Antifibrotic Effects of Amyloid-Beta and Its Loss in Cirrhotic Liver


 ,
,  , ,
, ,  , , , ,
, , , , 
Abstract
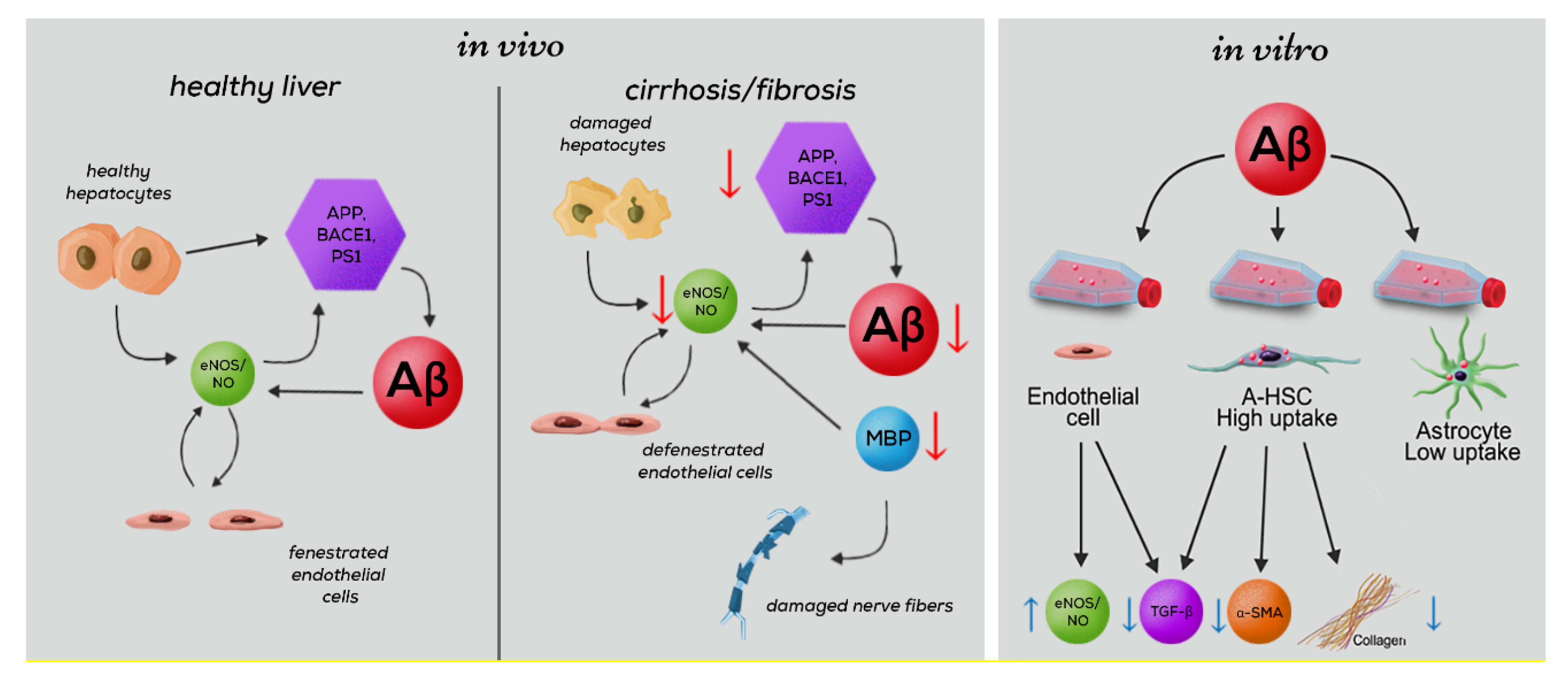
:1. Introduction
2. Materials and Methods
2.1. Human Liver Tissue Samples
2.2. Animal Models
2.3. Cell Culture
2.4. Western Blot Analyses
2.5. Real-Time qPCR of Rat Liver Tissue and Cell Cultures
2.6. Real-Time qPCR of Human Liver Tissue
2.7. Additional Information to qRT-PCR Analysis and Use of Housekeeping Gene YWAHZ
2.8. Aβ Quantification in Cell Cultures
2.9. Quantification of TGF-β1 in Cell Culture Supernatants
2.10. Quantification of Aβ Peptides in Liver and Brain Homogenates
2.11. Immunofluorescence Staining
2.12. Collagen 1 ELISA of Cell Culture Supernatants
2.13. Statistical Analyses
2.14. Ethics Approval Statements
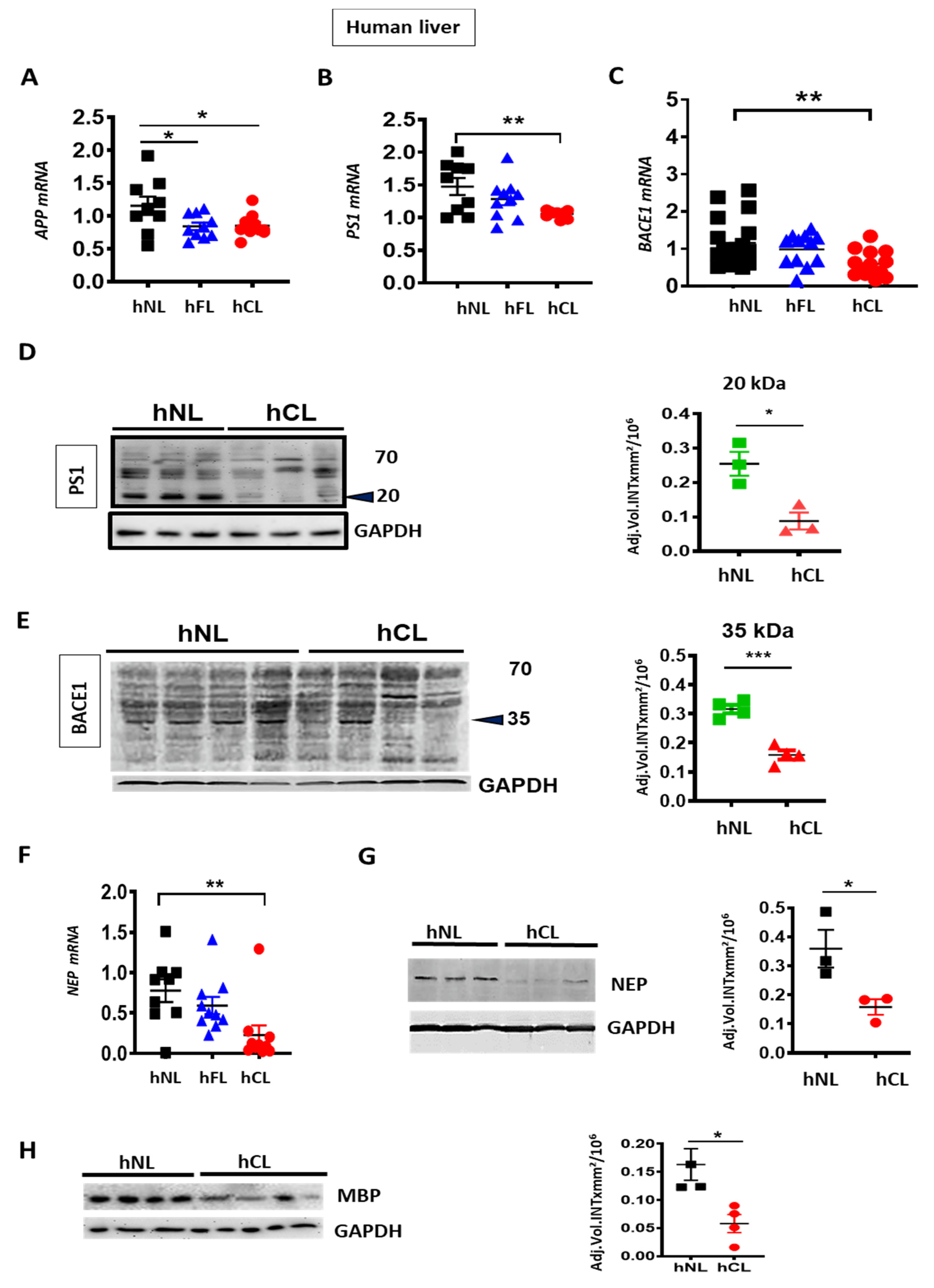
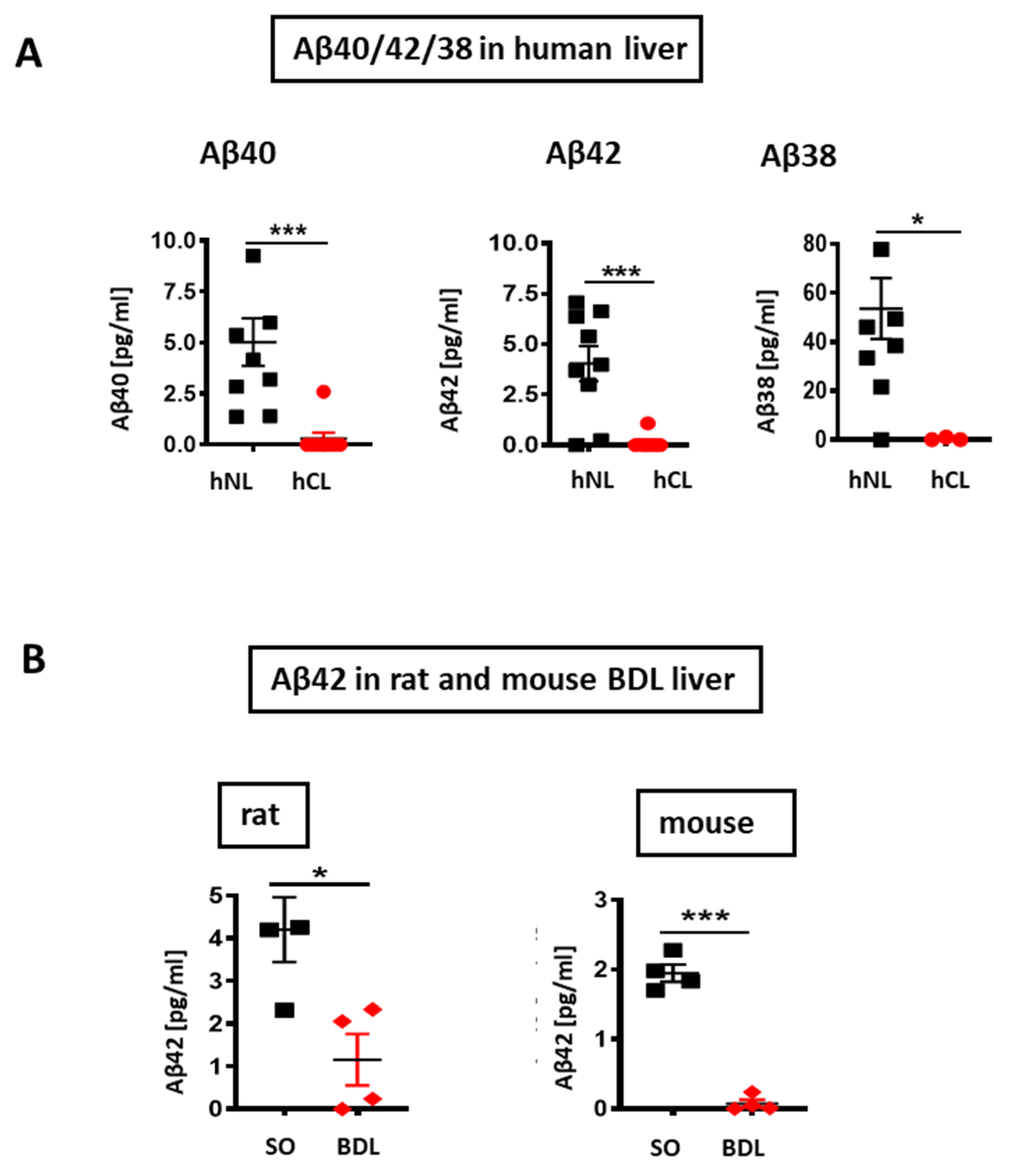
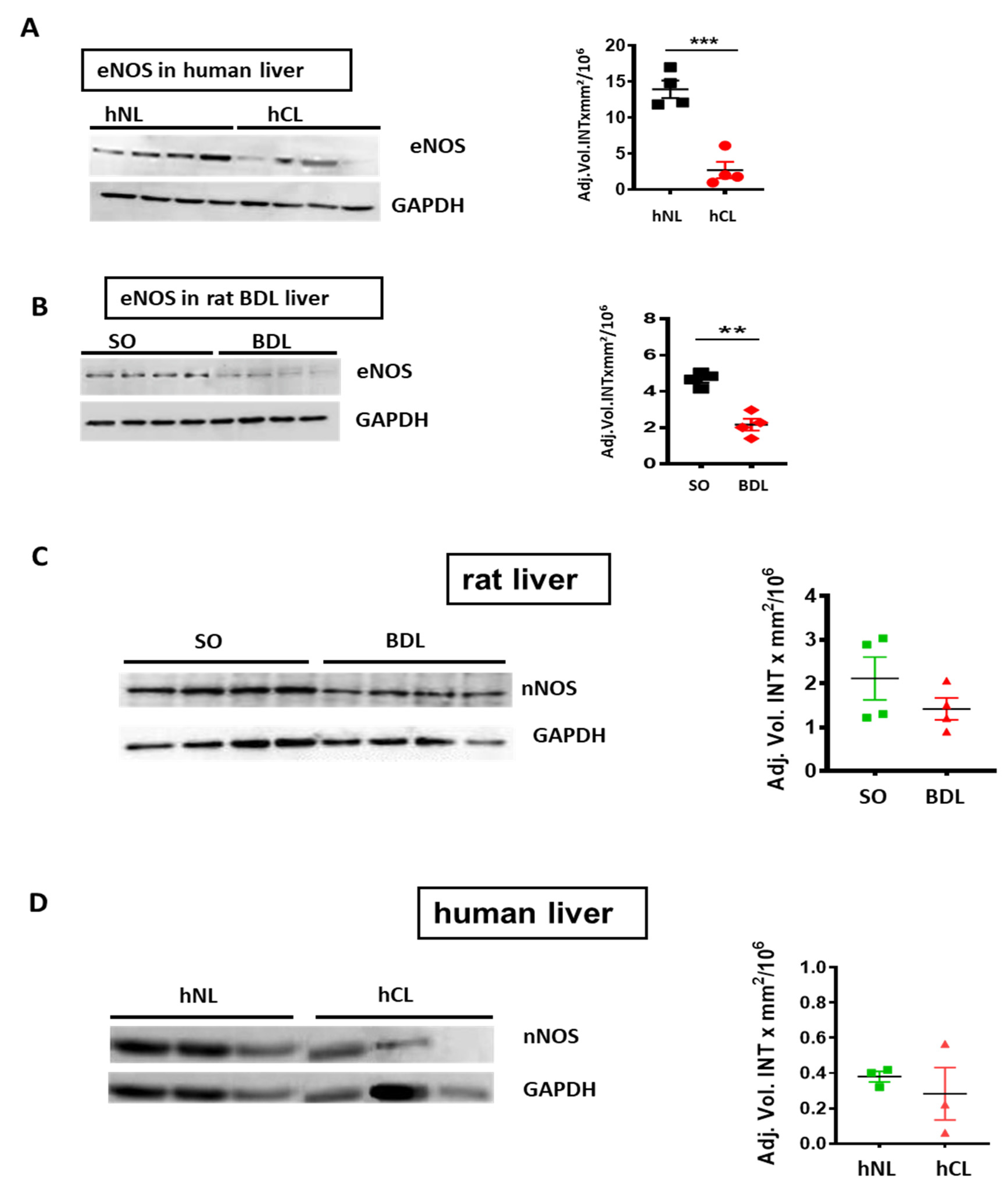
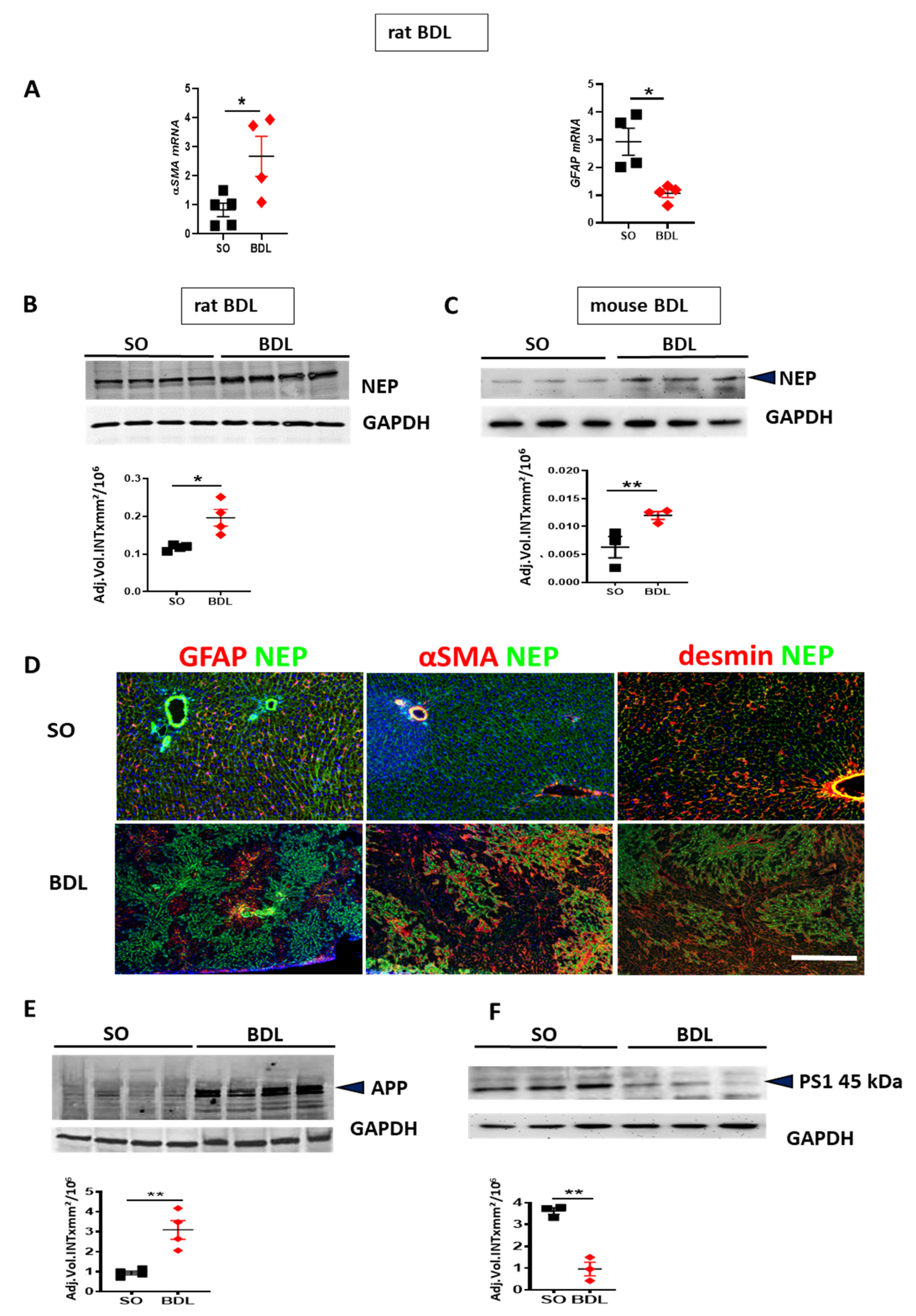
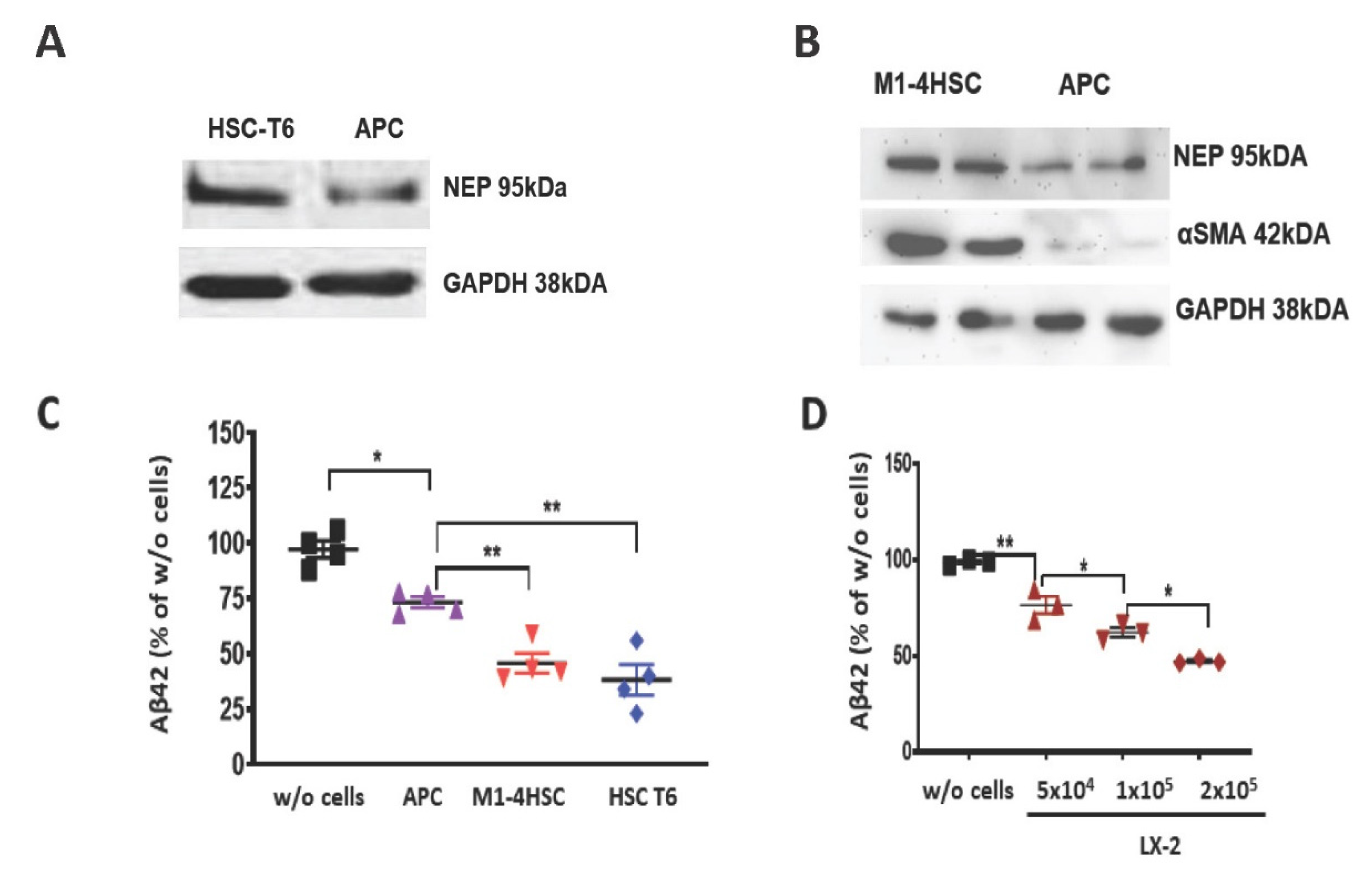
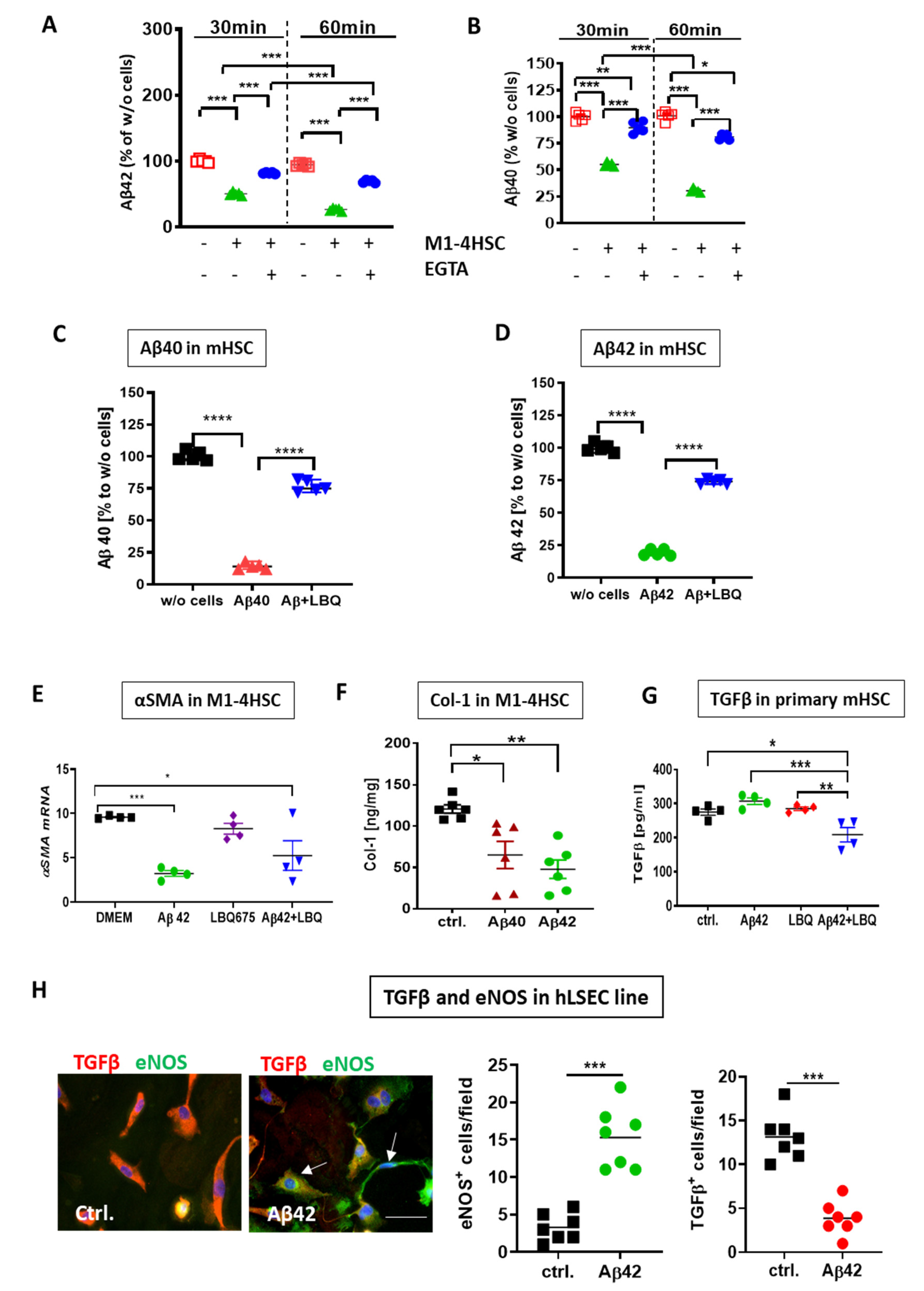
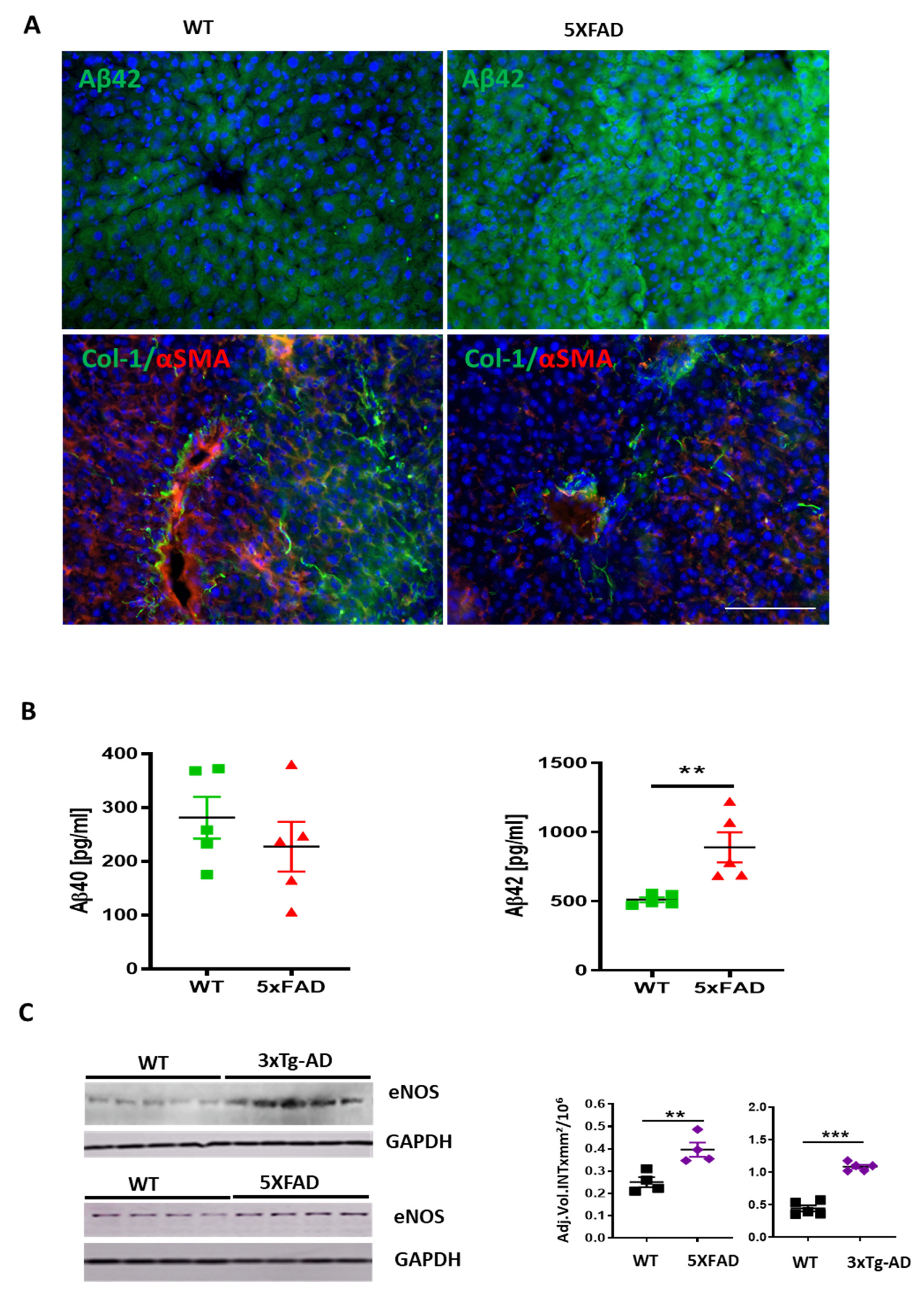
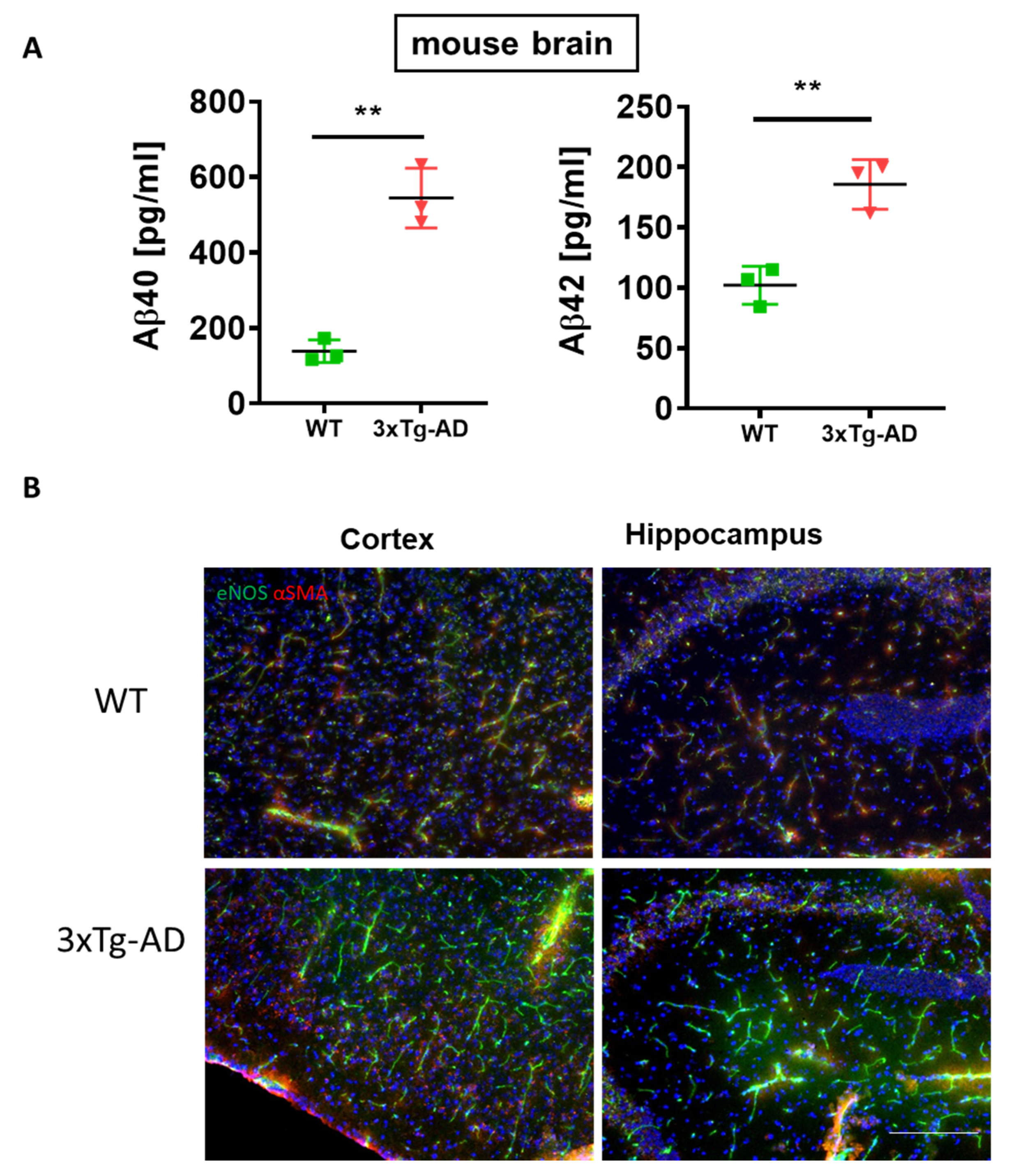
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghiso, J.; Shayo, M.; Calero, M.; Ng, D.; Tomidokoro, Y.; Gandy, S.; Rostagno, A.; Frangione, B. Systemic catabolism of Alzheimer’s Aβ40 and Aβ42. J. Biol. Chem. 2004, 279, 45897–45908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Buniatian, G.H.; Gebhardt, R.; Mecke, D.; Traub, P.; Wiesinger, H. Common myofibroblastic features of newborn rat astrocytes and cirrhotic rat liver stellate cells in early cultures and in vivo. Neurochem. Int. 1999, 35, 317–327. [Google Scholar] [CrossRef]
- Buniatian, G.H.; Hartmann, H.J.; Traub, P.; Weser, U.; Wiesinger, H.; Gebhardt, R. Acquisition of blood-tissue barrier-supporting features by hepatic stellate cells and astrocytes of myofibroblastic phenotype. Inverse dynamics of metallothionein and glial fibrillary acidic protein expression. Neurochem. Int. 2001, 38, 373–383. [Google Scholar] [CrossRef]
- Pihlaja, R.; Koistinaho, J.; Kauppinen, R.; Sandholm, J.; Tanila, H.; Koistinaho, M. Multiple cellular and molecular mechanisms Are involved in human Aβ clearance by transplanted adult astrocytes. Glia 2011, 59, 1643–1657. [Google Scholar] [CrossRef]
