The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review


Abstract
:1. Introduction
2. Genetic Risk Factor of RA
2.1. HLA
2.2. PTPN22
2.3. PADI4
2.4. Important Considerations for a Genetic Study
3. Cigarette Smoking as the Most Robust Environmental Risk of RA
3.1. Effects of Intensity and Duration of Cigarette Smoking and Smoking Cessation
3.2. Effects of Passive Smoking
3.3. Effects of Cigarette Smoking on RA-Related Autoantibody Production
3.4. Other Environmetal Risks Augmented by Cigarette Smoking
3.4.1. Occupational Silica Exposure
3.4.2. Alcohol Consumption
3.4.3. Sugar-Sweetened Soda Consumption
3.4.4. High Salt Intake
4. Impacts of Cigarette Smoking on RA Pathogenesis
4.1. Effects of Cigarette Smoking on Immune Systems
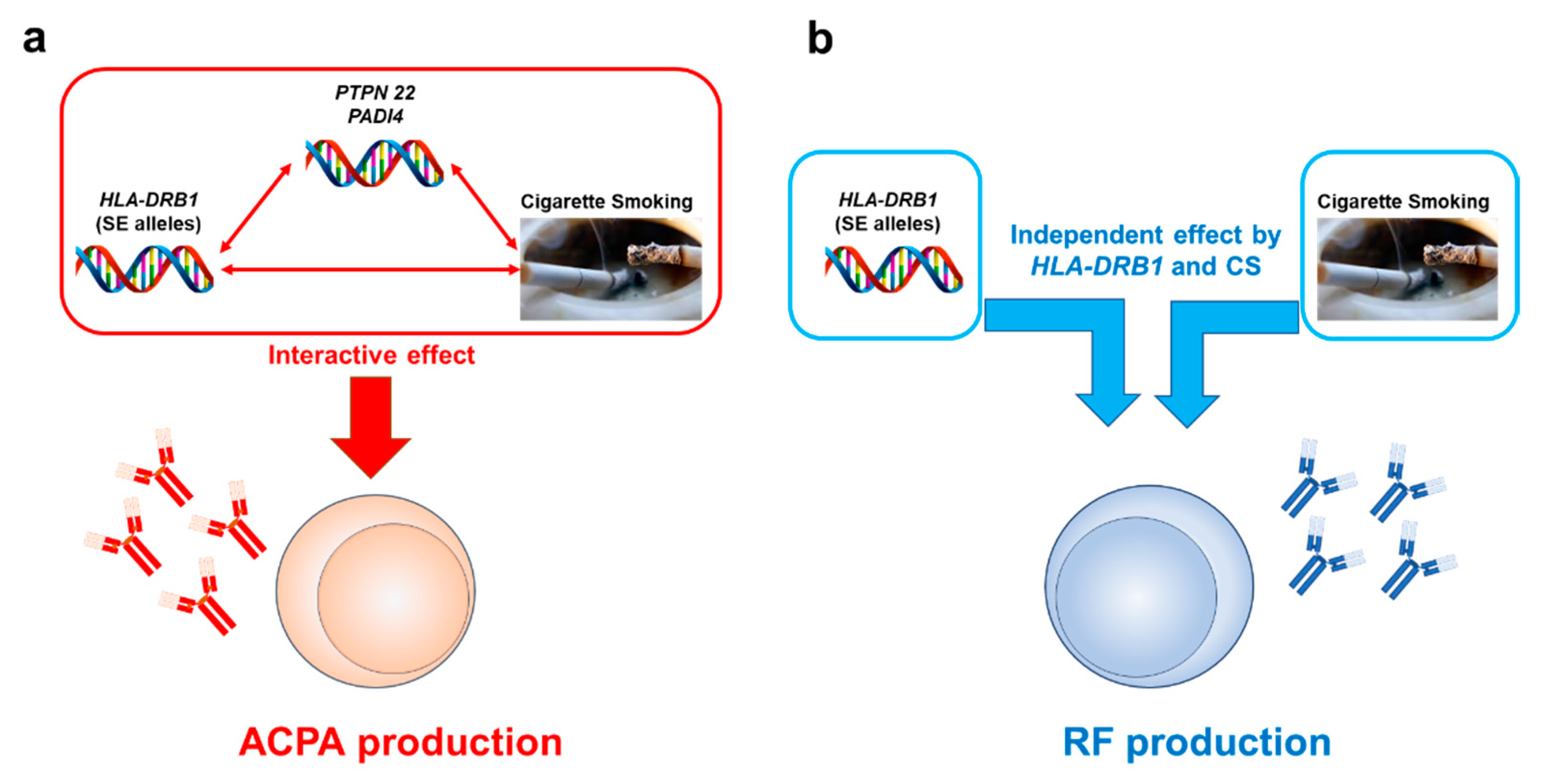
4.2. Interactive Effects of Cigarette Smoking and Genetic Components
4.2.1. Interactive Effects between CS and the HLA-DRB1 Gene on RA Development
4.2.2. Interactive Effects between CS and the HLA-DRB1 Gene on RA-Related Autoantibody Production
4.2.3. Interactive Effects between CS and the PTPN22 Gene on RA Pathology
4.2.4. Interactive Effects between CS and the PADI4 Gene on RA Pathology
4.3. Effects of CS on Epigenetic Changes
4.4. Cigarette Smoking Modulates Periodontal Disease Leading to a Higher Risk of RA Development
4.5. Airway Inflammation
4.6. The Role of Passive Smoking in RA Pathogenesis and the Role of Nicotine
5. Concluding Remarks and Future Directions of Studies
Author Contributions
Conflicts of Interest
References
- Calabresi, E.; Petrelli, F.; Bonifacio, A.F.; Puxeddu, I.; Alunno, A. One year in review 2018: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2018, 36, 175–184. [Google Scholar] [PubMed]
- Okada, Y.; Eyre, S.; Suzuki, A.; Kochi, Y.; Yamamoto, K. Genetics of rheumatoid arthritis: 2018 status. Ann. Rheum. Dis. 2019, 78, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukhari, M.; Lunt, M.; Harrison, B.J.; Scott, D.G.; Symmons, D.P.; Silman, A.J. Rheumatoid factor is the major predictor of increasing severity of radiographic erosions in rheumatoid arthritis: Results from the Norfolk Arthritis Register Study, a large inception cohort. Arthritis Rheum. 2002, 46, 906–912. [Google Scholar] [CrossRef]
- Terao, C.; Yamakawa, N.; Yano, K.; Markusse, I.M.; Ikari, K.; Yoshida, S.; Furu, M.; Hashimoto, M.; Ito, H.; Fujii, T.; et al. Rheumatoid Factor Is Associated with the Distribution of Hand Joint Destruction in Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 3113–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syversen, S.W.; Goll, G.L.; van der Heijde, D.; Landewé, R.; Lie, B.A.; Odegård, S.; Uhlig, T.; Gaarder, P.I.; Kvien, T.K. Prediction of radiographic progression in rheumatoid arthritis and the role of antibodies against mutated citrullinated vimentin: Results from a 10-year prospective study. Ann. Rheum. Dis. 2010, 69, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, C.; Englbrecht, M.; Rech, J.; Schmidt, S.; Araujo, E.; Engelke, K.; Finzel, S.; Schett, G. Additive effect of anti-citrullinated protein antibodies and rheumatoid factor on bone erosions in patients with RA. Ann. Rheum. Dis. 2015, 74, 2151–2156. [Google Scholar] [CrossRef] [Green Version]
- Akdemir, G.; Verheul, M.K.; Heimans, L.; Wevers-de Boer, K.V.; Goekoop-Ruiterman, Y.P.; van Oosterhout, M.; Harbers, J.B.; Bijkerk, C.; Steup-Beekman, G.M.; Lard, L.R.; et al. Predictive factors of radiological progression after 2 years of remission-steered treatment in early arthritis patients: A post hoc analysis of the IMPROVED study. RMD Open 2016, 2, e000172. [Google Scholar] [CrossRef] [Green Version]
- Orsolini, G.; Caimmi, C.; Viapiana, O.; Idolazzi, L.; Fracassi, E.; Gatti, D.; Adami, G.; Rossini, M. Titer-Dependent Effect of Anti-Citrullinated Protein Antibodies on Systemic Bone Mass in Rheumatoid Arthritis Patients. Calcif. Tissue Int. 2017, 101, 17–23. [Google Scholar] [CrossRef]
- Bugatti, S.; Bogliolo, L.; Vitolo, B.; Manzo, A.; Montecucco, C.; Caporali, R. Anti-citrullinated protein antibodies and high levels of rheumatoid factor are associated with systemic bone loss in patients with early untreated rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 226. [Google Scholar] [CrossRef] [Green Version]
- Katchamart, W.; Koolvisoot, A.; Aromdee, E.; Chiowchanwesawakit, P.; Muengchan, C. Associations of rheumatoid factor and anti-citrullinated peptide antibody with disease progression and treatment outcomes in patients with rheumatoid arthritis. Rheumatol. Int. 2015, 35, 1693–1699. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Rönnelid, J.; Wick, M.C.; Lampa, J.; Lindblad, S.; Nordmark, B.; Klareskog, L.; van Vollenhoven, R.F. Longitudinal analysis of citrullinated protein/peptide antibodies (anti-CP) during 5 year follow up in early rheumatoid arthritis: Anti-CP status predicts worse disease activity and greater radiological progression. Ann. Rheum. Dis. 2005, 64, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, L.B.; Lillegraven, S.; Aga, A.B.; Sexton, J.; Olsen, I.C.; Lie, E.; Berner Hammer, H.; Uhlig, T.; van der Heijde, D.; Kvien, T.K.; et al. Comparing the disease course of patients with seronegative and seropositive rheumatoid arthritis fulfilling the 2010 ACR/EULAR classification criteria in a treat-to-target setting: 2-year data from the ARCTIC trial. RMD Open 2018, 4, e000752. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Raychaudhuri, S.; Gregersen, P.K. Recent Advances in Defining the Genetic Basis of Rheumatoid Arthritis. Annu. Rev. Genom. Hum. Genet. 2016, 17, 273–301. [Google Scholar] [CrossRef]
- Terao, C.; Ikari, K.; Nakayamada, S.; Takahashi, Y.; Yamada, R.; Ohmura, K.; Hashimoto, M.; Furu, M.; Ito, H.; Fujii, T.; et al. A twin study of rheumatoid arthritis in the Japanese population. Mod. Rheumatol. 2016, 26, 685–689. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000, 43, 30–37. [Google Scholar] [CrossRef]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef]
- Okada, Y.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Kawaguchi, T.; Stahl, E.A.; Kurreeman, F.A.; Nishida, N.; et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat. Genet. 2012, 44, 511–516. [Google Scholar] [CrossRef]
