The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats

 ,
, 
Abstract
:1. Introduction
2. Material and Methods
2.1. Animal Model
2.2. Evaluation of Cardiac Structure and Function
2.2.1. Echocardiographic Study
2.2.2. Morphological and Histological Evaluation
2.2.3. Isolation of Cardiac Mitochondria
2.2.4. Lipidomic Analysis
2.2.5. In Vivo PET-CT Imaging to Study Uptake of 18F-Fluorodeoxyglucose in the Heart
2.2.6. Measurement of Cardiac Reactive Oxygen Species (ROS) Production
2.2.7. Intracellular Lipid Droplet Evaluation
2.3. Western Blot Analysis
2.4. Cell Culture Studies
2.5. Statistical Analysis
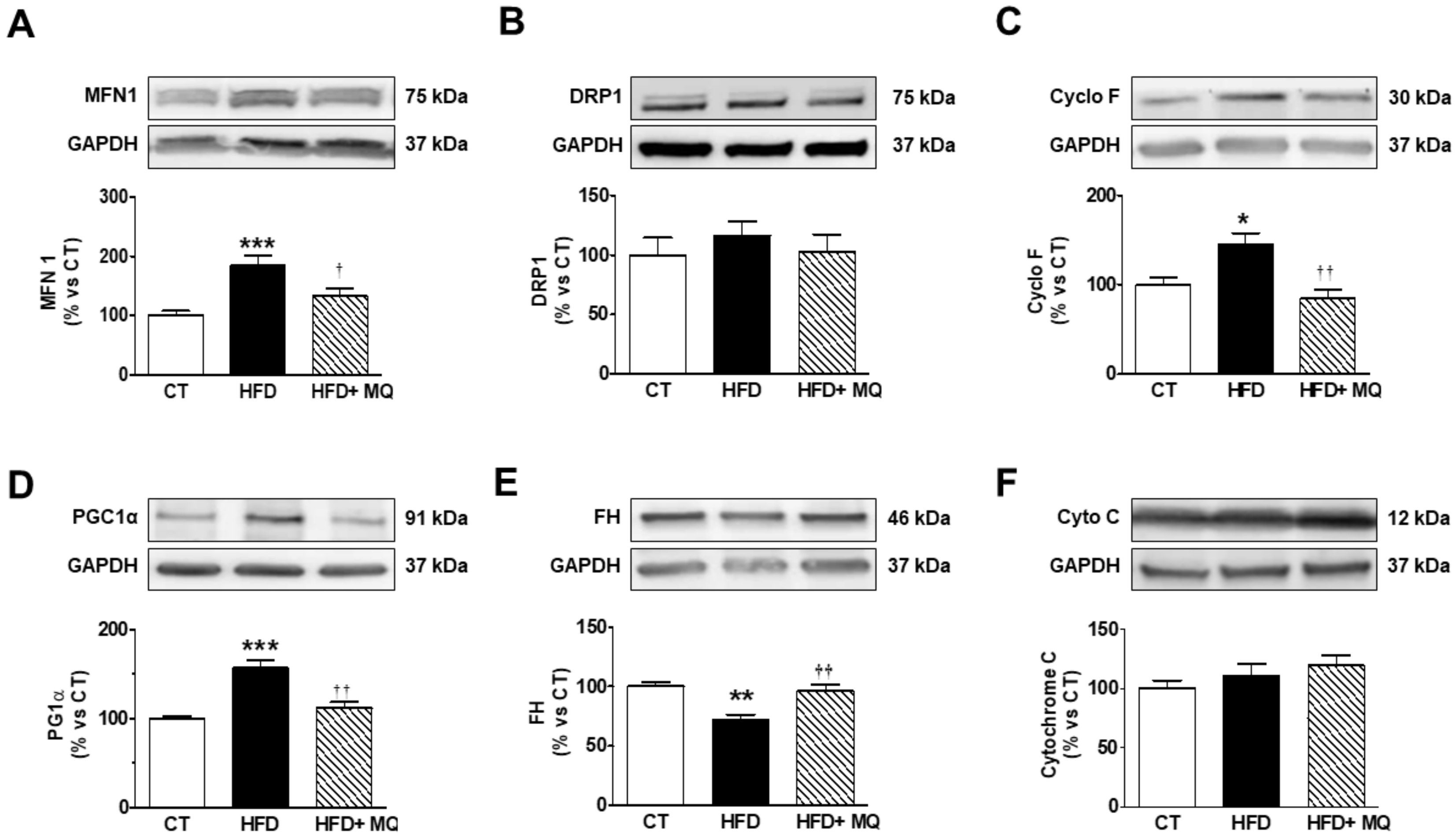
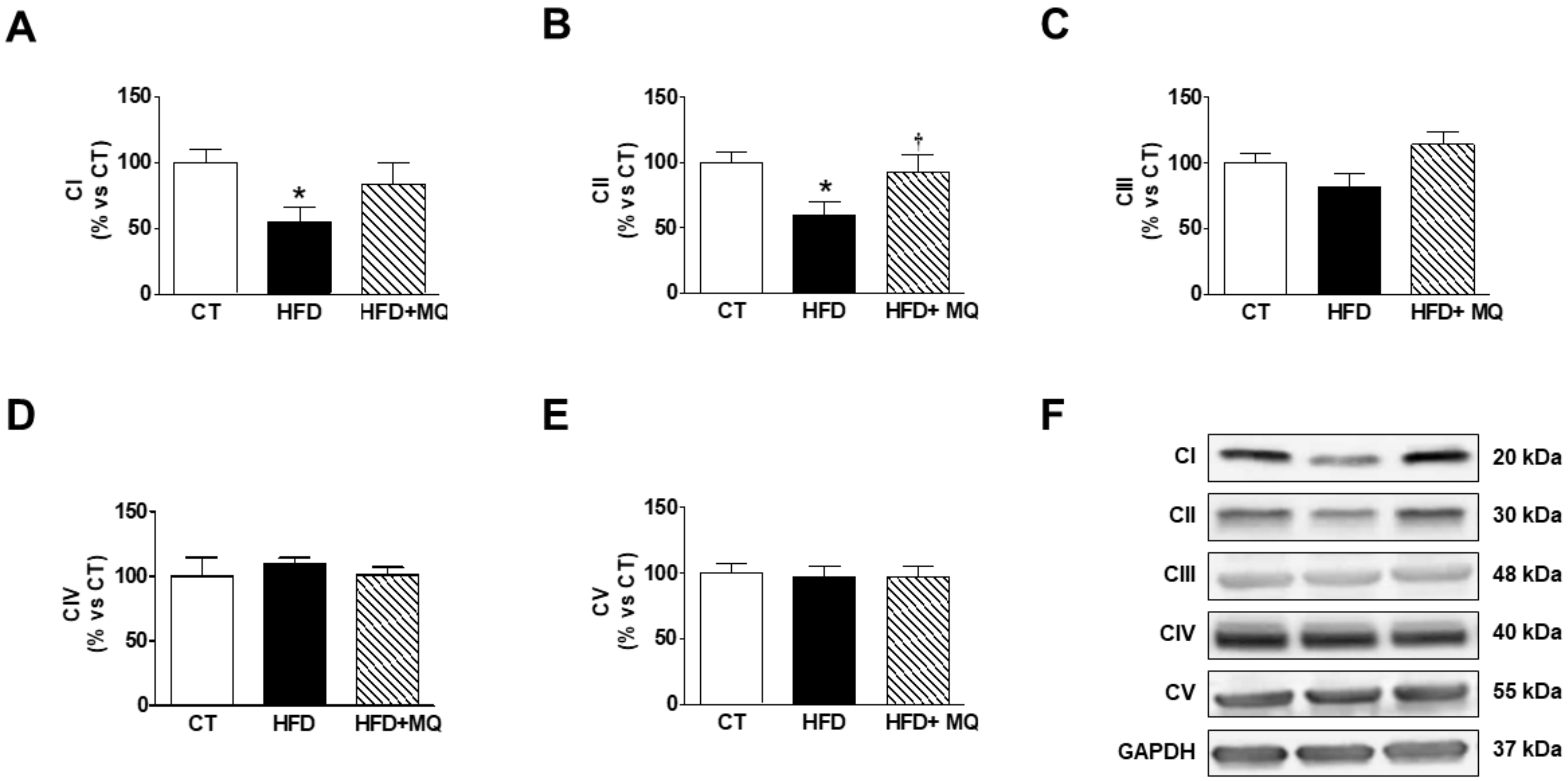
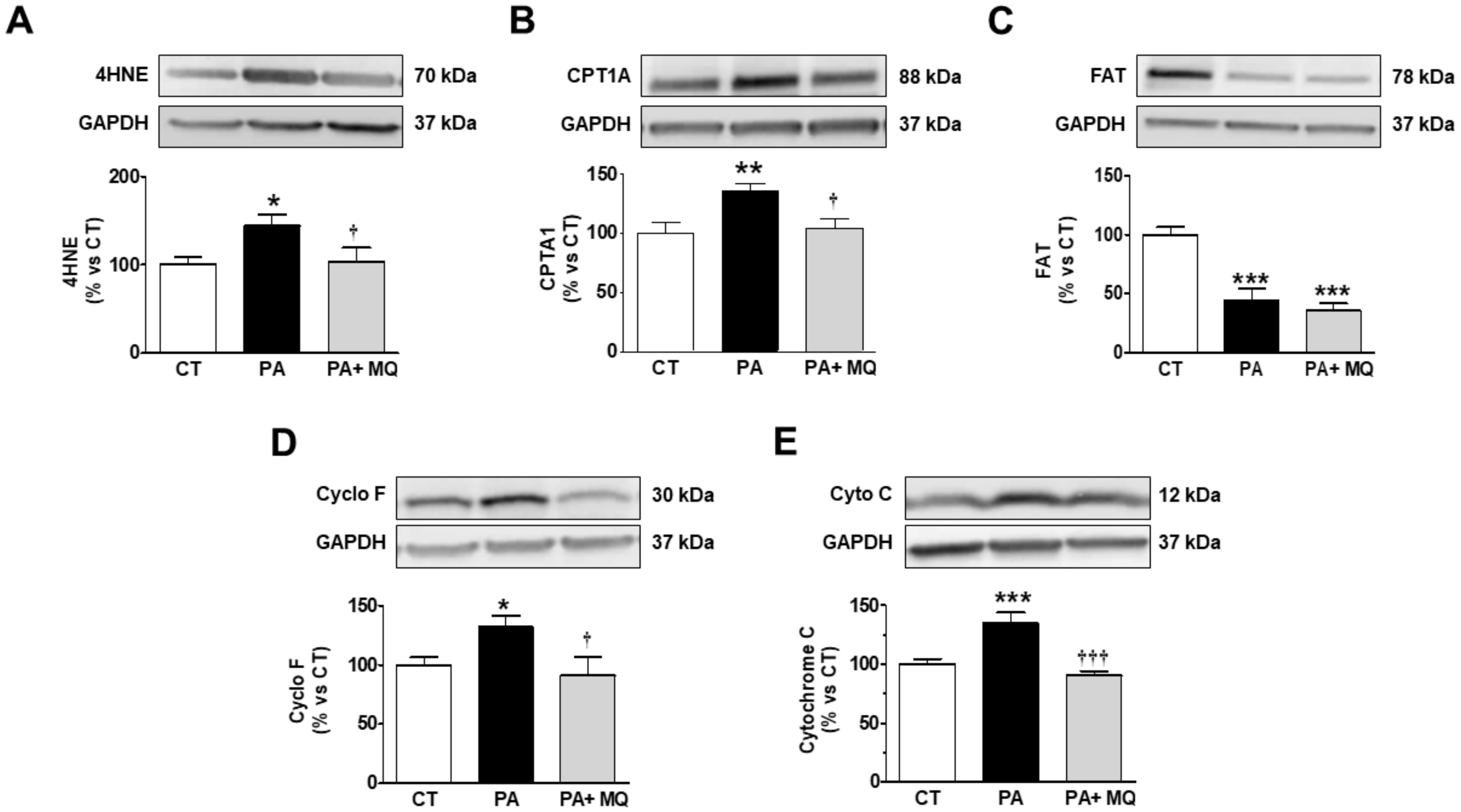
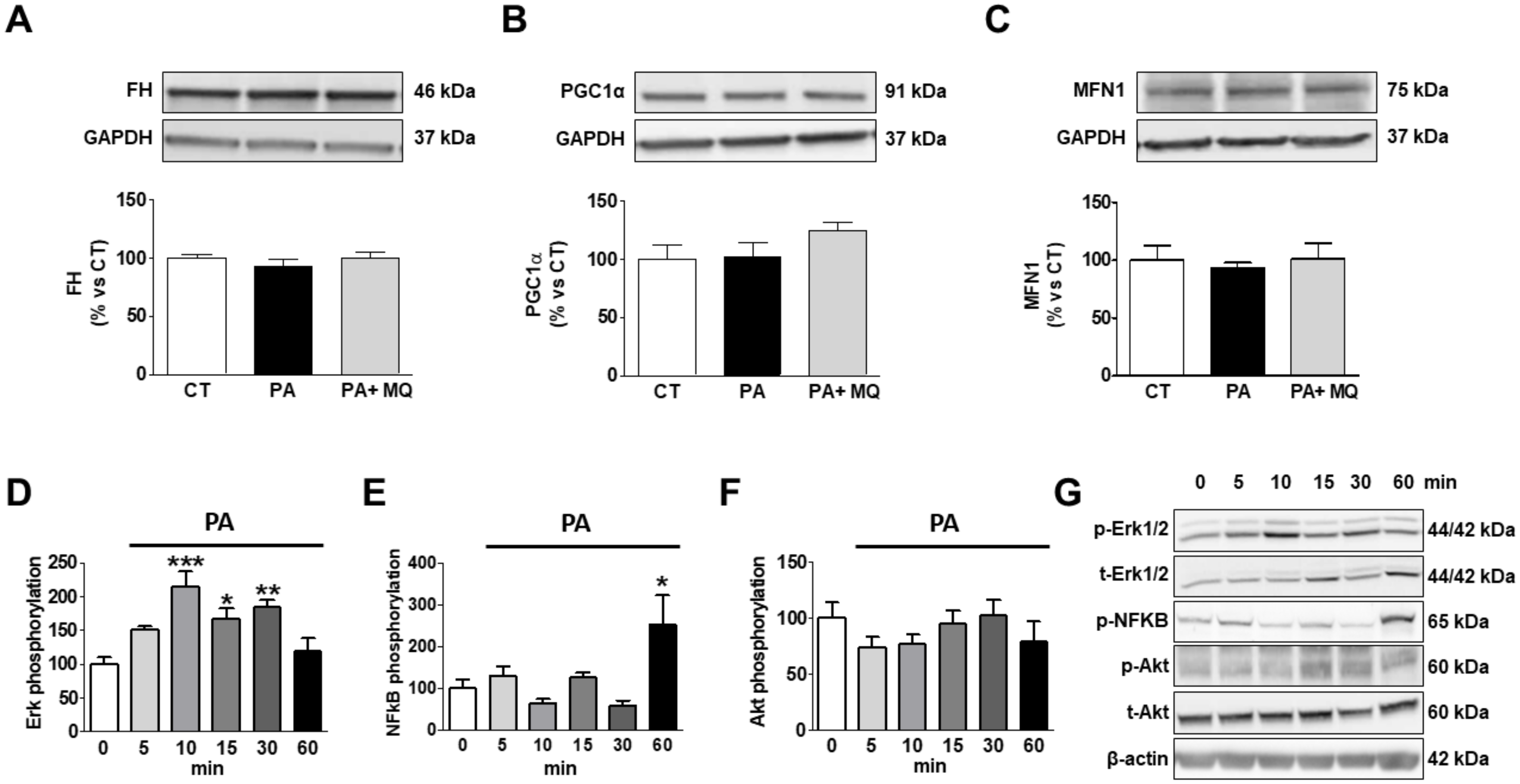
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Sulistyoningrum, D.C.; Glier, M.B.; Verchere, C.B.; Devlin, A.M. Altered glutathione homeostasis in heart augments cardiac lipotoxicity associated with diet-induced obesity in mice. J. Biol. Chem. 2011, 286, 42483–42493. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, T.; Kajio, H. Abdominal Obesity Is Associated With an Increased Risk of All-Cause Mortality in Patients with HFpEF. J. Am. Coll. Cardiol. 2017, 70, 2739–2749. [Google Scholar] [CrossRef]
- Oreopoulos, A.; Padwal, R.; Kalantar-Zadeh, K.; Fonarow, G.C.; Norris, C.M.; McAlister, F.A. Body mass index and mortality in heart failure: A meta-analysis. Am. Heart J. 2008, 156, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Marin-Royo, G.; Ortega-Hernandez, A.; Martinez-Martinez, E.; Jurado-Lopez, R.; Luaces, M.; Islas, F.; Gomez-Garre, D.; Delgado-Valero, B.; Lagunas, E.; Ramchandani, B.; et al. The Impact of Cardiac Lipotoxicity on Cardiac Function and Mirnas Signature in Obese and Non-Obese Rats with Myocardial Infarction. Sci. Rep. 2019, 9, 444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cedars, A.; Jenkins, C.M.; Mancuso, D.J.; Gross, R.W. Calcium-independent phospholipases in the heart: Mediators of cellular signaling, bioenergetics, and ischemia-induced electrophysiologic dysfunction. J. Cardiovasc. Pharmacol. 2009, 53, 277–289. [Google Scholar] [PubMed]