- Huse, J.T.; Byant, D.; Yang, Y.; Pijak, D.S.; D’Souza, I.; Lah, J.J.; Lee, V.M.Y.; Doms, R.W.; Cook, D.G. Endoproteolysis of β-secretase (β-site amyloid precursor protein-cleaving enzyme) within its catalytic domain: A potential mechanism for regulation. J. Biol. Chem. 2003, 278, 17141–17149. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Boyd-Kimball, D. Amyloid β-Peptide (1-42) Contributes to the oxidative stress and neurodegeneration found in Alzheimer disease brain. Brain Pathol. 2006, 14, 426–432. [Google Scholar] [CrossRef]
- Sutcliffe, J.G.; Hedlund, P.B.; Thomas, E.A.; Bloom, F.E.; Hilbush, B.S. Peripheral reduction of β-amyloid is sufficient to reduce brain β-amyloid: Implications for Alzheimer’s disease. J. Neurosci. Res. 2011, 89, 808–814. [Google Scholar] [CrossRef]
- Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Castaño, E.M.; Van Vickle, G.D.; Kalback, W.M.; Patton, R.L.; Luehrs, D.C.; Daugs, I.D.; Kuo, Y.M.; et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimer’s Dement. 2009, 5, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Shirotani, K.; Tsubuki, S.; Iwata, N.; Takaki, Y.; Harigaya, W.; Maruyama, K.; Kiryu-Seo, S.; Kiyama, H.; Iwata, H.; Tomita, T.; et al. Neprilysin degrades both amyloid β peptides 1-40 and 1-42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J. Biol. Chem. 2001, 276, 21895–21901. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T. Changes in expression of bile canalicular CD10 and sinusoidal CD105 (endoglin) in peritumoral hepatic tissue. Tumori 2009, 95, 495–500. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funyu, J.; Mochida, S.; Inao, M.; Matsui, A.; Fujiwara, K. VEGF can act as vascular permeability factor in the hepatic sinusoids through upregulation of porosity of endothelial cells. Biochem. Biophys. Res. Commun. 2001, 280, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Invest. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartz, A.M.S.; Bauer, B.; Soldner, E.L.B.; Wolf, A.; Boy, S.; Backhaus, R.; Mihaljevic, I.; Bogdahn, U.; Klünemann, H.H.; Schuierer, G.; et al. Amyloid-β contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 2012, 43, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, V.E.; Audus, K.L. Nitric oxide and blood-brain barrier integrity. Antioxidants Redox Signal. 