- Eyre, S.; Bowes, J.; Diogo, D.; Lee, A.; Barton, A.; Martin, P.; Zhernakova, A.; Stahl, E.; Viatte, S.; McAllister, K.; et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 2012, 44, 1336–1340. [Google Scholar] [CrossRef]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Kurreeman, F.A.; Stahl, E.A.; Okada, Y.; Liao, K.; Diogo, D.; Raychaudhuri, S.; Freudenberg, J.; Kochi, Y.; Patsopoulos, N.A.; Gupta, N.; et al. Use of a multiethnic approach to identify rheumatoid- arthritis-susceptibility loci, 1p36 and 17q12. Am. J. Hum. Genet. 2012, 90, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Bang, S.Y.; Ikari, K.; Yoo, D.H.; Cho, S.K.; Choi, C.B.; Sung, Y.K.; Kim, T.H.; Jun, J.B.; Kang, Y.M.; et al. Association-heterogeneity mapping identifies an Asian-specific association of the GTF2I locus with rheumatoid arthritis. Sci. Rep. 2016, 6, 27563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Muramatsu, T.; Suita, N.; Kanai, M.; Kawakami, E.; Iotchkova, V.; Soranzo, N.; Inazawa, J.; Tanaka, T. Significant impact of miRNA-target gene networks on genetics of human complex traits. Sci. Rep. 2016, 6, 22223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaue, S.; Hirata, J.; Maeda, Y.; Kawakami, E.; Nii, T.; Kishikawa, T.; Ishigaki, K.; Terao, C.; Suzuki, K.; Akiyama, M.; et al. Integration of genetics and miRNA-target gene network identified disease biology implicated in tissue specificity. Nucleic Acids Res. 2018, 46, 11898–11909. [Google Scholar] [CrossRef] [Green Version]
- De Silvestri, A.; Capittini, C.; Poddighe, D.; Marseglia, G.L.; Mascaretti, L.; Bevilacqua, E.; Scotti, V.; Rebuffi, C.; Pasi, A.; Martinetti, M.; et al. HLA-DRB1 alleles and juvenile idiopathic arthritis: Diagnostic clues emerging from a meta-analysis. Autoimmun. Rev. 2017, 16, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Nigrovic, P.A.; Raychaudhuri, S.; Thompson, S.D. Review: Genetics and the Classification of Arthritis in Adults and Children. Arthritis Rheumatol. 2018, 70, 7–17. [Google Scholar] [CrossRef] [Green Version]
- van der Woude, D.; Houwing-Duistermaat, J.J.; Toes, R.E.; Huizinga, T.W.; Thomson, W.; Worthington, J.; van der Helm-van Mil, A.H.; de Vries, R.R. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 2009, 60, 916–923. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- Raychaudhuri, S.; Sandor, C.; Stahl, E.A.; Freudenberg, J.; Lee, H.S.; Jia, X.; Alfredsson, L.; Padyukov, L.; Klareskog, L.; Worthington, J.; et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012, 44, 291–296. [Google Scholar] [CrossRef]
- Han, B.; Diogo, D.; Eyre, S.; Kallberg, H.; Zhernakova, A.; Bowes, J.; Padyukov, L.; Okada, Y.; González-Gay, M.A.; Rantapää-Dahlqvist, S.; et al. Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am. J. Hum. Genet. 2014, 94, 522–532. [Google Scholar] [CrossRef] [Green Version]
- Ohmura, K.; Terao, C.; Maruya, E.; Katayama, M.; Matoba, K.; Shimada, K.; Murasawa, A.; Honjo, S.; Takasugi, K.; Tohma, S.; et al. Anti-citrullinated peptide antibody-negative RA is a genetically distinct subset: A definitive study using only bone-erosive ACPA-negative rheumatoid arthritis. Rheumatology 2010, 49, 2298–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terao, C.; Ikari, K.; Ohmura, K.; Suzuki, T.; Iwamoto, T.; Takasugi, K.; Saji, H.; Taniguchi, A.; Momohara, S.; Yamanaka, H.; et al. Quantitative effect of HLA-DRB1 alleles to ACPA levels in Japanese rheumatoid arthritis: No strong genetic impact of shared epitope to ACPA levels after stratification of HLA-DRB1*09:01. Ann. Rheum. Dis. 2012, 71, 1095–1097. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Suzuki, A.; Ikari, K.; Kochi, Y.; Ohmura, K.; Katayama, M.; Nakabo, S.; Yamamoto, N.; Suzuki, T.; Iwamoto, T.; et al. An association between amino acid position 74 of HLA-DRB1 and anti-citrullinated protein antibody levels in Japanese patients with anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Hiwa, R.; Ikari, K.; Ohmura, K.; Nakabo, S.; Matsuo, K.; Saji, H.; Yurugi, K.; Miura, Y.; Maekawa, T.; Taniguchi, A.; et al. Analysis Identified a Genetically Unique Subset within Rheumatoid Arthritis and Distinct Genetic Background of Rheumatoid Factor Levels from Anticyclic Citrullinated Peptide Antibodies. J. Rheumatol. 2018, 45, 470–480. [Google Scholar] [CrossRef]
- Kochi, Y.; Yamada, R.; Kobayashi, K.; Takahashi, A.; Suzuki, A.; Sekine, A.; Mabuchi, A.; Akiyama, F.; Tsunoda, T.; Nakamura, Y.; et al. Analysis of single-nucleotide polymorphisms in Japanese rheumatoid arthritis patients shows additional susceptibility markers besides the classic shared epitope susceptibility sequences. Arthritis Rheum. 2004, 50, 63–71. [Google Scholar] [CrossRef]
- Padyukov, L.; Seielstad, M.; Ong, R.T.; Ding, B.; Rönnelid, J.; Seddighzadeh, M.; Alfredsson, L.; Klareskog, L.; Epidemiological Investigation of Rheumatoid Arthritis (EIRA) Study Group. A genome-wide association study suggests contrasting associations in ACPA-positive versus ACPA-negative rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 259–265. [Google Scholar] [CrossRef]
- Okada, Y.; Kim, K.; Han, B.; Pillai, N.E.; Ong, R.T.; Saw, W.Y.; Luo, M.; Jiang, L.; Yin, J.; Bang, S.Y.; et al. Risk for ACPA-positive rheumatoid arthritis is driven by shared HLA amino acid polymorphisms in Asian and European populations. Hum. Mol. Genet. 2014, 23, 6916–6926. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Suzuki, A.; Ikari, K.; Terao, C.; Kochi, Y.; Ohmura, K.; Higasa, K.; Akiyama, M.; Ashikawa, K.; Kanai, M.; et al. Contribution of a Non-classical HLA Gene, HLA-DOA, to the Risk of Rheumatoid Arthritis. Am. J. Hum. Genet. 2016, 99, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Vang, T.; Miletic, A.V.; Bottini, N.; Mustelin, T. Protein tyrosine phosphatase PTPN22 in human autoimmunity. Autoimmunity 2007, 40, 453–461. [Google Scholar] [CrossRef]
- Burn, G.L.; Svensson, L.; Sanchez-Blanco, C.; Saini, M.; Cope, A.P. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? FEBS Lett. 2011, 585, 3689–3698. [Google Scholar] [CrossRef] [Green Version]
- Nabi, G.; Akhter, N.; Wahid, M.; Bhatia, K.; Mandal, R.K.; Dar, S.A.; Jawed, A.; Haque, S. Meta-analysis reveals PTPN22 1858C/T polymorphism confers susceptibility to rheumatoid arthritis in Caucasian but not in Asian population. Autoimmunity 2016, 49, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K.; Lee, H.S.; Batliwalla, F.; Begovich, A.B. PTPN22: Setting thresholds for autoimmunity. Semin. Immunol. 2006, 18, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Rieck, M.; Arechiga, A.; Onengut-Gumuscu, S.; Greenbaum, C.; Concannon, P.; Buckner, J.H. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J. Immunol. 2007, 179, 4704–4710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vang, T.; Landskron, J.; Viken, M.K.; Oberprieler, N.; Torgersen, K.M.; Mustelin, T.; Tasken, K.; Tautz, L.; Rickert, R.C.; Lie, B.A. The autoimmune-predisposing variant of lymphoid tyrosine phosphatase favors T helper 1 responses. Hum. Immunol. 2013, 74, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milla, V.; Mahfuz, G.A.; Tyyne, V.; Emmi-Leena, I.; Ilse, E.; Jorma, T.; Mikael, K.; Riitta, V.; Jorma, I.; Johanna, L.; et al. Type 1 diabetes linked PTPN22 gene polymorphism is associated with the frequency of circulating regulatory T cells. Eur. J. Immunol. 2019. [Google Scholar] [CrossRef]