- Lim, H.Y.; Wang, W.; Wessells, R.J.; Ocorr, K.; Bodmer, R. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes Dev. 2011, 25, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Chiu, H.C.; Kovacs, A.; Ford, D.A.; Hsu, F.F.; Garcia, R.; Herrero, P.; Saffitz, J.E.; Schaffer, J.E. A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Investig. 2001, 107, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Abel, E.D.; O’Shea, K.M.; Ramasamy, R. Insulin resistance: Metabolic mechanisms and consequences in the heart. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2068–2076. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Tenorio, J.; Marin-Royo, G.; Martinez-Martinez, E.; Martin, R.; Miana, M.; Lopez-Andres, N.; Jurado-Lopez, R.; Gallardo, I.; Luaces, M.; San Roman, J.A.; et al. The role of oxidative stress in the crosstalk between leptin and mineralocorticoid receptor in the cardiac fibrosis associated with obesity. Sci. Rep. 2017, 7, 16802. [Google Scholar] [CrossRef] [Green Version]
- Joseph, L.C.; Subramanyam, P.; Radlicz, C.; Trent, C.M.; Iyer, V.; Colecraft, H.M.; Morrow, J.P. Mitochondrial oxidative stress during cardiac lipid overload causes intracellular calcium leak and arrhythmia. Heart Rhythm 2016, 13, 1699–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Royo, G.; Rodriguez, C.; Le Pape, A.; Jurado-Lopez, R.; Luaces, M.; Antequera, A.; Martinez-Gonzalez, J.; Souza-Neto, F.V.; Nieto, M.L.; Martinez-Martinez, E.; et al. The role of mitochondrial oxidative stress in the metabolic alterations in diet-induced obesity in rats. FASEB J. 2019, 33, 12060–12072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zozina, V.I.; Covantev, S.; Goroshko, O.A.; Krasnykh, L.M.; Kukes, V.G. Coenzyme Q10 in Cardiovascular and Metabolic Diseases: Current State of the Problem. Curr. Cardiol. Rev. 2018, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.; Gambardella, J.; Shu, J.; Santulli, G. Dietary fat is a key determinant in balancing mitochondrial dynamics in heart failure: A novel mechanism underlying the obesity paradox. Cardiovasc. Res. 2018, 114, 925–927. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Barahona, A.; Alonso-Barroso, E.; Perez, B.; Murphy, M.P.; Richard, E.; Desviat, L.R. Treatment with antioxidants ameliorates oxidative damage in a mouse model of propionic acidemia. Mol. Genet. Metab. 2017, 122, 43–50. [Google Scholar] [CrossRef]