2001, 3, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. β-Amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, A.; Dal Pra, I.; Menapace, L.; Pacchiana, R.; Whitfield, J.F.; Armato, U. Soluble amyloid beta-peptide and myelin basic protein strongly stimulate, alone and in synergism with combined proinflammatory cytokines, the expression of functional nitric oxide synthase-2 in normal adult human astrocytes. Int. J. Mol. Med. 2005, 16, 801–807. [Google Scholar]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles. Neuron 2004, 39, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Cholongitas, E.; Marelli, L.; Shusang, V.; Senzolo, M.; Rolles, K.; Patch, D.; Burroughs, A.K. A systematic review of the performance of the Model for End-Stage Liver Disease (MELD) in the setting of liver transplantation. Liver Transplant. 2006, 12, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Arias, M.; Sauer-Lehnen, S.; Treptau, J.; Janoschek, N.; Theuerkauf, I.; Buettner, R.; Gressner, A.M.; Weiskirchen, R. Adenoviral expression of a transforming growth factor-β1 antisense mRNA is effective in preventing liver fibrosis in bile-duct ligated rats. BMC Gastroenterol. 2003, 3, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tag, C.G.; Sauer-Lehnen, S.; Weiskirchen, S.; Borkham-Kamphorst, E.; Tolba, R.H.; Tacke, F.; Weiskirchen, R. Bile duct ligation in mice: Induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J. Vis. Exp. 2015, 96, e52438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tag, C.; Weiskirchen, S.; Hittatiya, K.; Tacke, F.; Tolba, R.; Weiskirchen, R. Induction of experimental obstructive cholestasis in mice. Lab. Anim. 2015, 49, 70–80. [Google Scholar] [CrossRef]
- Proell, V.; Mikula, M.; Fuchs, E.; Mikulits, W. The plasticity of p19ARF null hepatic stellate cells and the dynamics of activation. Biochim. Biophys. Acta-Mol. Cell Res. 2005, 1744, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.; Piantedosi, R.; Frank, J.; Lalazar, A.; Rockey, D.C.; Friedman, S.L.; Blaner, W.S. An immortalized rat liver stellate cell line (HSC-T6): A new cell model for the study of retinoid metabolism in vitro. J. Lipid Res. 2000, 41, 882–893. [Google Scholar]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Lourhmati, A.; Buniatian, G.H.; Paul, C.; Verleysdonk, S.; Buecheler, R.; Buadze, M.; Proksch, B.; Schwab, M.; Gleiter, C.H.; Danielyan, L. Age-dependent astroglial vulnerability to hypoxia and glutamate: The role for erythropoietin. PLoS ONE 2013, 8, e77182. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Huang, H.; Bihaqi, S.W.; Cui, L.; Zawia, N.H. In vitro Pb exposure disturbs the balance between Aβ production and elimination: The role of AβPP and neprilysin. Neurotoxicology 2011, 32, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G.; et al. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1α. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.D.; Sihota, K.K.; Byrne, C.D.; Cagampang, F.R. The housekeeping gene YWHAZ remains stable in a model of developmentally primed non-alcoholic fatty liver disease. Liver Int. 2012, 32, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, S.; Tag, C.G.; Sauer-Lehnen, S.; Tacke, F.; Weiskirchen, R. Isolation and culture of primary murine hepatic stellate cells. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2017; Volume 1627, pp. 165–191. [Google Scholar]
- Liao, M.C.; Ahmed, M.; Smith, S.O.; Van Nostrand, W.E. Degradation of amyloid β protein by purified myelin basic protein. J. Biol. Chem. 2009, 284, 28917–28925. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.W.; Song, H.J.; Back, S.J.; Kim, T.H.; Cho, S.H.; Han, J.-Y.; Yoo, K.; Lee, Y.S.; Chung, K.W. Decreased hepatic nerve fiber innervation in patients with liver cirrhosis. Gut Liver 2010, 1, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takase, S.; Leo, M.A.; Nouchi, T.; Lieber, C.S. Desmin distinguishes cultured fat-storing cells from myofibroblasts, smooth muscle cells and fibroblasts in the rat. J. Hepatol. 1988, 6, 267–276. [Google Scholar] [CrossRef]
- Buniatian, G.; Gebhardt, R.; Schrenk, D.; Hamprecht, B. Colocalization of three types of intermediate filament proteins in perisinusoidal stellate cells: Glial fibrillary acidic protein as a new cellular marker. Eur. J. Cell Biol. 1996, 70, 23–32. [Google Scholar]
- Chauhan, A.; Ray, I.; Chauhan, V.P.S. Interaction of amyloid beta-protein with anionic phospholipids: Possible involvement of Lys28 and C-terminus aliphatic amino acids. Neurochem. Res. 2000, 25, 423–429. [Google Scholar] [CrossRef]