- Wang, Y.; Ewart, D.; Crabtree, J.N.; Yamamoto, A.; Baechler, E.C.; Fazeli, P.; Peterson, E.J. PTPN22 Variant R620W Is Associated with Reduced Toll-like Receptor 7-Induced Type I Interferon in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2015, 67, 2403–2414. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.; Dwivedi, N.; Nicholas, A.P.; Ho, I.C. The W620 Polymorphism in PTPN22 Disrupts Its Interaction with Peptidylarginine Deiminase Type 4 and Enhances Citrullination and NETosis. Arthritis Rheumatol. 2015, 67, 2323–2334. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamada, R.; Chang, X.; Tokuhiro, S.; Sawada, T.; Suzuki, M.; Nagasaki, M.; Nakayama-Hamada, M.; Kawaida, R.; Ono, M.; et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 2003, 34, 395–402. [Google Scholar] [CrossRef]
- Engström, M.; Eriksson, K.; Lee, L.; Hermansson, M.; Johansson, A.; Nicholas, A.P.; Gerasimcik, N.; Lundberg, K.; Klareskog, L.; Catrina, A.I.; et al. Increased citrullination and expression of peptidylarginine deiminases independently of P. gingivalis and A. actinomycetemcomitans in gingival tissue of patients with periodontitis. J. Transl. Med. 2018, 16, 214. [Google Scholar] [CrossRef]
- Cantaert, T.; De Rycke, L.; Bongartz, T.; Matteson, E.L.; Tak, P.P.; Nicholas, A.P.; Baeten, D. Citrullinated proteins in rheumatoid arthritis: Crucial … but not sufficient! Arthritis Rheum. 2006, 54, 3381–3389. [Google Scholar] [CrossRef]
- Di Giuseppe, D.; Discacciati, A.; Orsini, N.; Wolk, A. Cigarette smoking and risk of rheumatoid arthritis: A dose-response meta-analysis. Arthritis Res. Ther. 2014, 16, R61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedström, A.K.; Stawiarz, L.; Klareskog, L.; Alfredsson, L. Smoking and susceptibility to rheumatoid arthritis in a Swedish population-based case-control study. Eur. J. Epidemiol. 2018, 33, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrente-Segarra, V.; Bergstra, S.A.; Solomon-Escoto, K.; Da Silva, J.; Veale, D.J.; Al-Emadi, S.; Huizinga, T. Is current smoking status and its relationship to anti-cyclic citrullinated peptide antibodies a predictor of worse response to biological therapies in rheumatoid arthritis patients? Scand. J. Rheumatol. 2018, 47, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Rydell, E.; Forslind, K.; Nilsson, J.; Jacobsson, L.T.H.; Turesson, C. Smoking, body mass index, disease activity, and the risk of rapid radiographic progression in patients with early rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivas, F.; Yurdakul, F.G.; Kiliçarslan, A.; Duran, S.; Başkan, B.; Bodur, H. Relationship Between Smoking and Structural Damage, Autoimmune Antibodies, and Disability in Rheumatoid Arthritis Patients. Arch. Rheumatol. 2018, 33, 45–51. [Google Scholar] [CrossRef]
- Vittecoq, O.; Richard, L.; Banse, C.; Lequerré, T. The impact of smoking on rheumatoid arthritis outcomes. Joint Bone Spine 2018, 85, 135–138. [Google Scholar] [CrossRef]
- Hedström, A.K.; Klareskog, L.; Alfredsson, L. Exposure to passive smoking and rheumatoid arthritis risk: Results from the Swedish EIRA study. Ann. Rheum. Dis. 2018, 77, 970–972. [Google Scholar] [CrossRef]
- Seror, R.; Henry, J.; Gusto, G.; Aubin, H.J.; Boutron-Ruault, M.C.; Mariette, X. Passive smoking in childhood increases the risk of developing rheumatoid arthritis. Rheumatology 2018. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Choe, J.Y. Passive Smoking is Responsible for Disease Activity in Female Patients with Rheumatoid Arthritis. Arch. Rheumatol. 2018, 33, 143–149. [Google Scholar] [CrossRef]
- Hammam, N.; Gheita, T.A. Impact of secondhand smoking on disease activity in women with rheumatoid arthritis. Clin. Rheumatol. 2017, 36, 2415–2420. [Google Scholar] [CrossRef]
- Stolt, P.; Bengtsson, C.; Nordmark, B.; Lindblad, S.; Lundberg, I.; Klareskog, L.; Alfredsson, L. Quantification of the influence of cigarette smoking on rheumatoid arthritis: Results from a population based case-control study, using incident cases. Ann. Rheum. Dis. 2003, 62, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Kronzer, V.L.; Crowson, C.S.; Sparks, J.A.; Vassallo, R.; Davis, J.M. Investigating Asthma, Allergic Disease, Passive Smoke Exposure, and Risk of Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1217–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wesemael, T.J.; Ajeganova, S.; Humphreys, J.; Terao, C.; Muhammad, A.; Symmons, D.P.; MacGregor, A.J.; Hafström, I.; Trouw, L.A.; van der Helm-van Mil, A.H.; et al. Smoking is associated with the concurrent presence of multiple autoantibodies in rheumatoid arthritis rather than with anti-citrullinated protein antibodies per se: A multicenter cohort study. Arthritis Res. Ther. 2016, 18, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Ikari, K.; Hashimoto, M.; Ohmura, K.; Tanaka, M.; Ito, H.; Taniguchi, A.; Yamanaka, H.; Mimori, T.; Terao, C. Shared epitope defines distinct associations of cigarette smoking with levels of anticitrullinated protein antibody and rheumatoid factor. Ann. Rheum. Dis. 2019, 78, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Yahya, A.; Bengtsson, C.; Källberg, H.; Rönnelid, J.; Lundberg, I.; Klareskog, L.; Alfredsson, L.; Group, E.S. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1072–1076. [Google Scholar] [CrossRef] [Green Version]
- Blanc, P.D.; Järvholm, B.; Torén, K. Prospective risk of rheumatologic disease associated with occupational exposure in a cohort of male construction workers. Am. J. Med. 2015, 128, 1094–1101. [Google Scholar] [CrossRef]
- Pollard, K.M. Silica, Silicosis, and Autoimmunity. Front. Immunol. 2016, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, I.M.; Verdrengh, M.; Brisslert, M.; Lindblad, S.; Bokarewa, M.; Islander, U.; Carlsten, H.; Ohlsson, C.; Nandakumar, K.S.; Holmdahl, R.; et al. Ethanol prevents development of destructive arthritis. Proc. Natl. Acad. Sci. USA 2007, 104, 258–263. [Google Scholar] [CrossRef] [Green Version]
- Scott, I.C.; Tan, R.; Stahl, D.; Steer, S.; Lewis, C.M.; Cope, A.P. The protective effect of alcohol on developing rheumatoid arthritis: A systematic review and meta-analysis. Rheumatology 2013, 52, 856–867. [Google Scholar] [CrossRef] [Green Version]
- Nissen, M.J.; Gabay, C.; Scherer, A.; Finckh, A.; Swiss Clinical Quality Management Project in Rheumatoid Arthritis. The effect of alcohol on radiographic progression in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 1265–1272. [Google Scholar] [CrossRef]