- Marin-Royo, G.; Gallardo, I.; Martinez-Martinez, E.; Gutierrez, B.; Jurado-Lopez, R.; Lopez-Andres, N.; Gutierrez-Tenorio, J.; Rial, E.; Bartolome, M.A.V.; Nieto, M.L.; et al. Inhibition of galectin-3 ameliorates the consequences of cardiac lipotoxicity in a rat model of diet-induced obesity. Dis. Model Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Deleye, S.; Verhaeghe, J.; Wyffels, L.; Dedeurwaerdere, S.; Stroobants, S.; Staelens, S. Towards a reproducible protocol for repetitive and semi-quantitative rat brain imaging with (18) F-FDG: Exemplified in a memantine pharmacological challenge. Neuroimage 2014, 96, 276–287. [Google Scholar] [CrossRef]
- Malin, S.K.; Kashyap, S.R. Type 2 Diabetes Treatment in the Patient with Obesity. Endocrinol. Metab. Clin. N. Am. 2016, 45, 553–564. [Google Scholar] [CrossRef]
- Buchanan, J.; Mazumder, P.K.; Hu, P.; Chakrabarti, G.; Roberts, M.W.; Yun, U.J.; Cooksey, R.C.; Litwin, S.E.; Abel, E.D. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005, 146, 5341–5349. [Google Scholar] [CrossRef] [Green Version]
- Borisov, A.B.; Ushakov, A.V.; Zagorulko, A.K.; Novikov, N.Y.; Selivanova, K.F.; Edwards, C.A.; Russell, M.W. Intracardiac lipid accumulation, lipoatrophy of muscle cells and expansion of myocardial infarction in type 2 diabetic patients. Micron 2008, 39, 944–951. [Google Scholar] [CrossRef]
- Chitraju, C.; Walther, T.C.; Farese, R.V., Jr. The triglyceride synthesis enzymes DGAT1 and DGAT2 have distinct and overlapping functions in adipocytes. J. Lipid Res. 2019, 60, 1112–1120. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Shi, X.; Bharadwaj, K.G.; Ikeda, S.; Yamashita, H.; Yagyu, H.; Schaffer, J.E.; Yu, Y.H.; Goldberg, I.J. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J. Biol. Chem. 2009, 284, 36312–36323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.C.; Stone, S.J.; Zhou, P.; Buhman, K.K.; Farese, R.V., Jr. Dissociation of obesity and impaired glucose disposal in mice overexpressing acyl coenzyme a:diacylglycerol acyltransferase 1 in white adipose tissue. Diabetes 2002, 51, 3189–3195. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, J.; Zhao, Y.; Zhang, X.; Dai, B.; Zhan, J.; Yin, Z.; Nie, X.; Fu, X.D.; Chen, C.; et al. Nuclear miR-320 Mediates Diabetes-Induced Cardiac Dysfunction by Activating Transcription of Fatty Acid Metabolic Genes to Cause Lipotoxicity in the Heart. Circ. Res. 2019, 125, 1106–1120. [Google Scholar] [CrossRef] [PubMed]
- Chess, D.J.; Stanley, W.C. Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc. Res. 2008, 79, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiller, S.; Bluher, M.; Hoffmann, R. Plasma levels of free fatty acids correlate with type 2 diabetes mellitus. Diabetes Obes. Metab. 2018, 20, 2661–2669. [Google Scholar] [CrossRef]
- Thakker, G.D.; Frangogiannis, N.G.; Zymek, P.T.; Sharma, S.; Raya, J.L.; Barger, P.M.; Taegtmeyer, H.; Entman, M.L.; Ballantyne, C.M. Increased myocardial susceptibility to repetitive ischemia with high-fat diet-induced obesity. Obesity 2008, 16, 2593–2600. [Google Scholar] [CrossRef] [Green Version]
- Muthuramu, I.; Amin, R.; Postnov, A.; Mishra, M.; Jacobs, F.; Gheysens, O.; Van Veldhoven, P.P.; De Geest, B. Coconut Oil Aggravates Pressure Overload-Induced Cardiomyopathy without Inducing Obesity, Systemic Insulin Resistance, or Cardiac Steatosis. Int. J. Mol. Sci. 2017, 18, 1565. [Google Scholar] [CrossRef] [Green Version]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated Fatty acids. J. Nutr. Metab. 2012, 2012, 539426. [Google Scholar] [CrossRef]
- Van Bilsen, M.; Planavila, A. Fatty acids and cardiac disease: Fuel carrying a message. Acta Physiol. 2014, 211, 476–490. [Google Scholar] [CrossRef]