- Friedman, S.L.; Roll, F.J.; Boyles, J.; Bissell, D.M. Hepatic lipocytes: The principal collagen-producing cells of normal rat liver. Proc. Natl. Acad. Sci. USA 1985, 82, 8681–8685. [Google Scholar] [CrossRef] [Green Version]
- Karrar, A.; Broomé, U.; Uzunel, M.; Qureshi, A.R.; Sumitran-Holgersson, S. Human liver sinusoidal endothelial cells induce apoptosis in activated T cells: A role in tolerance induction. Gut 2007, 56, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Ou-Yang, M.H.; Van Nostrand, W.E. The absence of myelin basic protein promotes neuroinflammation and reduces amyloid β-protein accumulation in Tg-5xFAD mice. J. Neuroinflammation 2013, 10, 134. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.M.; Lee, S.; Yang, S.H.; Kim, H.Y.; Lee, M.J.; Kim, H.V.; Kim, J.; Baek, S.; Yun, J.; Kim, D.; et al. Age-dependent inverse correlations in CSF and plasma amyloid-β(1-42) concentrations prior to amyloid plaque deposition in the brain of 3×Tg-AD mice. Sci. Rep. 2016, 6, 20185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhanam, A.V.R.; D’Uscio, L.V.; He, T.; Das, P.; Younkin, S.G.; Katusic, Z.S. Uncoupling of endothelial nitric oxide synthase in cerebral vasculature of Tg2576 mice. J. Neurochem. 2015, 134, 1129–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Broome, U.; Uzunel, M.; Nava, S.; Ge, X.; Kumagai-Braesch, M.; Hultenby, K.; Christensson, B.; Ericzon, B.G.; Holgersson, J.; et al. Capillarization of hepatic sinusoid by liver endothelial cell-reactive autoantibodies in patients with cirrhosis and chronic hepatitis. Am. J. Pathol. 2003, 163, 1275–1289. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.S.; Van Nostrand, W.E. Humanin rescues human cerebrovascular smooth muscle cells from Aβ-induced toxicity. J. Neurochem. 2003, 84, 266–272. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, L.; Puttagunta, L.; Martinez-Cuesta, M.A.; Kneteman, N.; Mayers, I.; Moqbel, R.; Hamid, Q.; Radomski, M.W. Distribution of nitric oxide synthase in normal and cirrhotic human liver. Proc. Natl. Acad. Sci. USA 2002, 99, 17161–17166. [Google Scholar] [CrossRef] [Green Version]
- Duong, H.T.T.; Dong, Z.; Su, L.; Boyer, C.; George, J.; Davis, T.P.; Wang, J. The use of nanoparticles to deliver nitric oxide to hepatic stellate cells for treating liver fibrosis and portal hypertension. Small 2015, 11, 2291–2304. [Google Scholar] [CrossRef]