- Di Giuseppe, D.; Wallin, A.; Bottai, M.; Askling, J.; Wolk, A. Long-term intake of dietary long-chain n-3 polyunsaturated fatty acids and risk of rheumatoid arthritis: A prospective cohort study of women. Ann. Rheum. Dis. 2014, 73, 1949–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Källberg, H.; Jacobsen, S.; Bengtsson, C.; Pedersen, M.; Padyukov, L.; Garred, P.; Frisch, M.; Karlson, E.W.; Klareskog, L.; Alfredsson, L. Alcohol consumption is associated with decreased risk of rheumatoid arthritis: Results from two Scandinavian case-control studies. Ann. Rheum. Dis. 2009, 68, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Costenbader, K.H.; Gao, X.; Al-Daabil, M.; Sparks, J.A.; Solomon, D.H.; Hu, F.B.; Karlson, E.W.; Lu, B. Sugar-sweetened soda consumption and risk of developing rheumatoid arthritis in women. Am. J. Clin. Nutr. 2014, 100, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Sopori, M. Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2002, 2, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Harrison, O.J.; Foley, J.; Bolognese, B.J.; Long, E.; Podolin, P.L.; Walsh, P.T. Airway infiltration of CD4+ CCR6+ Th17 type cells associated with chronic cigarette smoke induced airspace enlargement. Immunol. Lett. 2008, 121, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Cozen, W.; Diaz-Sanchez, D.; James Gauderman, W.; Zadnick, J.; Cockburn, M.G.; Gill, P.S.; Masood, R.; Hamilton, A.S.; Jyrala, M.; Mack, T.M. Th1 and Th2 cytokines and IgE levels in identical twins with varying levels of cigarette consumption. J. Clin. Immunol. 2004, 24, 617–622. [Google Scholar] [CrossRef]
- Whetzel, C.A.; Corwin, E.J.; Klein, L.C. Disruption in Th1/Th2 immune response in young adult smokers. Addict. Behav. 2007, 32, 1–8. [Google Scholar] [CrossRef]
- Reckner Olsson, A.; Skogh, T.; Wingren, G. Comorbidity and lifestyle, reproductive factors, and environmental exposures associated with rheumatoid arthritis. Ann. Rheum. Dis. 2001, 60, 934–939. [Google Scholar] [CrossRef] [Green Version]
- Kero, J.; Gissler, M.; Hemminki, E.; Isolauri, E. Could TH1 and TH2 diseases coexist? Evaluation of asthma incidence in children with coeliac disease, type 1 diabetes, or rheumatoid arthritis: A register study. J. Allergy Clin. Immunol. 2001, 108, 781–783. [Google Scholar] [CrossRef]
- Hemminki, K.; Li, X.; Sundquist, J.; Sundquist, K. Subsequent autoimmune or related disease in asthma patients: Clustering of diseases or medical care? Ann. Epidemiol. 2010, 20, 217–222. [Google Scholar] [CrossRef]
- Karatay, S.; Yildirim, K.; Ugur, M.; Senel, K.; Erdal, A.; Durmus, B.; Baysal, O.; Altay, Z.; Sarac, A.J.; Gur, A.; et al. Prevalence of atopic disorders in rheumatic diseases. Mod. Rheumatol. 2013, 23, 351–356. [Google Scholar] [CrossRef] [PubMed]
- de Roos, A.J.; Cooper, G.S.; Alavanja, M.C.; Sandler, D.P. Personal and family medical history correlates of rheumatoid arthritis. Ann. Epidemiol. 2008, 18, 433–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, W.U.; Keaney, N.P.; Holland, C.D.; Kelly, C.A. Bronchial reactivity and airflow obstruction in rheumatoid arthritis. Ann. Rheum. Dis. 1994, 53, 511–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provenzano, G.; Donato, G.; Brai, G.; Rinaldi, F. Prevalence of allergic respiratory diseases in patients with RA. Ann. Rheum. Dis. 2002, 61, 281. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.A.; Park, S.H.; Cha, H.S.; Park, W.; Kim, H.A.; Yoo, D.H.; Baek, H.J.; Lee, S.G.; Lee, Y.J.; Park, Y.B.; et al. Prevalence of co-morbidities and evaluation of their monitoring in Korean patients with rheumatoid arthritis: Comparison with the results of an international, cross-sectional study (COMORA). Int. J. Rheum. Dis. 2018, 21, 1414–1422. [Google Scholar] [CrossRef] [Green Version]
- Dougados, M.; Soubrier, M.; Antunez, A.; Balint, P.; Balsa, A.; Buch, M.H.; Casado, G.; Detert, J.; El-Zorkany, B.; Emery, P.; et al. Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: Results of an international, cross-sectional study (COMORA). Ann. Rheum. Dis. 2014, 73, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Hekking, P.P.; Bel, E.H. Developing and emerging clinical asthma phenotypes. J. Allergy Clin. Immunol. Pract. 2014, 2, 671–680. [Google Scholar] [CrossRef]
- Talbot, J.; Peres, R.S.; Pinto, L.G.; Oliveira, R.D.R.; Lima, K.A.; Donate, P.B.; Silva, J.R.; Ryffel, B.; Cunha, T.M.; Alves-Filho, J.C.; et al. Smoking-induced aggravation of experimental arthritis is dependent of aryl hydrocarbon receptor activation in Th17 cells. Arthritis Res. Ther. 2018, 20, 119. [Google Scholar] [CrossRef] [Green Version]
- Ruschpler, P.; Stiehl, P. Shift in Th1 (IL-2 and IFN-gamma) and Th2 (IL-10 and IL-4) cytokine mRNA balance within two new histological main-types of rheumatoid-arthritis (RA). Cell. Mol. Biol. 2002, 48, 285–293. [Google Scholar]
- Harel-Meir, M.; Sherer, Y.; Shoenfeld, Y. Tobacco smoking and autoimmune rheumatic diseases. Nat. Clin. Pract. Rheumatol. 2007, 3, 707–715. [Google Scholar] [CrossRef]
- Klareskog, L.; Padyukov, L.; Alfredsson, L. Smoking as a trigger for inflammatory rheumatic diseases. Curr. Opin. Rheumatol. 2007, 19, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Tanaka, T. Interleukin 6 and rheumatoid arthritis. Biomed. Res. Int. 2014, 2014, 698313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnson, Y.; Shoenfeld, Y.; Amital, H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J. Autoimmun. 2010, 34, J258–J265. [Google Scholar] [CrossRef] [PubMed]
- Glossop, J.R.; Dawes, P.T.; Mattey, D.L. Association between cigarette smoking and release of tumour necrosis factor alpha and its soluble receptors by peripheral blood mononuclear cells in patients with rheumatoid arthritis. Rheumatology 2006, 45, 1223–1229. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Pérez, I.V.; Sánchez-Hernández, P.E.; Muñoz-Valle, J.F.; Martínez-Bonilla, G.E.; García-Iglesias, T.; González-Díaz, V.; García-Arellano, S.; Cerpa-Cruz, S.; Polanco-Cruz, J.; Ramírez-Dueñas, M.G. Cytokines (IL-15, IL-21, and IFN-γ) in rheumatoid arthritis: Association with positivity to autoantibodies (RF, anti-CCP, anti-MCV, and anti-PADI4) and clinical activity. Clin. Rheumatol. 2019, 38, 3061–3071. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 Update. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Vanoni, F.; Minoia, F.; Malattia, C. Biologics in juvenile idiopathic arthritis: A narrative review. Eur. J. Pediatr. 2017, 176, 1147–1153. [Google Scholar] [CrossRef]
- Tollerud, D.J.; Weiss, S.T.; Leung, D.Y. Elevated soluble interleukin-2 receptors in young healthy cigarette smokers: Lack of association with atopy or airways hyperresponsiveness. Int. Arch. Allergy Immunol. 1992, 97, 25–30. [Google Scholar] [CrossRef]
- Tollerud, D.J.; Kurman, C.C.; Nelson, D.L.; Brown, L.M.; Maloney, E.M.; Blattner, W.A. Racial variation in serum-soluble interleukin-2 receptor levels: A population-based study of healthy smokers and nonsmokers. Clin. Immunol. Immunopathol. 1994, 70, 274–279. [Google Scholar] [CrossRef]