- Siddique, M.M.; Li, Y.; Chaurasia, B.; Kaddai, V.A.; Summers, S.A. Dihydroceramides: From Bit Players to Lead Actors. J. Biol. Chem. 2015, 290, 15371–15379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikman, B.T.; Summers, S.A. Ceramides as modulators of cellular and whole-body metabolism. J. Clin. Investig. 2011, 121, 4222–4230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaurasia, B.; Tippetts, T.S.; Mayoral Monibas, R.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 2019, 365, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Yazici, D.; Sezer, H. Insulin Resistance, Obesity and Lipotoxicity. Adv. Exp. Med. Biol. 2017, 960, 277–304. [Google Scholar] [CrossRef]
- Ji, R.; Akashi, H.; Drosatos, K.; Liao, X.; Jiang, H.; Kennel, P.J.; Brunjes, D.L.; Castillero, E.; Zhang, X.; Deng, L.Y.; et al. Increased de novo ceramide synthesis and accumulation in failing myocardium. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Shi, Y.; Lin, P.; Wang, X.; Zou, G.; Li, K. Sphingomyelin phosphodiesterase 1 (SMPD1) mediates the attenuation of myocardial infarction-induced cardiac fibrosis by astaxanthin. Biochem. Biophys. Res. Commun. 2018, 503, 637–643. [Google Scholar] [CrossRef]
- Martinez-Martinez, E.; Jurado-Lopez, R.; Valero-Munoz, M.; Bartolome, M.V.; Ballesteros, S.; Luaces, M.; Briones, A.M.; Lopez-Andres, N.; Miana, M.; Cachofeiro, V. Leptin induces cardiac fibrosis through galectin-3, mTOR and oxidative stress: Potential role in obesity. J. Hypertens. 2014, 32, 1104–1114. [Google Scholar] [CrossRef]
- Goldenberg, J.R.; Carley, A.N.; Ji, R.; Zhang, X.; Fasano, M.; Schulze, P.C.; Lewandowski, E.D. Preservation of Acyl Coenzyme A Attenuates Pathological and Metabolic Cardiac Remodeling Through Selective Lipid Trafficking. Circulation 2019, 139, 2765–2777. [Google Scholar] [CrossRef]
- Butler, T.J.; Ashford, D.; Seymour, A.M. Western diet increases cardiac ceramide content in healthy and hypertrophied hearts. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 991–998. [Google Scholar] [CrossRef]
- Summers, S.A. Could Ceramides Become the New Cholesterol? Cell. Metab. 2018, 27, 276–280. [Google Scholar] [CrossRef]