- Ishii, K.; Muelhauser, F.; Liebl, U.; Picard, M.; Kühl, S.; Penke, B.; Bayer, T.; Wiessler, M.; Hennerici, M.; Beyreuther, K.; et al. Subacute NO generation induced by Alzheimer’s β-amyloid in the living brain: Reversal by inhibition of the inducible NO synthase. FASEB J. 2018, 14, 1485–1489. [Google Scholar]
- Nagai, N.; Ito, Y.; Shibata, T.; Kubo, E.; Sasaki, H. A positive feedback loop between nitric oxide and amyloid β (1-42) accelerates mitochondrial damage in human lens epithelial cells. Toxicology 2017, 381, 19–30. [Google Scholar] [CrossRef]
- Wang, Y.R.; Wang, Q.H.; Zhang, T.; Liu, Y.H.; Yao, X.Q.; Zeng, F.; Li, J.; Zhou, F.Y.; Wang, L.; Yan, J.C.; et al. Associations between hepatic functions and plasma amyloid-beta levels—implications for the capacity of liver in peripheral amyloid-beta clearance. Mol. Neurobiol. 2017, 54, 2338–2344. [Google Scholar] [CrossRef]
- Herath, C.B.; Lubel, J.S.; Jia, Z.; Velkoska, E.; Casley, D.; Brown, L.; Tikellis, C.; Burrell, L.M.; Angus, P.W. Portal pressure responses and angiotensin peptide production in rat liver are determined by relative activity of ACE and ACE2. Am. J. Physiol. Liver Physiol. 2009, 297, G98–G106. [Google Scholar] [CrossRef] [Green Version]
- Ferro, C.J.; Spratt, J.C.; Haynes, W.G.; Webb, D.J. Inhibition of neutral endopeptidase causes vasoconstriction of human resistance vessels in vivo. Circulation 1998, 97, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Haass, C. Intramembrane proteolysis by presenilins. Nat. Rev. Mol. Cell Biol. 2000, 1, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Drummond, E. Developing therapeutic vaccines against Alzheimers disease. Expert Rev. Vaccines 2016, 15, 401–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, P.C.; Willis, B.A.; Lowe, S.L.; Dean, R.A.; Monk, S.A.; Cocke, P.J.; Audia, J.E.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; et al. The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 2015, 35, 1199–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; DeLong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A Plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef] [Green Version]