- Kuuliala, A.; Nissinen, R.; Kautiainen, H.; Repo, H.; Leirisalo-Repo, M. Low circulating soluble interleukin 2 receptor level predicts rapid response in patients with refractory rheumatoid arthritis treated with infliximab. Ann. Rheum. Dis. 2006, 65, 26–29. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.M.; Crowson, C.S.; Knutson, K.L.; Achenbach, S.J.; Strausbauch, M.A.; Therneau, T.M.; Matteson, E.L.; Gabriel, S.E.; Wettstein, P.J. Longitudinal relationships between rheumatoid factor and cytokine expression by immunostimulated peripheral blood lymphocytes from patients with rheumatoid arthritis: New insights into B-cell activation. Clin. Immunol. 2020, 108342. [Google Scholar] [CrossRef] [PubMed]
- Bidkar, M.; Vassallo, R.; Luckey, D.; Smart, M.; Mouapi, K.; Taneja, V. Cigarette Smoke Induces Immune Responses to Vimentin in both, Arthritis-Susceptible and -Resistant Humanized Mice. PLoS ONE 2016, 11, e0162341. [Google Scholar] [CrossRef] [PubMed]
- Nadigel, J.; Préfontaine, D.; Baglole, C.J.; Maltais, F.; Bourbeau, J.; Eidelman, D.H.; Hamid, Q. Cigarette smoke increases TLR4 and TLR9 expression and induces cytokine production from CD8(+) T cells in chronic obstructive pulmonary disease. Respir. Res. 2011, 12, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saetta, M.; Di Stefano, A.; Turato, G.; Facchini, F.M.; Corbino, L.; Mapp, C.E.; Maestrelli, P.; Ciaccia, A.; Fabbri, L.M. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998, 157, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.Q.; Liu, X.S.; Wang, J.M.; Xu, Y.J. CD8(+) Tc-lymphocytes immunodeviation in peripheral blood and airway from patients of chronic obstructive pulmonary disease and changes after short-term smoking cessation. Chin. Med. J. 2013, 126, 3608–3615. [Google Scholar]
- Chen, G.; Zhou, M.; Chen, L.; Meng, Z.J.; Xiong, X.Z.; Liu, H.J.; Xin, J.B.; Zhang, J.C. Cigarette Smoke Disturbs the Survival of CD8+ Tc/Tregs Partially through Muscarinic Receptors-Dependent Mechanisms in Chronic Obstructive Pulmonary Disease. PLoS ONE 2016, 11, e0147232. [Google Scholar] [CrossRef]
- Koch, A.; Gaczkowski, M.; Sturton, G.; Staib, P.; Schinköthe, T.; Klein, E.; Rubbert, A.; Bacon, K.; Wassermann, K.; Erdmann, E. Modification of surface antigens in blood CD8+ T-lymphocytes in COPD: Effects of smoking. Eur. Respir. J. 2007, 29, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Wasén, C.; Erlandsson, M.C.; Bossios, A.; Ekerljung, L.; Malmhäll, C.; Töyrä Silfverswärd, S.; Pullerits, R.; Lundbäck, B.; Bokarewa, M.I. Smoking Is Associated with Low Levels of Soluble PD-L1 in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1677. [Google Scholar] [CrossRef]
- Wasén, C.; Turkkila, M.; Bossios, A.; Erlandsson, M.; Andersson, K.M.; Ekerljung, L.; Malmhäll, C.; Brisslert, M.; Töyrä Silfverswärd, S.; Lundbäck, B.; et al. Smoking activates cytotoxic CD8. J. Autoimmun. 2017, 78, 101–110. [Google Scholar] [CrossRef]
- Wang, J.; Urbanowicz, R.A.; Tighe, P.J.; Todd, I.; Corne, J.M.; Fairclough, L.C. Differential activation of killer cells in the circulation and the lung: A study of current smoking status and chronic obstructive pulmonary disease (COPD). PLoS ONE 2013, 8, e58556. [Google Scholar] [CrossRef]
- Stolberg, V.R.; Martin, B.; Mancuso, P.; Olszewski, M.A.; Freeman, C.M.; Curtis, J.L.; Chensue, S.W. Role of CC chemokine receptor 4 in natural killer cell activation during acute cigarette smoke exposure. Am. J. Pathol. 2014, 184, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tollerud, D.J.; Clark, J.W.; Brown, L.M.; Neuland, C.Y.; Mann, D.L.; Pankiw-Trost, L.K.; Blattner, W.A.; Hoover, R.N. Association of cigarette smoking with decreased numbers of circulating natural killer cells. Am. Rev. Respir. Dis. 1989, 139, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Moszczyński, P.; Rutowski, J.; Słowiński, S. The effect of cigarettes smoking on the blood counts of T and NK cells in subjects with occupational exposure to organic solvents. Cent. Eur. J. Public Health 1996, 4, 164–168. [Google Scholar] [PubMed]
- Mian, M.F.; Lauzon, N.M.; Stämpfli, M.R.; Mossman, K.L.; Ashkar, A.A. Impairment of human NK cell cytotoxic activity and cytokine release by cigarette smoke. J. Leukoc. Biol. 2008, 83, 774–784. [Google Scholar] [CrossRef]
- Arimilli, S.; Damratoski, B.E.; Prasad, G.L. Combustible and non-combustible tobacco product preparations differentially regulate human peripheral blood mononuclear cell functions. Toxicol. In Vitro 2013, 27, 1992–2004. [Google Scholar] [CrossRef]
- Mian, M.F.; Pek, E.A.; Mossman, K.L.; Stämpfli, M.R.; Ashkar, A.A. Exposure to cigarette smoke suppresses IL-15 generation and its regulatory NK cell functions in poly I:C-augmented human PBMCs. Mol. Immunol. 2009, 46, 3108–3116. [Google Scholar] [CrossRef]
- de Lima, C.A.D.; Rushansky, E.; Adelino, J.E.; de Oliveira Souza, A.P.; d’Emery Alves Santos, P.; de Araújo Mariano, M.H.Q.; Crovella, S.; de Azevêdo Silva, J.; Sandrin-Garcia, P. Are key cytokines genetic and serum levels variations related to rheumatoid arthritis clinical severity? Gene 2020, 722, 144098. [Google Scholar] [CrossRef]
- Ospelt, C.; Bang, H.; Feist, E.; Camici, G.; Keller, S.; Detert, J.; Krämer, A.; Gay, S.; Ghannam, K.; Burmester, G.R. Carbamylation of vimentin is inducible by smoking and represents an independent autoantigen in rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1176–1183. [Google Scholar] [CrossRef] [Green Version]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Källberg, H.; Bengtsson, C.; Grunewald, J.; Rönnelid, J.; Harris, H.E.; Ulfgren, A.K.; Rantapää-Dahlqvist, S.; et al. A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006, 54, 38–46. [Google Scholar] [CrossRef]
- Too, C.L.; Yahya, A.; Murad, S.; Dhaliwal, J.S.; Larsson, P.T.; Muhamad, N.A.; Abdullah, N.A.; Mustafa, A.N.; Klareskog, L.; Alfredsson, L.; et al. Smoking interacts with HLA-DRB1 shared epitope in the development of anti-citrullinated protein antibody-positive rheumatoid arthritis: Results from the Malaysian Epidemiological Investigation of Rheumatoid Arthritis (MyEIRA). Arthritis Res. Ther. 2012, 14, R89. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Irigoyen, P.; Kern, M.; Lee, A.; Batliwalla, F.; Khalili, H.; Wolfe, F.; Lum, R.F.; Massarotti, E.; Weisman, M.; et al. Interaction between smoking, the shared epitope, and anti-cyclic citrullinated peptide: A mixed picture in three large North American rheumatoid arthritis cohorts. Arthritis Rheum. 2007, 56, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.Y.; Lee, K.H.; Cho, S.K.; Lee, H.S.; Lee, K.W.; Bae, S.C. Smoking increases rheumatoid arthritis susceptibility in individuals carrying the HLA-DRB1 shared epitope, regardless of rheumatoid factor or anti-cyclic citrullinated peptide antibody status. Arthritis Rheum. 2010, 62, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.; Mattey, D.; Hutchinson, D. Anti-citrullinated protein antibody positive rheumatoid arthritis is primarily determined by rheumatoid factor titre and the shared epitope rather than smoking per se. PLoS ONE 2017, 12, e0180655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Helm-van Mil, A.H.; Verpoort, K.N.; le Cessie, S.; Huizinga, T.W.; de Vries, R.R.; Toes, R.E. The HLA-DRB1 shared epitope alleles differ in the interaction with smoking and predisposition to antibodies to cyclic citrullinated peptide. Arthritis Rheum. 2007, 56, 425–432. [Google Scholar] [CrossRef]