- Wang, D.D.; Toledo, E.; Hruby, A.; Rosner, B.A.; Willett, W.C.; Sun, Q.; Razquin, C.; Zheng, Y.; Ruiz-Canela, M.; Guasch-Ferre, M.; et al. Plasma Ceramides, Mediterranean Diet, and Incident Cardiovascular Disease in the PREDIMED Trial (Prevencion con Dieta Mediterranea). Circulation 2017, 135, 2028–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menuz, V.; Howell, K.S.; Gentina, S.; Epstein, S.; Riezman, I.; Fornallaz-Mulhauser, M.; Hengartner, M.O.; Gomez, M.; Riezman, H.; Martinou, J.C. Protection of C. elegans from anoxia by HYL-2 ceramide synthase. Science 2009, 324, 381–384. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell. Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Yang, J.; Cheng, H.; Yang, K.; Abendschein, D.R.; Gross, R.W. Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry 2005, 44, 16684–16694. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, J.; Yang, K.; Zhao, Z.; Abendschein, D.R.; Gross, R.W. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: A shotgun lipidomics study. Biochemistry 2007, 46, 6417–6428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croston, T.L.; Shepherd, D.L.; Thapa, D.; Nichols, C.E.; Lewis, S.E.; Dabkowski, E.R.; Jagannathan, R.; Baseler, W.A.; Hollander, J.M. Evaluation of the cardiolipin biosynthetic pathway and its interactions in the diabetic heart. Life Sci. 2013, 93, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, M.; Phoon, C.K.; Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 2014, 55, 1–16. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Claypool, S.M.; Koehler, C.M. The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 2012, 37, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Chen, T.; Wanagat, J.; Laflamme, M.; Marcinek, D.J.; Emond, M.J.; Ngo, C.P.; Prolla, T.A.; Rabinovitch, P.S. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.F.; Chen, T.; Szeto, H.; Nieves-Cintron, M.; Kutyavin, V.; Santana, L.F.; Rabinovitch, P.S. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 2011, 58, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelenik, T.; Flogel, U.; Alvarez-Hernandez, E.; Scheiber, D.; Zweck, E.; Ding, Z.; Rothe, M.; Mastrototaro, L.; Kohlhaas, V.; Kotzka, J.; et al. Insulin Resistance and Vulnerability to Cardiac Ischemia. Diabetes 2018, 67, 2695–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarrola, J.; Sadaba, R.; Garcia-Pena, A.; Arrieta, V.; Martinez-Martinez, E.; Alvarez, V.; Fernandez-Celis, A.; Gainza, A.; Santamaria, E.; Fernandez-Irigoyen, J.; et al. A role for fumarate hydratase in mediating oxidative effects of galectin-3 in human cardiac fibroblasts. Int. J. Cardiol. 2018, 258, 217–223. [Google Scholar] [CrossRef]