- Bansal, R.; Prakash, J.; Post, E.; Beljaars, L.; Schuppan, D.; Poelstra, K. Novel engineered targeted interferon-gamma blocks hepatic fibrogenesis in mice. Hepatology 2011, 54, 586–596. [Google Scholar] [CrossRef]
- Ng, C.T.; Fong, L.Y.; Sulaiman, M.R.; Moklas, M.A.M.; Yong, Y.K.; Hakim, M.N.; Ahmad, Z. Interferon-Gamma Increases Endothelial Permeability by Causing Activation of p38 MAP Kinase and Actin Cytoskeleton Alteration. J. Interf. Cytokine Res. 2015, 35, 513–522. [Google Scholar] [CrossRef]
- Durán, W.N.; Beuve, A.V.; Sánchez, F.A. Nitric oxide, S-Nitrosation, and endothelial permeability. IUBMB Life 2013, 65, 819–826. [Google Scholar] [CrossRef] [Green Version]
- Tai, L.M.; Holloway, K.A.; Male, D.K.; Loughlin, A.J.; Romero, I.A. Amyloid-β-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J. Cell. Mol. Med. 2010, 14, 1101–1112. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human Liver Tissue Sample a | Histological Score (0/1/2/3/4)a | MELD Score b,c | Steatosis (0/1/2/3) d | Gender (m/f) | Age | AST [U/L] | ALT | AP [U/L] |
|---|---|---|---|---|---|---|---|---|
| normal (n = 15) | (15/0/0/0/0) | 7.3 ± 1.3 | (15/0/0/0/0) | (4/11) | 57.1 ± 12.8 | 30.9 ± 13.7 | 27.6 ± 15.5 | 111.3 ± 59.2 |
| fibrosis (n = 15) | (0/1/6/8/0) | 8.8 ± 3.3 | (10/4//1/0) | (12/3) | 63.0 ± 11.9 | 48.5 ± 42.4 | 46.7 ± 26.9 | 200.9 ± 190.8 |
| cirrhosis (n = 14) | (0/0/0/0/14) | 15.6 ± 7.3 | (10/1/3) | (5/9) | 51.6 ± 7.9 | 122.0±139.8 | 66.7 ± 80.4 | 211.1 ± 117.1 |
| Antibody | Cat. No. | Dilution | Supplier |
|---|---|---|---|
| APP, rabbit pab | SAB3500274 | 1:1000 | Sigma-Aldrich, Taufkirchen, Germany |
| α-SMA, rabbit pab | ab5694 | 1:1000 | Abcam, Cambridge, UK |
| eNOS, rabbit pab | ab95254 | 1:500 | Abcam |
| Neprilysin, rabbit mab, clone EPR2997 | ab79423 | 1:1000 | Abcam |
| MBP, mouse mab clone MBP101 | ab62631 | 1:500 | Abcam |
| nNOS, rabbit pab | ab5586 | 1:1000 | Abcam |
| PS1, rabbit pab | PA5-20376 | 1:750 | Thermo Fisher Scientific, MA, USA |
| BACE1, rabbit pab | PA1-757 | 1:1000 | Thermo Fisher Scientific |
| GAPDH, mouse mab, clone 6C5 | MAB374 | 1:500 | EMD Millipore, Billerica |
| Target Gene | Gene Symbol | RefSeq | Assay ID |
|---|---|---|---|
| α-SMA | Acta2 | NM_031004.2 | Rn01759928_g1 |
| GFAP | Gfap | NM_017009.2 | Rn00566603_m1 |
| α-SMA | Acta2 | NM_007392.3 | Mm01204962_gH |