- Pedersen, M.; Jacobsen, S.; Garred, P.; Madsen, H.O.; Klarlund, M.; Svejgaard, A.; Pedersen, B.V.; Wohlfahrt, J.; Frisch, M. Strong combined gene-environment effects in anti-cyclic citrullinated peptide-positive rheumatoid arthritis: A nationwide case-control study in Denmark. Arthritis Rheum. 2007, 56, 1446–1453. [Google Scholar] [CrossRef]
- Padyukov, L.; Silva, C.; Stolt, P.; Alfredsson, L.; Klareskog, L. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004, 50, 3085–3092. [Google Scholar] [CrossRef]
- Mattey, D.L.; Dawes, P.T.; Clarke, S.; Fisher, J.; Brownfield, A.; Thomson, W.; Hajeer, A.H.; Ollier, W.E. Relationship among the HLA-DRB1 shared epitope, smoking, and rheumatoid factor production in rheumatoid arthritis. Arthritis Rheum. 2002, 47, 403–407. [Google Scholar] [CrossRef]
- Hedström, A.K.; Rönnelid, J.; Klareskog, L.; Alfredsson, L. Complex Relationships of Smoking, HLA-DRB1 Genes, and Serologic Profiles in Patients with Early Rheumatoid Arthritis: Update from a Swedish Population-Based Case-Control Study. Arthritis Rheumatol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Lundström, E.; Källberg, H.; Alfredsson, L.; Klareskog, L.; Padyukov, L. Gene-environment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis: All alleles are important. Arthritis Rheum. 2009, 60, 1597–1603. [Google Scholar] [CrossRef] [Green Version]
- Bang, S.Y.; Lee, H.S.; Lee, K.W.; Bae, S.C. Interaction of HLA-DRB1*09:01 and *04:05 with smoking suggests distinctive mechanisms of rheumatoid arthritis susceptibility beyond the shared epitope. J. Rheumatol. 2013, 40, 1054–1062. [Google Scholar] [CrossRef]
- Westra, H.J.; Martínez-Bonet, M.; Onengut-Gumuscu, S.; Lee, A.; Luo, Y.; Teslovich, N.; Worthington, J.; Martin, J.; Huizinga, T.; Klareskog, L.; et al. Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet. 2018, 50, 1366–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahdi, H.; Fisher, B.A.; Källberg, H.; Plant, D.; Malmström, V.; Rönnelid, J.; Charles, P.; Ding, B.; Alfredsson, L.; Padyukov, L.; et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat. Genet. 2009, 41, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, K.; Bengtsson, C.; Kharlamova, N.; Reed, E.; Jiang, X.; Kallberg, H.; Pollak-Dorocic, I.; Israelsson, L.; Kessel, C.; Padyukov, L.; et al. Genetic and environmental determinants for disease risk in subsets of rheumatoid arthritis defined by the anticitrullinated protein/peptide antibody fine specificity profile. Ann. Rheum. Dis. 2013, 72, 652–658. [Google Scholar] [CrossRef] [Green Version]
- Willemze, A.; van der Woude, D.; Ghidey, W.; Levarht, E.W.; Stoeken-Rijsbergen, G.; Verduyn, W.; de Vries, R.R.; Houwing-Duistermaat, J.J.; Huizinga, T.W.; Trouw, L.A.; et al. The interaction between HLA shared epitope alleles and smoking and its contribution to autoimmunity against several citrullinated antigens. Arthritis Rheum. 2011, 63, 1823–1832. [Google Scholar] [CrossRef]
- Fisher, B.A.; Bang, S.Y.; Chowdhury, M.; Lee, H.S.; Kim, J.H.; Charles, P.; Venables, P.; Bae, S.C. Smoking, the HLA-DRB1 shared epitope and ACPA fine-specificity in Koreans with rheumatoid arthritis: Evidence for more than one pathogenic pathway linking smoking to disease. Ann. Rheum. Dis. 2014, 73, 741–747. [Google Scholar] [CrossRef]
- Kochi, Y.; Thabet, M.M.; Suzuki, A.; Okada, Y.; Daha, N.A.; Toes, R.E.; Huizinga, T.W.; Myouzen, K.; Kubo, M.; Yamada, R.; et al. PADI4 polymorphism predisposes male smokers to rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 512–515. [Google Scholar] [CrossRef]
- Zeilinger, S.; Kühnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef]
- Meng, W.; Zhu, Z.; Jiang, X.; Too, C.L.; Uebe, S.; Jagodic, M.; Kockum, I.; Murad, S.; Ferrucci, L.; Alfredsson, L.; et al. DNA methylation mediates genotype and smoking interaction in the development of anti-citrullinated peptide antibody-positive rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 71. [Google Scholar] [CrossRef] [Green Version]
- Ballestar, E. Epigenetic alterations in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2011, 7, 263–271. [Google Scholar] [CrossRef]
- Viatte, S.; Plant, D.; Raychaudhuri, S. Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 141–153. [Google Scholar] [CrossRef]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Eriksson, K.; Nise, L.; Alfredsson, L.; Catrina, A.I.; Askling, J.; Lundberg, K.; Klareskog, L.; Yucel-Lindberg, T. Seropositivity combined with smoking is associated with increased prevalence of periodontitis in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1236–1238. [Google Scholar] [CrossRef]
- Albandar, J.M.; Streckfus, C.F.; Adesanya, M.R.; Winn, D.M. Cigar, pipe, and cigarette smoking as risk factors for periodontal disease and tooth loss. J. Periodontol. 2000, 71, 1874–1881. [Google Scholar] [CrossRef]
- Potempa, J.; Mydel, P.; Koziel, J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 606–620. [Google Scholar] [CrossRef]
- Janssen, K.M.; Vissink, A.; de Smit, M.J.; Westra, J.; Brouwer, E. Lessons to be learned from periodontitis. Curr. Opin. Rheumatol. 2013, 25, 241–247. [Google Scholar] [CrossRef]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef]
- Lappin, D.F.; Apatzidou, D.; Quirke, A.M.; Oliver-Bell, J.; Butcher, J.P.; Kinane, D.F.; Riggio, M.P.; Venables, P.; McInnes, I.B.; Culshaw, S. Influence of periodontal disease, Porphyromonas gingivalis and cigarette smoking on systemic anti-citrullinated peptide antibody titres. J. Clin. Periodontol. 2013, 40, 907–915. [Google Scholar] [CrossRef]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef] [Green Version]