- Tian, H.; Yu, D.; Hu, Y.; Zhang, P.; Yang, Y.; Hu, Q.; Li, M. Angiotensin II upregulates cyclophilin A by enhancing ROS production in rat cardiomyocytes. Mol. Med. Rep. 2018, 18, 4349–4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.Y.; Hausenloy, D.J.; Arjun, S.; Price, A.N.; Davidson, S.M.; Lythgoe, M.F.; Yellon, D.M. Mitochondrial cyclophilin-D as a potential therapeutic target for post-myocardial infarction heart failure. J. Cell. Mol. Med. 2011, 15, 2443–2451. [Google Scholar] [CrossRef]
- Cao, M.; Yuan, W.; Peng, M.; Mao, Z.; Zhao, Q.; Sun, X.; Yan, J. Role of CyPA in cardiac hypertrophy and remodeling. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.M.; Hsu, Y.M.; Ying, M.C.; Tsai, F.J.; Tsai, C.H.; Chung, J.G.; Yang, J.S.; Tang, C.H.; Cheng, L.Y.; Su, P.H.; et al. High-density lipoprotein ameliorates palmitic acid-induced lipotoxicity and oxidative dysfunction in H9c2 cardiomyoblast cells via ROS suppression. Nutr. Metab. 2019, 16, 36. [Google Scholar] [CrossRef]
- Shen, C.J.; Kong, B.; Shuai, W.; Liu, Y.; Wang, G.J.; Xu, M.; Zhao, J.J.; Fang, J.; Fu, H.; Jiang, X.B.; et al. Myeloid differentiation protein 1 protected myocardial function against high-fat stimulation induced pathological remodelling. J. Cell Mol. Med. 2019, 23, 5303–5316. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CT | MitoQ | HFD | HFD-MitoQ | |
|---|---|---|---|---|
| HW/TL (g/cm) | 0.20 ± 0.003 | 0.21 ± 0.006 | 0.24 ± 0.007 *** | 0.22 ± 0.004 *† |
| CSA (µm2) | 301.2 ± 7.8 | 308.1 ± 7.7 | 380.3 ± 11.9 *** | 322.7 ± 10.8 ††† |
| IVTs (mm) | 0.130 ± 0.008 | 0.137 ± 0.006 | 0.143 ± 0.02 | 0.135 ± 0.008 |
| PWTs (mm) | 0.190 ± 0.013 | 0.140 ± 0.008 | 0.171 ± 0.012 | 0.167 ± 0.019 |
| ESD (mm) | 0.352 ± 0.034 | 0.403 ± 0.012 | 0.293 ± 0.030 | 0.349 ± 0.032 |
| EDD (mm) | 0.653 ± 0.043 | 0.670 ± 0.022 | 0.615 ± 0.023 | 0.613 ± 0.032 |
