g., spatial spread of cytokines, immune infiltration of the joint, etc.), and how can these components affect the spatial erosion of the cartilage. They can also be used to investigate the spread of the therapeutic drug into the affected tissue. | |
| Disadvantages: The numerical simulations of these models are more complex (compared to the simulations of ODEs). It is also more difficult to parametrise these types of mathematical models using patient data. As with the ODEs, since these models are deterministic, they cannot capture the variability in the cytokine levels between different patients [20]. Due to the complexity of these models (coupled sometimes with a large number of variables modelled), it can be more difficult to investigate these models analytically. | |
| Stochastic/Probabilistic models | Mathematical and computational models describe the interactions between the different components of the system, or the transitions between different states of these components as probabilistic. Such models have been mainly applied in the context of treatment decisions [75]. Very few models have been used to describe the probability of RA occurrence. |
| Advantages: These models can reproduce more accurately the randomness of certain aspects in the evolution of RA (e.g., transition between different states as a result of different levels of pro-inflammatory cytokines, etc.). | |
| Disadvantages: Due to slightly more complex numerical simulations, until now, these models have been used to describe the temporal dynamics of different variables involved in the evolution of RA. As far as we know, probabilistic approaches have not been combined yet with spatio-temporal models to investigate the variability in the spatial dynamics of different pro-inflammatory and/or anti-inflammatory immune responses. Since these are mostly computational models, analytical investigations are inexistent. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macfarlane, F.R.; Chaplain, M.A.J.; Eftimie, R. Quantitative Predictive Modelling Approaches to Understanding Rheumatoid Arthritis: A Brief Review. Cells 2020, 9, 74. https://doi.org/10.3390/cells9010074
Macfarlane FR, Chaplain MAJ, Eftimie R. Quantitative Predictive Modelling Approaches to Understanding Rheumatoid Arthritis: A Brief Review. Cells. 2020; 9(1):74. https://doi.org/10.3390/cells9010074
Chicago/Turabian StyleMacfarlane, Fiona R., Mark A. J. Chaplain, and Raluca Eftimie. 2020. "Quantitative Predictive Modelling Approaches to Understanding Rheumatoid Arthritis: A Brief Review" Cells 9, no. 1: 74. https://doi.org/10.3390/cells9010074
APA StyleMacfarlane, F. R., Chaplain, M. A. J., & Eftimie, R. (2020). Quantitative Predictive Modelling Approaches to Understanding Rheumatoid Arthritis: A Brief Review. Cells, 9(1), 74. https://doi.org/10.3390/cells9010074