- Terao, C.; Asai, K.; Hashimoto, M.; Yamazaki, T.; Ohmura, K.; Yamaguchi, A.; Takahashi, K.; Takei, N.; Ishii, T.; Kawaguchi, T.; et al. Significant association of periodontal disease with anti-citrullinated peptide antibody in a Japanese healthy population—The Nagahama study. J. Autoimmun. 2015, 59, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Bello-Gualtero, J.M.; Lafaurie, G.I.; Hoyos, L.X.; Castillo, D.M.; De-Avila, J.; Munevar, J.C.; Unriza, S.; Londoño, J.; Valle-Oñate, R.; Romero-Sánchez, C. Periodontal Disease in Individuals with a Genetic Risk of Developing Arthritis and Early Rheumatoid Arthritis: A Cross-Sectional Study. J. Periodontol. 2016, 87, 346–356. [Google Scholar] [CrossRef]
- Golub, L.M.; Payne, J.B.; Reinhardt, R.A.; Nieman, G. Can systemic diseases co-induce (not just exacerbate) periodontitis? A hypothetical “two-hit” model. J. Dent. Res. 2006, 85, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Marotte, H.; Farge, P.; Gaudin, P.; Alexandre, C.; Mougin, B.; Miossec, P. The association between periodontal disease and joint destruction in rheumatoid arthritis extends the link between the HLA-DR shared epitope and severity of bone destruction. Ann. Rheum. Dis. 2006, 65, 905–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargiulo, A.V.; Robinson, J.; Toto, P.D.; Gargiulo, A.W. Identification of rheumatoid factor in periodontal disease. J. Periodontol. 1982, 53, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Demoruelle, M.K.; Weisman, M.H.; Simonian, P.L.; Lynch, D.A.; Sachs, P.B.; Pedraza, I.F.; Harrington, A.R.; Kolfenbach, J.R.; Striebich, C.C.; Pham, Q.N.; et al. Brief report: Airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: Early injury or initiating site of autoimmunity? Arthritis Rheum. 2012, 64, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Sun, H.; Wang, X.; Koike, T.; Mishima, H.; Ikeda, K.; Watanabe, T.; Ochiai, N.; Fan, J. Association of increased expression of macrophage elastase (matrix metalloproteinase 12) with rheumatoid arthritis. Arthritis Rheum. 2004, 50, 3112–3117. [Google Scholar] [CrossRef] [PubMed]
- Bracke, K.; Cataldo, D.; Maes, T.; Gueders, M.; Noël, A.; Foidart, J.M.; Brusselle, G.; Pauwels, R.A. Matrix metalloproteinase-12 and cathepsin D expression in pulmonary macrophages and dendritic cells of cigarette smoke-exposed mice. Int. Arch. Allergy Immunol. 2005, 138, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Raitio, A.; Tuomas, H.; Kokkonen, N.; Salo, T.; Sorsa, T.; Hanemaaijer, R.; Oikarinen, A. Levels of matrix metalloproteinase-2, -9 and -8 in the skin, serum and saliva of smokers and non-smokers. Arch. Dermatol. Res. 2005, 297, 242–248. [Google Scholar] [CrossRef]
- Xue, M.; McKelvey, K.; Shen, K.; Minhas, N.; March, L.; Park, S.Y.; Jackson, C.J. Endogenous MMP-9 and not MMP-2 promotes rheumatoid synovial fibroblast survival, inflammation and cartilage degradation. Rheumatology 2014, 53, 2270–2279. [Google Scholar] [CrossRef] [Green Version]
- Burke, H.; Leonardi-Bee, J.; Hashim, A.; Pine-Abata, H.; Chen, Y.; Cook, D.G.; Britton, J.R.; McKeever, T.M. Prenatal and passive smoke exposure and incidence of asthma and wheeze: Systematic review and meta-analysis. Pediatrics 2012, 129, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Alfredsson, L.; Klareskog, L.; Bengtsson, C. Smokeless tobacco (moist snuff) use and the risk of developing rheumatoid arthritis: Results from a case-control study. Arthritis Care Res. 2014, 66, 1582–1586. [Google Scholar] [CrossRef]
- Zhou, Y.; Zuo, X.; Li, Y.; Wang, Y.; Zhao, H.; Xiao, X. Nicotine inhibits tumor necrosis factor-α induced IL-6 and IL-8 secretion in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Rheumatol. Int. 2012, 32, 97–104. [Google Scholar] [CrossRef]
- Wu, S.; Luo, H.; Xiao, X.; Zhang, H.; Li, T.; Zuo, X. Attenuation of collagen induced arthritis via suppression on Th17 response by activating cholinergic anti-inflammatory pathway with nicotine. Eur. J. Pharmacol. 2014, 735, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Yang, H.; Ulloa, L.; Al-Abed, Y.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- van Maanen, M.A.; Lebre, M.C.; van der Poll, T.; LaRosa, G.J.; Elbaum, D.; Vervoordeldonk, M.J.; Tak, P.P. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum. 2009, 60, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yang, Y.H.; Rajaiah, R.; Moudgil, K.D. Nicotine-induced differential modulation of autoimmune arthritis in the Lewis rat involves changes in interleukin-17 and anti-cyclic citrullinated peptide antibodies. Arthritis Rheum. 2011, 63, 981–991. [Google Scholar] [CrossRef]
- Vardavas, C.I.; Plada, M.; Tzatzarakis, M.; Marcos, A.; Warnberg, J.; Gomez-Martinez, S.; Breidenassel, C.; Gonzalez-Gross, M.; Tsatsakis, A.M.; Saris, W.H.; et al. Passive smoking alters circulating naïve/memory lymphocyte T-cell subpopulations in children. Pediatr. Allergy Immunol. 2010, 21, 1171–1178. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| First Authors | Study Type | Outcomes | Effects and Effect Sizes | Interaction between CS and Genetic Components | Stratifications | Population, Country, Study Period |
|---|---|---|---|---|---|---|
| Di Giuseppe | Meta-analysis | RF (+) or (−) RA development | Dose-dependent increase of RR (1.26–2.07) up to 40 pack-years; RR 2.47 and 1.58 for RF (+) and (−) RA, respectively | NA | Pack-years; RF | Three cohorts and seven case-control studies; a total of 4552 RA cases |
| Hedström | Case-control | ACPA (+) or (−) RA development | OR 1.9 and 1.3 for ACPA (+) and (−) RA, respectively; a dose-response association (p for trend < 0.0001); cessation > 20 years diminishes the risk of ACPA (−) RA | NA | Never-, ever-, past, current smokers; duration; intensity; pack-years; ACPA | 3655 cases and 5883 matched controls in Sweden |
| Hedström | Case-control | ACPA (+) or (−) RA development | No association between passive smoking and RA risk (OR ~ 1.0 for both ACPA (+) and (−) RA) | NA | Duration of exposure; ACPA | 589 cases and 1764 controls without smoking history |
| Seror | cohort | RA development | Only a suggestive risk of passive smoking (HR1.4–1.7) | NA | Never- or ever-smokers w/or w/o passive CS during childhood | 71,248 French female volunteers prospectively followed since 1990; 371 RA cases |
| Kim | intra-case | Clinical response | Better clinical response in never-smokers than in passive smokers | NA | Never, current, ex-, and passive smokers | 191 female RA cases in South Korea |
| Torrente-Segarra | intra-case | Clinical response | Better clinical response in never- than in passive smokers, which does not result in better drug survival | NA | Smoking status, ACPA | 1349 RA cases from METEOR database between 2006 and 2016 |
| Rydell | intra-case | Radiographic progression | OR 3.17 for RRP in ever-smokers | NA | Never-, current, ever-, and previous smokers | 233 early RA cases during 1995–2005 in Sweden |
| Sivas | intra-case | Disease activity, radiographic score | Higher erosion and joint space narrowing scores in smokers; no correlation of smoking with disease activity | NA | Never-, long-term, and new smokers | 165 Turkish RA cases (129 females) followed between January 2015 and February 2016 |