| EF (%) | 77.8 ± 3.7 | 73.2 ± 2.8 | 82.3 ± 1.9 | 77.3 ± 5.3 |
| FS (%) | 44.0 ± 3.3 | 41.4 ± 2.0 | 49.3 ± 4.7 | 43.2 ± 4.8 |
| E/A | 1.05 ± 0.08 | 1.20 ± 0.03 | 1.07 ± 0.05 | 1.20 ± 0.02 |
| SBP (mmHg) | 133.7 ± 2.5 | 130.6 ± 3.9 | 138.1 ± 2.1 | 133.9 ± 2.6 |
| Cardiac Fibrosis | Cardiac Hypertrophy | 18-FDG Uptake | ||||
|---|---|---|---|---|---|---|
| r | p | r | p | r | p | |
| Cardiac fibrosis | - | - | 0.792 | <0.0001 | −0.636 | 0.003 |
| Cardiac hypertrophy | 0.792 | <0.0001 | - | - | −0.661 | 0.002 |
| Mitochondrial TGs | 0.586 | 0.007 | 0.641 | 0.003 | −0.578 | 0.01 |
| Mitochondrial FFA | 0.759 | <0.0001 | 0.487 | 0.048 | - | - |
| Mitochondrial CL | −0.555 | 0.017 | - | - | - | - |
| Mitochondrial Ceramides | 0.547 | 0.015 | 0.624 | 0.006 | −0.460 | 0.041 |
| MFN1 | - | - | 0.549 | 0.018 | −0.581 | 0.011 |
| FH | −0.564 | 0.01 | −0.469 | 0.037 | −0.596 | 0.006 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-González, S.; Marín-Royo, G.; Jurado-López, R.; Bartolomé, M.V.; Romero-Miranda, A.; Luaces, M.; Islas, F.; Nieto, M.L.; Martínez-Martínez, E.; Cachofeiro, V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. https://doi.org/10.3390/cells9020451
Jiménez-González S, Marín-Royo G, Jurado-López R, Bartolomé MV, Romero-Miranda A, Luaces M, Islas F, Nieto ML, Martínez-Martínez E, Cachofeiro V. The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells. 2020; 9(2):451. https://doi.org/10.3390/cells9020451
Chicago/Turabian StyleJiménez-González, Sara, Gema Marín-Royo, Raquel Jurado-López, María Visitación Bartolomé, Ana Romero-Miranda, María Luaces, Fabián Islas, María Luisa Nieto, Ernesto Martínez-Martínez, and Victoria Cachofeiro. 2020. "The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats" Cells 9, no. 2: 451. https://doi.org/10.3390/cells9020451
APA StyleJiménez-González, S., Marín-Royo, G., Jurado-López, R., Bartolomé, M. V., Romero-Miranda, A., Luaces, M., Islas, F., Nieto, M. L., Martínez-Martínez, E., & Cachofeiro, V. (2020). The Crosstalk between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells, 9(2), 451. https://doi.org/10.3390/cells9020451