| GAPDH | Gapdh | NM_008084.3/NM_001289726.1 | Mm99999915_g1 |
| Antibody | Cat. No | Dilution | Supplier |
|---|---|---|---|
| Desmin mouse, mab, clone DE-U-10 | D 1033 | 1:80 | Sigma-Aldrich, Taufkirchen, Germany |
| GFAP mouse, mab, clone GF12.24 | GF 12.24 | 1:10 | ProGen, Heidelberg, Germany |
| GFAP rabbit, pab | Z 0334 | 1:300 | Dako, Glostrup, Denmark |
| NEP rabbit, mab, clone EPR2997 | ab 79423 | 1:200 | Abcam, Cambridge, UK |
| α-SMA mouse, mab, clone ASM-1 | 61001 | 1:100 | ProGen |
| eNOS, rabbit pab | ab 95254 | 1:150 | Abcam |
| Amyloid β42, mab clone D3E10 | 12843 | 1:500 | Cell Signaling, Frankfurt am Main, Germany |
| FITC-conjugated goat antirabbit IgG | 111-095-144 | 1:100 | Dianova, Hamburg, Germany |
| FITC-conjugated goat antimouse IgG | 115-095-003 | 1:100 | Dianova |
| Cy3-conjugated goat antimouse IgG | 111-165-144 | 1:800 | Dianova |
| Cy3-conjugated goat antirabbit IgG | 115-165-166 | 1:800 | Dianova |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buniatian, G.H.; Weiskirchen, R.; Weiss, T.S.; Schwinghammer, U.; Fritz, M.; Seferyan, T.; Proksch, B.; Glaser, M.; Lourhmati, A.; Buadze, M.; et al. Antifibrotic Effects of Amyloid-Beta and Its Loss in Cirrhotic Liver. Cells 2020, 9, 452. https://doi.org/10.3390/cells9020452
Buniatian GH, Weiskirchen R, Weiss TS, Schwinghammer U, Fritz M, Seferyan T, Proksch B, Glaser M, Lourhmati A, Buadze M, et al. Antifibrotic Effects of Amyloid-Beta and Its Loss in Cirrhotic Liver. Cells. 2020; 9(2):452. https://doi.org/10.3390/cells9020452
Chicago/Turabian StyleBuniatian, Gayane Hrachia, Ralf Weiskirchen, Thomas S. Weiss, Ute Schwinghammer, Martin Fritz, Torgom Seferyan, Barbara Proksch, Michael Glaser, Ali Lourhmati, Marine Buadze, and et al. 2020. "Antifibrotic Effects of Amyloid-Beta and Its Loss in Cirrhotic Liver" Cells 9, no. 2: 452. https://doi.org/10.3390/cells9020452
APA StyleBuniatian, G. H., Weiskirchen, R., Weiss, T. S., Schwinghammer, U., Fritz, M., Seferyan, T., Proksch, B., Glaser, M., Lourhmati, A., Buadze, M., Borkham-Kamphorst, E., Gaunitz, F., Gleiter, C. H., Lang, T., Schaeffeler, E., Tremmel, R., Cynis, H., Frey, W. H., II, Gebhardt, R., ... Danielyan, L. (2020). Antifibrotic Effects of Amyloid-Beta and Its Loss in Cirrhotic Liver. Cells, 9(2), 452. https://doi.org/10.3390/cells9020452