| van Wesemael | Case-control | RF, ACPA, and anti-CarP Ab presence | Smoking was associated with multiple autoantibody positivity both in non-RA and RA cases (OR 1.32–2.95) | NA | Never- and ever-smokers; ACPA, RF, anit-CarP Ab | 9575 Japanese non-RA subjects; early RA cases from the Netherlands (n = 678), UK (n = 761), and Sweden (n = 795) |
| Ishikawa | intra-case | RF or ACPA positivities and levels | OR of CS 2.06 and 1.29 for high levels of RF and ACPA, respectively | Interactive effect of CS and SE on ACPA levels but not those of RF | Never-smokers, ex- or active smokers at the onset; SE; ACPA; RF | 6239 Japanese RA cases |
| Klareskog | Case-control | ACPA (+) or (−) RA development | Dose-dependent effect of CS on ACPA (+) RA development | Interactive effect between CS and SE on ACPA (+) RA | Never and ever-smokers; pack-years; numbers of SE; RF; ACPA | 913 early RA cases and 1357 controls, Sweden |
| Too | Case-control | ACPA (+) or (−) RA development | OR of CS 4.1 and OR of SE 4.7 for ACPA (+) RA development | Interactive effect between CS and SE on ACPA (+) RA | Never- and ever-smokers; SE; ACPA; RF | 1076 early RA cases and 1612 matched controls, Malaysia, 2005–2009 |
| Lee | intra-case | ACPA (+) or (−) RA development | Correlation between CS and ACPA (+) RA was observed in 2 out 3 cohorts. | Weak interaction between CS and SE for ACPA only in one cohort | Never- and ever-smokers; SE; ACPA; RF | A total of 2476 white patients with RA from three different cohorts, North America |
| Bang | Case-control | ACPA or RF (+) or (−) RA development | OR of ever-smoking 2.22 for ACPA (+) and 2.80 for ACPA (−) RA | Interactive effect of CS and SE both on ACPA (+) and ACPA (−) subsets | Never- and ever-smokers; SE; DRB1*09:01; ACPA; RF | 1482 RA cases and 1119 control subjects, Korea |
| Murphy | intra-case | ACPA or RF (+) or (−) RA development | Strong association between ACPA and RF but not ACPA and CS; no association of CS and ACPA in RF (−) cases | No interaction between CS and SE | Never- and ever-smokers; Pack-years; SE; ACPA; RF | Two different UK RA cohorts (n = 658 and 409) |
| van der Helm-van Mil | cohort | ACPA (+) or (−) RA development | HLA–DRB1*0401, *0404, *0405, or *0408 SE alleles conferred the highest risk of ACPA development | Strongest interaction between CS and *01:01 or *01:02 and *10:01 alleles | Current and past smokers; SE and subsets; ACPA | 977 undifferentiated arthritis cases, Netherland |
| Pedersen | Case-control | ACPA (+) or (−) RA development | No significant effect of CS on SE (−) subjects | Strong interaction between CS and SE for ACPA (+) but not ACPA (−) RA | SE; ACPA; never- and ever-smokers; pack-years; coffee or alcohol consumption; oral contraceptive use | 445 RA cases and 533 age- and sex-matched controls, Denmark, 2002–2004 |
| Padyukov | Case-control | RF (+) or (−) RA development | Neither CS nor SE genes nor the combination increased the risk of RF (−) RA development | Significant interaction between CS and any SE genes on RF (+) RA | Gender, smoking status, and HLA-DRB1 genotypes, RF | RA 858 cases and 1048 controls recruited during 1996 to 2001, Sweden |
| Mattey | intra-case | RF (+) or (−) RA development | OR of ever-smoker for RF (+) RA development 2.2 in ever-smokers | independent effects of CS and SE, HLA-DRB1*04:01, on RF (+) RA | Never-, ever-, current smokers; SE and subsets; RF | 371 RA cases, UK |
| Hedström | Case-control | ACPA or RF (+) or (−) RA development | An independent effect of CS on RF (+) but not on RF (−) RA regardless of ACPA status | Significant interaction between CS and SE on ACPA (+) RA | Never-, ever-, current smokers; SE; ACPA, RF | 3645 cases, 5883 matched controls, Sweden; follow-up on Ref. 17 |
| Lundström | Case-control | ACPA (+) or (−) RA development | Lack of an independent effect of CS on ACPA (+) RA | Significant interaction of CS with all SE genes tested on ACPA (+) RA | Never- or ever-smokers; SE (DRB1*04, *01, and *10); ACPA | RA 1319 cases and 943 controls recruited during 1996 to 2005, Sweden; partially overlapped with Ref. 119 |
| Bang | Case-control | ACPA (+) or (−) RA; ACPA levels | Smokers had a trend of higher ACPA levels than never-smokers without significant difference | Significant interaction of CS with SE but not with *09:01 on ACPA (+) RA | Never- or ever-smokers; SE; DRB1*09:01; ACPA | 1924 RA cases and 1119 control subjects, Korea; partially overlapped with Ref. 115 |
| Mahdi | Intra case and case–control | Anti-CEP-1 Ab response | 43–63% of ACPA (+) cases were anti-CEP-1 Ab (+), and this subset was preferentially linked to HLA-DRB1*04. | Combined effect of CS, PTPN22, and SE on anti-CEP (+) response | Never- or ever-smokers; SE; PTPN22; ACPA; anti-CEP | 1497 cases, Sweden and UK; 1000 cases and 872 controls, Sweden (cases were overlapped) |
| Lundberg | Case-control | Specific ACPA responses | The strongest association of SE, PTPN22, and CS for the RA subset anti-CEP-1 (+) or anti-cVim Ab (+) subsets of RA | Never-, past, and current smokers; SE; PTPN22; ACPA subsets | 1985 cases and 2252 matched controls, Sweden overlapped with Refs. 17, 121 | |
| Willemze | intra-case | Specific ACPA responses | A significant interaction between CS and SE for the presence of ACPA, not restricted to specific citrullinated antigens | Never- and ever-smokers; SE; ACPA subsets; RF; ANA | 661 cases with recent onset (< 2 years), Netherland | |
| Fisher | Case-control | Specific ACPA responses, erosion | CS-SE interaction was associated with all the ACPA (+) subgroups; highest OR in an anti-CCP (+) cVim (+) subset | Never- and ever-smokers; SE and DRB1*09:01; ACPA subsets | 513 cases and 1101 controls, Korea overlapped with Ref. 115 | |
| Kochi | Case-control | RA development | PADI4 SNP (rs1748033) conferred a higher risk in men (OR 1.39) and in ever-smokers (OR 1.25) | The highest risk in male ever-smokers (OR 1.46) | Never- and ever-smokers; PADI4 SNP genotypes; gender; ACPA | 1019 cases/907 controls and 999 cases/1128 controls, Japan; 635 cases/391 controls, Netherland |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. https://doi.org/10.3390/cells9020475
Ishikawa Y, Terao C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells. 2020; 9(2):475. https://doi.org/10.3390/cells9020475
Chicago/Turabian StyleIshikawa, Yuki, and Chikashi Terao. 2020. "The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review" Cells 9, no. 2: 475. https://doi.org/10.3390/cells9020475
APA StyleIshikawa, Y., & Terao, C. (2020). The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells, 9(2), 475. https://doi.org/10.3390/cells9020475